


Abstract
Genomic structural variations (SVs) are pervasive in many types of cancers. Characterizing their underlying mechanisms and potential molecular consequences is crucial for understanding the basic biology of tumorigenesis. Here, we engineered a local assembly-based algorithm (laSV) that detects SVs with high accuracy from paired-end high-throughput genomic sequencing data and pinpoints their breakpoints at single base-pair resolution. By applying laSV to 97 tumor-normal paired genomic sequencing datasets across six cancer types produced by The Cancer Genome Atlas Research Network, we discovered that non-allelic homologous recombination is the primary mechanism for generating somatic SVs in acute myeloid leukemia. This finding contrasts with results for the other five types of solid tumors, in which non-homologous end joining and microhomology end joining are the predominant mechanisms. We also found that the genes recursively mutated by single nucleotide alterations differed from the genes recursively mutated by SVs, suggesting that these two types of genetic alterations play different roles during cancer progression. We further characterized how the gene structures of the oncogene JAK1 and the tumor suppressors KDM6A and RB1 are affected by somatic SVs and discussed the potential functional implications of intergenic SVs.
INTRODUCTION
Genomic structural variations (SVs) such as deletions, insertions, inversions, translocations and tandem duplications are an important class of genetic variations that underlies genomic diversity in a population (1). A deep and comprehensive understanding of the formation mechanism, genomic distribution and functional impacts of SVs is crucial for studying complex diseases such as cancer.
Performing a comprehensive survey of different SV formation mechanisms and their relative contributions across different cancer types is difficult because it entails precise characterization of the sequences across the breakpoints. Despite extensive efforts, accurately detecting SVs with a high resolution still remains a challenge. Most existing SV discovery methods take advantage of three types of signals that are indicative of SVs between the reference genome and the sample genome: changes in the coverage of read pile-up, suggesting copy number alterations (read depth); discordant read pairs with a distance or orientation between the two reads that is inconsistent with the reference genome (read pair); and reads that can be split into parts that align to discontiguous loci in the reference genome (split reads) (2–8). It is algorithmically challenging to integrate information from these sources; furthermore, reads (or parts of a read) that can be aligned to multiple loci in the reference genome may result in spurious SV calls. Some methods such as TIGRA (9) try to pinpoint the breakpoints of predicted SVs by assembling reads mapped to the loci. This approach does not avoid the mapping ambiguities since both the SV predictions and the read selection for assembly are based on aligning short reads to the reference genome. A potential alternative is to perform a reference-free de novo assembly of the sequencing reads first and then compare the contigs with the reference genome. However, conventional de novo assembly methods are not designed for the purpose of SV discovery, especially for samples with a high degree of heterogeneity such as tumor samples. These tools assume that all the reads originate from a single underlying genome and therefore only detect homozygous SVs (10). In this report, we described a de novo local assembly-based SV discovery algorithm, designated laSV, which is able to pinpoint SV breakpoints at a single-nucleotide resolution and estimate the allele frequencies of the detected SVs in the sample.
Double-stranded breaks (DSBs) in genomic DNA are detrimental to the cell, and several DSB repair pathways have therefore evolved to protect the cell from such catastrophic events. These pathways do not repair DSBs perfectly and erroneous repairs are believed to be an important source of SVs (11), especially in cancers. Homologous recombination (HR) is the mechanism most widely used by the cell to repair DSBs, and it requires long stretches of homologous sequences at the breakpoints. When HR occurs between non-allelic regions with high sequence similarity, termed non-allelic homologous recombination (NAHR), structural alterations may ensue (12–14). Mutations in genes that are key components of the HR pathway, such as BRCA1 and BRCA2, are observed in many types of cancers and deemed the major driving force of genomic instability in these cancers. Nonhomologous end joining (NHEJ), however, does not require sequence homology and often generates very short deletions or insertions at the breakpoint. Key players in this pathway include XRCC5/6 and TP53 (15,16). An alternative pathway, known as microhomology-mediated end joining (MMEJ), plays an active role in some cancers (17,18). MMEJ relies on relatively short stretches of homologous sequence (≤25 bp) at the breakpoint (19). The molecular details of this pathway are much less well understood compared with NAHR and NHEJ, although it is known to share the initial end resection step with NAHR (19). In another DNA replication-associated repair mechanism, known as fork stalling and template switching (FoSTeS), it has been proposed that when a replication fork is stalled during replication, the polymerase is able to switch to a nearby locus and use it as the template to continue replicating, which often results in complex rearrangements (18,20).
Applying laSV to six cancer types, we discovered that in acute myeloid leukemia (AML), NAHR is the major mechanism for generating somatic SVs, while in the other five types of solid tumor, NHEJ and MMEJ are the predominant forces underlying somatic SVs. We further observed that such a preference for DSB repair pathway utilization could be ascribed to the differential expression of several key genes in the HR pathway among the evaluated cancer types. Moreover, we analyzed genes that were affected by somatic SVs and to our surprise we found that the genes frequently mutated by SVs tended to differ from the genes frequently mutated by single nucleotide alterations, which suggests different roles for the two types of mutations during cancer development. We also described in detail examples of complex genomic rearrangements and intragenic SVs disrupting known oncogenes and tumor suppressors. Finally we characterized the somatic SVs in intergenic regions and discussed the potential functional implications of SVs that overlap with genomic regulatory elements. The laSV package is freely available at https://github.com/JialiUMassWengLab/laSV/tree/master.
MATERIALS AND METHODS
Detection of putative SVs via de novo local assembly
The construction and storage of a de Bruijn graph is adopted from the CORTEX algorithm (21). After the construction of the de Bruijn graph from raw reads, laSV maps the reads to the branch sequences using the BWA MEM algorithm and identifies ‘connected’ branches as those covered by the same read or the same read pair (Supplementary Figure S1). The connections of branches are stored as a hash table in the RAM and used for extending contigs during traversing. Next, the de Bruijn graph is traversed in a breadth-first manner to output the ‘maximal unambiguous contigs’ (MUCs). MUCs are defined as the longest contigs that contain only the connected branches (Supplementary Figure S2). These MUCs are then mapped to the reference genome using BWA MEM, which performs a local alignment. Contig segments that can be mapped to multiple loci in the reference genome are discarded because laSV cannot determine their origin unequivocally. Contigs that can be split-mapped to discontiguous loci of the reference genome are classified as discordant. Discordant contigs are indicative of putative SVs and are retained for further analysis.
Genotyping and estimation of SV allele frequencies
laSV further validates the putative SVs by mapping the raw reads to sequences that represent both the putative SV alleles derived from the assembled contigs and the corresponding alleles in the reference genome using the BWA aln algorithm. SV and reference alleles are prepared by extending 500 bp from the breakpoints in both directions. SV calls with fewer than four read pairs mapping to the variant allele are most likely false positives and are discarded. Based on the number of reads mapped to the variant allele and the corresponding reference allele, laSV estimates the frequency of the variant allele using the formula 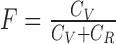 , with effective coverages
, with effective coverages 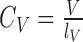 and
and 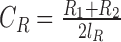 , where V, R1 and R2 represent the number of SV-supporting reads, the number of reads supporting reference locus 1 and the number of reads supporting reference locus 2, respectively (Supplementary Figure S3). Effective lengths
, where V, R1 and R2 represent the number of SV-supporting reads, the number of reads supporting reference locus 1 and the number of reads supporting reference locus 2, respectively (Supplementary Figure S3). Effective lengths 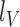 and
and 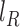 are given by
are given by 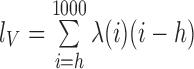 and
and 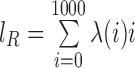 , where
, where 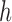 is the homologous sequence length and
is the homologous sequence length and 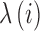 is the proportion of reads with fragment size
is the proportion of reads with fragment size 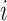 in the library (22).
in the library (22).
After performing de novo SV discovery in the cancer genomes, we genotyped all of the putative SVs in the matched normal genomes. The SVs present in the cancer genomes with a ≥ 10% allele frequency that were supported by ≥4 read pairs and were absent from the matched normal genomes were considered somatic SVs.
Validation of NA12878 SVs using long-read sequencing datasets
We validated the SV calls of laSV in an individual with European ancestry using the long-read sequencing datasets for the same individual provided by Moleculo and PacBio. The datasets were downloaded from the 1000 Genomes Project FTP site:
ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase3/integrated_sv_map/supporting/NA12878/moleculo/, and
ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase3/integrated_sv_map/supporting/NA12878/pacbio/.
An SV was considered validated if there are 2 PacBio reads or 1 Moleculo read that supported the same type of SV with a breakpoint within 6 nt of that identified by laSV.
Simulated datasets for comparing SV detection algorithms
To compare the performance of laSV with several other methods, we generated simulated datasets each with 100 deletions, inversions and tandem duplications randomly inserted across human chromosome 9 using the SV simulation tool RSVSim (23). Paired-end Illumina sequencing reads at 30× coverage with the mean and variance of the fragment size 400 and 50 bp respectively were then simulated using the pIRS software (24). The process was repeated for 100 times to produce 10 000 deletions, inversions and tandem duplications in total.
Classification of SV mechanisms
Our inference regarding the SV formation mechanism is based on the homology length, defined as the length of the homologous sequence between the two breakpoint loci (Supplementary Figure S4). We define breakpoints with a homology length ≤2 and ≥−10 (a negative homology length indicates insertion at the breakpoint) as being generated by NHEJ, those with a homology length >2 and ≤25 as being generated by MMEJ, and those with a homology length >25 as being generated by NAHR. Breakpoints with >10 nt insertion at the breakpoint are classified as non-template insertions.
Detection of complex rearrangements
We used breakpoint graphs, as described by Pevzner (25), for the detection of complex rearrangements. Briefly, each node in the graph represents a genomic position, and two nodes are connected by a ‘breakpoint edge’ if there is an SV bringing the two genomic positions together. Two nodes are connected by an ‘adjacency edge’ if the distance between the two genomic positions is shorter than 100 Kb, and the weight of the edge is defined as the genomic distance between the two positions. An alternating path in the graph is defined as a path consisting of adjacency edges and breakpoint edges in an alternating fashion. A shortest alternating path in the graph that contains at least two breakpoint edges represents a potential complex rearrangement. The shortest alternating path between all pairs of nodes can be computed using a variant of the Dijkstra algorithm, as described by Brown (26).
Whole-genome sequencing and RNA-seq datasets
All of the whole-genome sequencing and RNA-seq datasets used in this study were produced by The Cancer Genome Atlas (TCGA) Research Network. The full list of samples used is listed in Supplementary Table S1. The FASTQ raw sequence reads of genomic DNA were downloaded from CGHub (https://cghub.ucsc.edu/) and transcriptome RNA-seq data were obtained from the Data Portal of TCGA (https://tcga-data.nci.nih.gov/tcga/).
Analysis of intergenic SVs
Intergenic SVs (SVs that do not overlap with any genes) in BRCA, CESC, GBM, AML and UCEC were overlapped with the ENCODE DNaseI Hypersensitivity Uniform Peaks from the cell lines MCF-7, Helas3, Gliobla, K562 and Ishikawa, respectively. For enrichment simulation analysis, 20 000 random SV sets were generated for each of the five types of cancer, with each random set exhibiting exactly the same number of SVs and same SV length distribution as the observed set. Each random SV set was overlapped with the DNase HS peaks in the corresponding cell type and empirical P-values were computed as the fraction of random sets showing more overlap than the observed SV set.
RESULTS
Detection of SVs
The overall workflow of laSV is depicted in Figure 1. It first uses raw sequence reads in the FASTQ format as input and performs reference-free local assembly using de Bruijn graphs to generate contigs. Next, it aligns those contigs to the reference genome and detects all discordant alignments, i.e. different parts of the same contig mapped to discontiguous loci of the reference genome, which are indicative of putative SVs. Finally it maps the raw sequence reads to both the variant allele sequence (obtained from the assembled contigs) and the corresponding reference allele sequence and estimates the allele frequencies of the putative SVs based on the ratio of variant-supporting reads over reference-supporting reads. This approach naturally integrates read-pair and split-read information, and by producing contigs that are much longer than raw sequence reads, it avoids some mapping ambiguities and, hence, achieves higher accuracy. We use a local assembly approach to avoid aggressively pruning the de Bruijn graphs, preserving true SVs present at low allele frequencies in the sample. Moreover, the reference-free assembly makes it possible to capture novel sequences that are not present in the reference genome.
Figure 1.
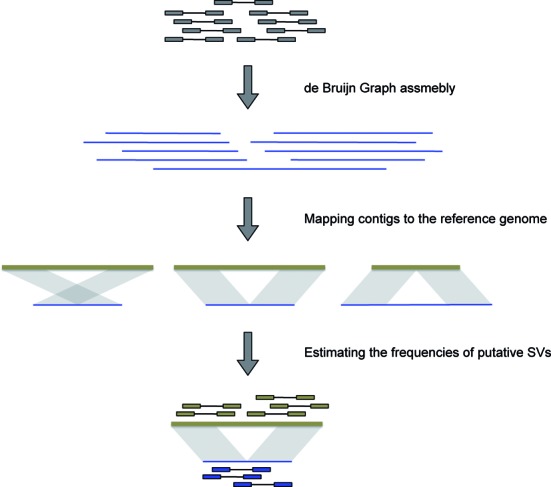
A schematic representation of the workflow of laSV.
To evaluate the accuracy of our method, we ran laSV on a high-coverage whole-genome DNA sequencing dataset from an individual of European descent (NA12878) produced by the 1000 Genomes Project and validated its results by comparing the calls with Moleculo and PacBio long-read sequencing datasets for the same individual. Among the SVs predicted by laSV with allele frequencies above 10%, 91.54% (1687 out of 1843) of the deletions and 94.93% (262 out of 276) of the non-template insertions were supported by the long-read sequencing datasets, suggesting that most of the laSV predictions were correct.
We also compared laSV and other SV detection methods CREST (6), pindel (7), delly (5) and lumpy (8) on both simulated datasets (see ‘Materials and Methods’ section) and the NA12878 sequencing dataset. On simulated datasets, laSV achieved 99.20, 99.46, 99.51% precision rates and 83.18, 85.98, 81.34% recall rates for deletions, inversions and tandem duplications, respectively. Compared with the other methods, laSV has high specificity while maintaining good sensitivity (Supplementary Figure S5). On the NA12878 dataset, laSV outperforms the other methods in specificity (Supplementary Figure S6). These results show that laSV is able to make reliable predictions for various SV types.
We applied laSV to 97 cancer-normal paired high-coverage whole-genome sequencing datasets across six cancer types: uterine corpus endometrial carcinoma (UCEC), glioblastoma multiforme (GBM) (27,28), sarcoma (SARC), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), breast invasive carcinoma (BRCA) (29) and AML (30), produced by The Cancer Genome Atlas (TCGA) Research Network (Supplementary Table S1). We identified somatic SVs as those that were present in the cancer sample but absent in the normal tissue of the corresponding individual. A total of 35 396 somatic SVs were detected, and we observed a high degree of heterogeneity in terms of the total number of somatic SVs, the composition of different SV types and the possible contributions of different SV mechanisms across samples, even within the same cancer type (Figure 2). An analysis of the SV length distribution reveals that most of the somatic SVs are very short (a few hundred base pairs) deletions and inversions (Supplementary Figure S7), which are possibly the product of error-prone DNA repairs and may have limited phenotypic impact.
Figure 2.
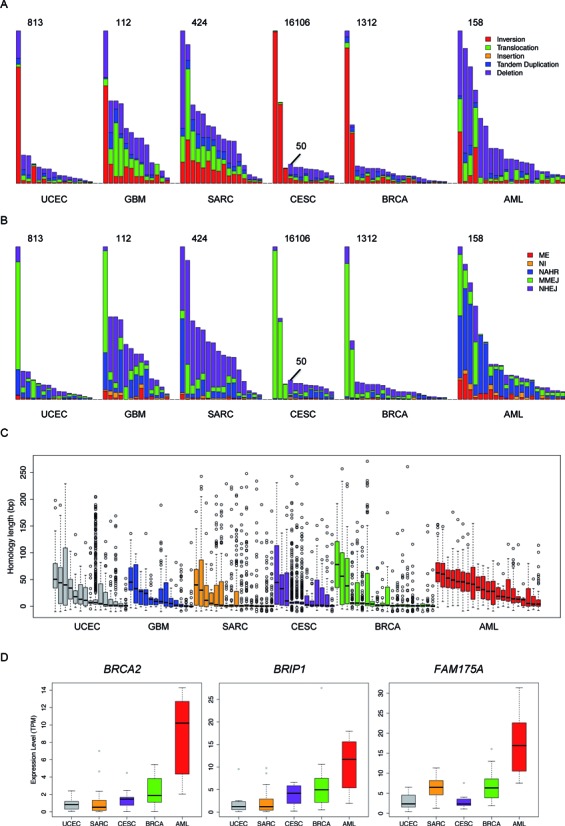
An overview of the SVs across all of the samples we analyzed. The distribution of (A) different types of SVs, (B) different breakpoint mechanisms and (C) breakpoint homology sequence lengths across all 97 samples. The evaluated cancer types are indicated at the bottom of each panel. For CESC, two different scales are used because three of the samples contain many more SVs than the remaining samples. (D) Expression levels of three key genes of the HR pathway across different cancer types. Samples within the same cancer type are ranked by the total number of somatic SVs in descending order in (A) and (B) and are ranked by the median homology sequence length in descending order in (C). TPM is transcripts per million, a means of gene expression quantification used by the RSEM algorithm, in which the total number of transcripts in a cell is normalized to one million. RNA-seq data were not available for the GBM samples.
We asked whether the SVs in CESC were due to human papillomavirus (HPV), which is a major cause for CESC. We aligned all the contigs assembled from CESC samples to the genomes of all 175 HPV strains downloaded from PaVE (http://pave.niaid.nih.gov/) using the BLAT algorithm (31). None of the contigs indicative of SVs could be aligned to the HPV genomes, suggesting that the SVs we identified were not caused by HPV.
A survey of molecular mechanisms underlying somatic SVs
Because laSV has the capability to pinpoint breakpoints with a single-nucleotide resolution, we were able to infer the molecular mechanisms underlying the somatic SVs we detected based on the sequence homology at breakpoints (Figure 2B). In all five of the solid tumor types we analyzed, NHEJ and MMEJ appear to be the predominant forces driving somatic SVs, which is consistent with previous reports (32,33). Surprisingly, in AML, most somatic SVs show long stretches of homologous sequences across breakpoints and are probably the result of NAHR. To ensure that this difference is not an artifact due to the choice of homology length cutoffs for classifying the three mechanisms, we plotted the distributions of sequence homology lengths at SV breakpoints across all of the samples we analyzed (Figure 2C). Despite substantial heterogeneity among samples within the same cancer type, AML samples generally exhibit longer sequence homology at breakpoints than the other cancer types (P-values are 1.46e-4, 1.20e-3, 8.69e-3, 2.36e-5 and 0.0153 versus BRCA, CESC, GBM, SARC and UCEC, respectively; Wilcoxon rank sum test).
What might be the reasons for such a differential preference for DSB repair pathways among cancer types? We found that for a third of the known genes in the HR pathway (6/18), the expression level is significantly higher in AML than in all the other cancer types (q-value < 0.01; Supplementary Figure S9). The genes that are more abundantly expressed in AML include BRCA2, FAM175A and BRIP1, which encode RAD51 mediators, proteins crucial for recruiting RAD51 to the damaged sites and initiating the HR pathway upon DNA damage (12). Perhaps these more highly expressed HR genes increase the activity of the HR pathway in AML and lead to a higher proportion of SVs produced by NAHR than in the other cancers.
Identification of complex genomic rearrangements
Complex genomic rearrangements are defined as SVs that are formed in a single event and involve multiple breakpoints. One class of replication-based mechanisms capable of generating such complex rearrangements is replication fork stalling and template switching (FoSTes) and more generally microhomology-mediated break-induced replication (MMBIR) (20). Another mechanism is chromothripsis, massive chromosomal rearrangements that occur during a single catastrophic event within a localized genomic region (34). To identify potential complex rearrangements, we developed a graph-based algorithm to connect breakpoints that are proximal to each other (see ‘Materials and Methods’ section for more details).
Figure 3A shows an example of complex rearrangement likely due to MMBIR. In the gene body of MEGF8, there is a 749 bp deletion and in its place is a segment of 5584 bp that includes the 3′ portion of PPR19 and the 5′ portion of TMEM145, two genes upstream of MEGF8. The two breakpoints exhibit 3 and 1-bp homology, respectively. This rearrangement effectively creates two fused genes, MEGF8–PPR19 with exons 1–19 of MEGF8 and TMEM145–MEGF8 with exons 20–42 of MEGF8. RNA-seq data from the same individual indicates that the expression level of exons 1–19 of MEGF8 is 1.24-fold higher than exons 20–42 of MEGF8 (Figure 3A). The TMEM145–MEGF8 chimeric transcript likely undergoes nonsense-mediated decay due to a premature stop codon caused by the fusion and the reads mapping to exons 20–42 of MEGF8 are from the wild-type copy of the MEGF8 gene in the sister chromosome.
Figure 3.
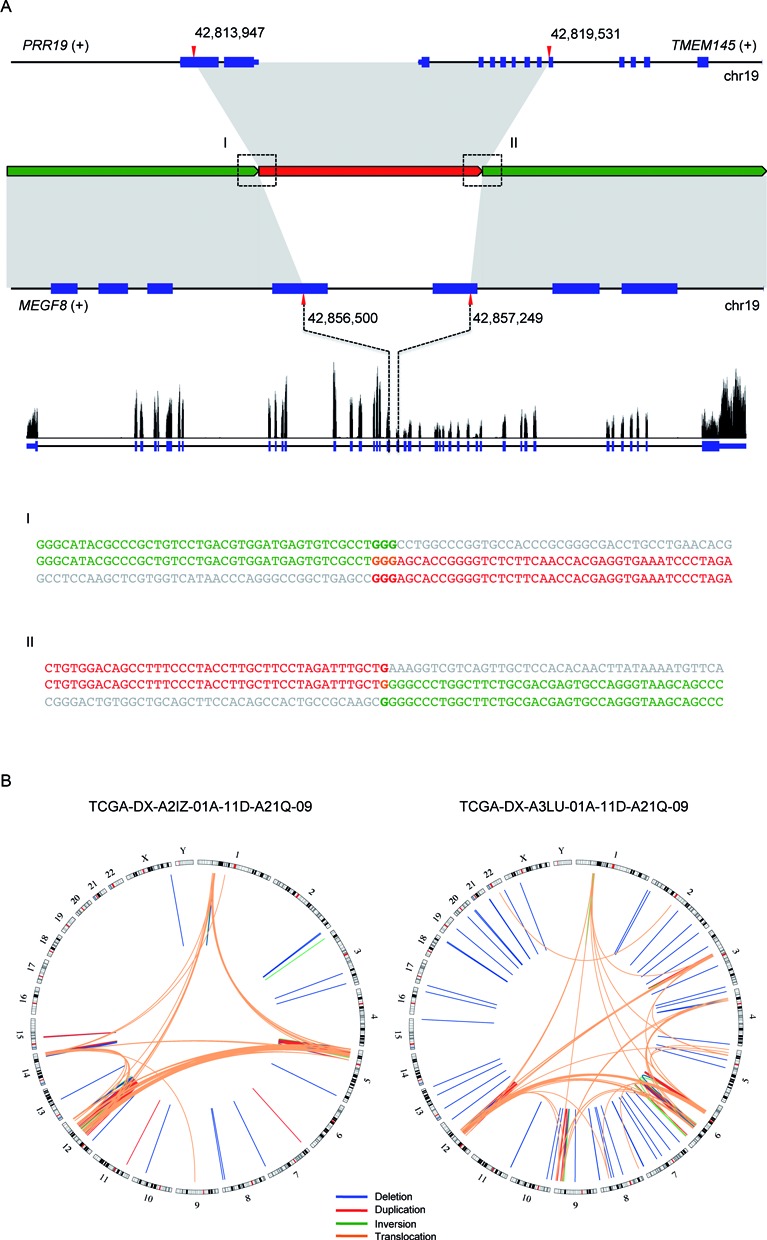
Examples of complex SVs. (A) An example of an MMBIR. Boxes (I) and (II) show the two breakpoint sequences. The characters in bold indicate homologous sequences. (B) Two examples of chromothripsis.
In addition, we noticed that in some of the samples, there are a large number of breakpoints concentrated within localized genomic regions. For instance, in one SARC sample, the vast majority of breakpoints fall within four narrow genomic regions in chromosomes 1, 5, 12 and 14 (Figure 3B; left). There are also many novel adjacencies connecting fragments of these four regions, suggesting extensive rearrangements possibly as a result of faulty DNA repairs in response to chromothripsis. Another SARC sample shows similar patterns with different genomic loci being affected (Figure 3B; right).
Somatic SVs that overlap protein-coding genes
In the samples we studied, there were a total of 17 184 protein-coding genes overlapping at least one SV in at least one sample (Supplementary Table S2). Most of those SVs probably do not confer a growth advantage to the tumor cells carrying them and, hence, are so-called ‘passenger mutations’. However, there was a significant enrichment of known oncogenes and tumor suppressors (35) (P-value = 0.013; hypergeometric test) among the genes affected by somatic SVs. Furthermore, when we restricted our analysis to somatic SVs present with a 20% or higher allele frequencies, the enrichment was more significant (P-value = 6.5e-3; hypergeometric test), suggesting that SVs having phenotypic consequences are more likely to cause a cancer subclone to be selected and increase in frequency.
When we performed gene ontology analysis on genes that are affected by SVs in multiple samples for each of the six cancer types (Supplementary Figure S10), we observed enrichment for processes such as immune responses, keratinization, metallopeptidase-related processes and cell–cell adhesion. Mutations in genes belonging to these biological processes and pathways are unlikely to cause tumorigenesis. Instead, they might confer a growth advantage on tumor cells and allow them to evade the immune system and metastasize.
Three of the cancer types we analyzed (BRCA, GBM and AML) have been extensively studied before by the TCGA consortium (29–30,27). Whole-exome sequencing was performed on hundreds of samples for each of these three cancer types to identify recurrently mutated genes. We compared the lists of genes showing recurrent single-nucleotide alterations (SNAs) and indels with the genes that we found to be affected by SVs and asked whether the same genes tended to harbor both SNAs/indels and SVs. In BRCA, the overlap between genes showing recurrent point mutations and genes affected by SVs was statistically significant (P-value = 0.0184, hypergeometric test). In GBM and AML, however, there was no significant overlap between recurrently point-mutated and SV-mutated genes (P-value = 0.378 and 0.093 for GBM and AML, respectively), suggesting that SNAs/indels and SVs may play different roles during cancer development.
For all of the genes harboring SVs or SNAs in a given cancer type (point-mutation data are not available for SARC), we correlated the number of samples where SV-induced mutations occurred with the number of samples where point mutations occurred. We observed negative correlations for all five cancer types, with Pearson correlation coefficients being −0.537, −0.785, −0.713, −0.293 and −0.697 (all P-values < 1e-100) for UCEC, GBM, CESC, BRCA and AML, respectively, indicating that genes show recurrent point mutations are less likely to harbor SV mutations (Figure 4A).
Figure 4.
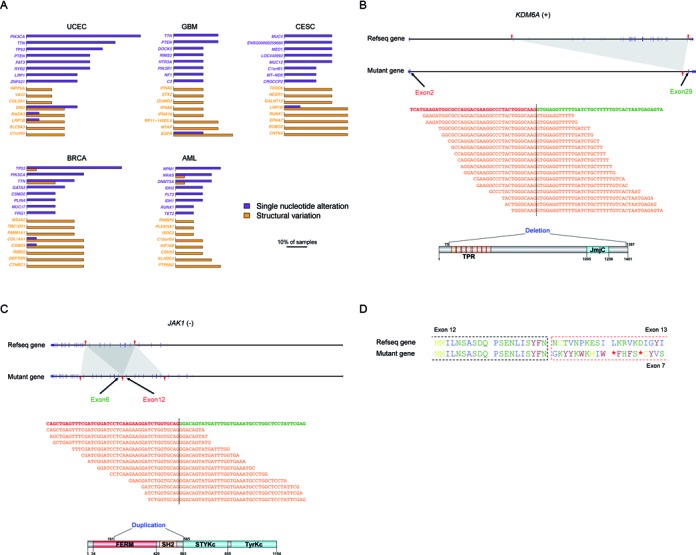
The impact of somatic SVs on protein-coding genes. (A) Comparison of genes frequently affected by point mutations with those frequently affected by SVs. Purple bars indicate the percentage of samples in which the gene carries SNAs while orange bars indicate the percentage of samples in which the gene carries SVs. The purple genes are the ones most frequently affected by SNAs within each cancer type; orange genes are the ones mostly frequently affected by SVs. (B) A deletion within the tumor suppressor KSDM6A. The black dashed line indicates the exon–exon junction. The orange sequences are representative RNA-seq reads that map across the junction. (C) A tandem duplication within the oncogene JAK1. (D) Amino acid sequences of the wild-type and duplicated versions of RB1. The red asterisks indicate stop codons.
To test whether this negative correlation was due to the decreased power of detecting SNAs in deleted regions, we assessed the relative impact of deletions in each cancer type. Since all SNAs are in coding regions (CDS), we compared the total length of deleted CDS with the length of duplicated CDS in each cancer type (Supplementary Table S3). Our results revealed no strong bias toward deletion over duplication and therefore the aforementioned negative correlations are unlikely to be the result of compromised SNA detection power due to deletions. Furthermore, since the breakpoints of the SVs fall predominantly in intergenic and intronic regions far away from CDS, it is also highly unlikely that the negative correlations are caused by the effect of SNAs on SV detection power.
The tumor suppressor KDM6A encodes a lysine-specific demethylase that catalyzes the demethylation of tri- and di-methylated H3K27. Missense and nonsense mutations in this gene have been reported in multiple cancer types (36). In one of the CESC samples, laSV detected a 148 495 bp deletion that eliminates exons 3–28 of KDM6A (Figure 4B). This deletion leads to a much shortened transcript, which if translated, encodes a nonfunctional protein because the JmjC catalytic domain is deleted. Based on RNA-seq data from the same individual, we observed 48 reads that map across the exon 2–exon 29 junction, indicating that the mutated version of the KDM6A gene was indeed transcribed.
The oncogene JAK1 encodes a non-receptor tyrosine kinase whose hyperactivity has been implicated in multiple cancer types, including breast cancer, colorectal cancer and lung cancer (37,38). We observed a 22 471 bp tandem duplication that includes exons 6–12 in one of the BRCA samples (Figure 4C). At the protein level, this duplication leads to an extra copy of a portion of the FERM domain, the entire SH2 domain, and the SH2-pseudokinase linker. Recent biochemical studies have shown that the FERM domain and the SH2 domain of JAK family proteins are crucial for binding to the cytoplasmic region of the cytokine receptors (39,40). Perhaps the duplication increases the affinity with which JAK1 binds to the cytokine receptor or shifts the relative position of the kinase domain with respect to the cytokine receptor, disrupting proper regulation.
In both of the above examples, the mutated genes are still translated in-frame. In other cases, SVs may also cause a frameshift and, thus, grossly alter the amino acid sequences of the protein product. RB1 is a negative regulator of the cell cycle and was the first discovered tumor suppressor (41). In one of the BRCA samples, we observed a tandem duplication that included exons 7–12 of the RB1 gene. This results in a frameshift that leads to a premature stop codon (Figure 4D). In the same individual, we observed a six-fold decrease in RB1 expression in the tumor tissue compared with the nearby normal tissue. The premature stop codon is located 2077 nt upstream of the last exon–exon junction and may have triggered nonsense-mediated decay, leading to the decreased RB1 level.
Some intergenic SVs may impact genomic regulation
SVs that do not overlap any gene are usually ignored due to the difficulty of evaluating their possible effects. For five of the six cancer types we analyzed (except SARC) we were able to find DNaseI sequencing data produced by the ENCODE consortium on cell types corresponding to the same tissue. We then intersected the intergenic SVs with DNase hypersensitive sites (DHSs) in the corresponding cell type. Overall, a background level of 1.10% (356/32455) intergenic SVs overlapped with DNase hypersensitive regions. Nevertheless, DHS-overlapping SVs have higher allele frequencies than the non-overlapping SVs (P-value = 3.73e-4, Wilcoxon Rank Sum test, Supplementary Figure S11), indicating that DHS-overlapping SVs are more likely to confer a growth advantage.
BCL9 is an oncogene that encodes an important component of the Wnt pathway. BCL9 interacts with β-catenin to enhance its transcriptional activity and is implicated in several types of cancer (42,43). In one of the BRCA samples, we observed a 22 847-bp duplication upstream of the BCL9 gene that overlaps with two DHSs in MCF-7 cells (Figure 5). One of the DHSs is bound by the transcription factors E2F1, CTCF, RAD21 and MAX. Moreover, the Pol II ChIA-PET data indicate that there is a chromatin interaction between the DHS and the promoter of the BCL9 gene, which suggests that the DHS may regulate BCL9 transcription. Indeed, we observed a 63.52% increase in BCL9 expression in the tumor sample compared with the matched normal sample. It is likely that the duplication of the regulatory DHS leads to an elevated expression level of BCL9.
Figure 5.
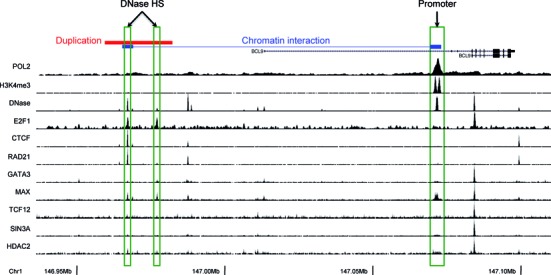
An example of somatic SV affecting intergenic gene regulatory elements. In one of the BRCA samples a tandem duplication spans a DNase hypersensitive site containing regulatory elements of oncogene BCL9.
DISCUSSION
Cancer is a group of complex diseases driven by various genetic and epigenetic alterations. Previous surveys on genetic alterations in cancer have mostly focused on single-nucleotide mutations in protein-coding sequences, fusion transcripts and copy number alterations of large genomic segments (35). In this article, we reported a novel algorithm, laSV, that is capable of detecting genomic SVs across a wide spectrum of sizes from highly heterogeneous tumor samples and pinpointing their breakpoints at a single-nucleotide resolution. Applying this algorithm to 97 high-coverage whole-genome sequencing datasets across six cancer types, we observed several interesting phenomena.
Because laSV supports nucleotide-resolution delineation of SV breakpoints, we examined the prevalence of different breakpoint formation mechanisms across all of the samples that we analyzed. To our knowledge, there have been two studies conducted thus far that have comprehensively surveyed breakpoint formation mechanisms across multiple cancer types (32,33). Both studies included only solid tumors and concluded that most breakpoints exhibit little or no homology and were therefore probably formed via NHEJ or MMEJ. We observed similar patterns in the five types of solid tumors that we analyzed. In AML, however, most of the breakpoints showed homologous sequences much longer than those needed for NHEJ and MMEJ, suggesting that NAHR is the predominant mechanism of breakpoint formation. Such a preference for different breakpoint formation mechanisms might provide important insight into the course of evolution taken by different cancer types and have implications for the development of cancer type-specific treatments.
At present laSV employs BWA to align assembled contigs to the reference and predict SV breakpoints. There are other methods, such as YAHA (44) and AGE (45), that specialize in aligning long sequences and detecting potential breakpoints. In the future it would be interesting to explore how laSV performs using these software for contigs alignment. In addition to reflecting the confidence level of the SV calls, the SV allele frequency computed by laSV could also be useful in some other applications, such as distinguishing driver mutations from passenger mutations since driver mutations tend to occur early on during the tumor development and therefore be present in most of the cells in the tissue. Furthermore, when samples from different stages of the tumor development or from different metastasized sites are available, it would be informative to compare the SV allele frequencies across those samples as they may reveal how the cancer progressed and adapted to new metastasized locations.
The fact that genes that show recurrent SNAs do not appear to be preferentially mutated by SVs is noteworthy. Considering that the spontaneous mutation rate for point mutations is much higher than for SVs (46), we hypothesize that tumorigenesis is often initiated by point mutations and that most SVs occur later during cancer development, when DNA repair mechanisms are compromised. Because additional SV mutations in a gene already disrupted by cancer-causing point mutations rarely enhance the cancer phenotype, they are unlikely to be selected for in the tumor tissues. The observation that genes that are recurrently affected by SVs are enriched for pathways such as cytoskeleton metabolism, immune response and cell–cell adhesion, which are unlikely to cause uncontrolled cell proliferation but may contribute to the migration, immune defense evasion and metastasis of cancer cells, further supports our hypothesis. The characterization of SVs in cancer lags behind that of SNAs/indels because whole-genome sequencing is more costly than exome sequencing. More cancer genomes need to be sequenced to more accurately identify genes recurrently affected by SV mutations for various cancer types. Our results indicated that in addition to point mutations, gains/losses of large genomic segments and transcript fusions, intragenic SVs can also have a significant impact on the expression levels and products of protein-coding genes, as demonstrated by the examples of KDM6A, JAK1 and RB1. Therefore, laSV, with its ability to accurately detect more subtle SVs, will be a valuable tool for future surveys of genetic alterations in cancers.
Previous reports on cancer research have mostly focused on genetic alterations within or including CDS. A recent study suggests that a large fraction of the non-coding portion of the human genome may contain regulatory elements (47). We found that some of the SVs in non-CDS regions overlap with DHSs and might have regulatory functions. The example of BCL9 that we described demonstrates how SV discovery in the non-CDS regions can, when considered in conjunction with the rich information accumulated by the ENCODE consortium, shed new light on regulatory alterations in cancer. With our rapidly expanding knowledge regarding the various regulatory elements in the human genome, further studies will be carried out to interrogate the roles of non-coding regulatory regions in cancer. The accurate identification of more subtle SVs and the precise determination of their breakpoints will be crucial for the success of such investigations.
Supplementary Material
Acknowledgments
We thank members of the Weng lab for stimulating discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [U41 HG007000]. Funding for open access charge: National Institutes of Health [U41 HG007000].
Conflict of interest statement. None declared.
REFERENCES
- 1.Alkan C., Coe B.P., Eichler E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011;12:363–375. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Escaramís G., Tornador C., Bassaganyas L., Rabionet R., Tubio J.M.C., Martínez-Fundichely A., Cáceres M., Gut M., Ossowski S., Estivill X. PeSV-Fisher: identification of somatic and non-somatic structural variants using next generation sequencing data. PLoS One. 2013;8:e63377. doi: 10.1371/journal.pone.0063377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinlan A.R., Clark R.A., Sokolova S., Leibowitz M.L., Zhang Y., Hurles M.E., Mell J.C., Hall I.M. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010;20:623–635. doi: 10.1101/gr.102970.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q., Locke D.P., et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Mullighan C.G., Easton J., Roberts S., Heatley S.L., Ma J., Rusch M.C., Chen K., Harris C.C., Ding L., et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Layer R.M., Chiang C., Quinlan A.R., Hall I.M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen K., Chen L., Fan X., Wallis J., Ding L., Weinstock G. TIGRA: a targeted iterative graph routing assembler for breakpoint assembly. Genome Res. 2013;24:310–317. doi: 10.1101/gr.162883.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Zheng H., Luo R., Wu H., Zhu H., Li R., Cao H., Wu B., Huang S., Shao H., et al. structural variation in two human genomes mapped at single-nucleotide resolution by whole genomede novo assembly. Nat. Biotechnol. 2011;29:725–732. doi: 10.1038/nbt.1904. [DOI] [PubMed] [Google Scholar]
- 11.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krejci L., Altmannova V., Spirek M., Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jasin M., Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta A., Haber J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weterings E., Chen D.J. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 16.Aparicio T., Baer R., Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst.) 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decottignies A. Alternative end-joining mechanisms: a historical perspective. Front. Genet. 2013;4:48. doi: 10.3389/fgene.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ottaviani D., Lecain M., Sheer D. The role of microhomology in genomic structural variation. Trends Genet. 2014;30:85–94. doi: 10.1016/j.tig.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Truong L.N., Li Y., Shi L.Z., Hwang P.Y.-H., He J., Wang H., Razavian N., Berns M.W., Wu X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iqbal Z., Caccamo M., Turner I., Flicek P., McVean G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 2012;44:226–232. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartenhagen C., Dugas M. RSVSim: an R/Bioconductor package for the simulation of structural variations. Bioinformatics. 2013;29:1679–1681. doi: 10.1093/bioinformatics/btt198. [DOI] [PubMed] [Google Scholar]
- 24.Hu X., Yuan J., Shi Y., Lu J., Liu B., Li Z., Chen Y., Mu D., Zhang H., Li N., et al. pIRS: profile-based Illumina pair-end reads simulator. Bioinformatics. 2012;28:1533–1535. doi: 10.1093/bioinformatics/bts187. [DOI] [PubMed] [Google Scholar]
- 25.Pevzner P. Computational Molecular Biology. Cambridge, MA: MIT Press; 2000. [Google Scholar]
- 26.Brown J.R. Shortest alternating path algorithms. Networks. 1974;4:311–334. [Google Scholar]
- 27.Brennan C.W., Verhaak R.G.W., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H., et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L., Luquette L.J., Gehlenborg N., Xi R., Haseley P.S., Hsieh C.-H., Zhang C., Ren X., Protopopov A., Chin L., et al. Diverse mechanismsof somatic structural variations in human cancer genomes. Cell. 2013;153:919–929. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra A., Lindberg M., Faust G.G., Leibowitz M.L., Clark R.A., Layer R.M., Quinlan A.R., Hall I.M. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 2013;23:762–776. doi: 10.1101/gr.143677.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stephens P.J., Greenman C.D., Fu B., Yang F., Bignell G.R., Mudie L.J., Pleasance E.D., Lau K.W., Beare D., Stebbings L.A., et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sengoku T., Yokoyama S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011;25:2266–2277. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren Y., Zhang Y., Liu R.Z., Fenstermacher D.A., Wright K.L., Teer J.K., Wu J. JAK1 truncating mutations in gynecologic cancer define new role of cancer-associated protein tyrosine kinase aberrations. Sci. Rep. 2013;3:3042. doi: 10.1038/srep03042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song L., Rawal B., Nemeth J.A., Haura E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011;10:481–494. doi: 10.1158/1535-7163.MCT-10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallweber H.J.A., Tam C., Franke Y., Starovasnik M.A., Lupardus P.J. Structural basis of recognition of interferon. Nature Structural & Molecular Biology. 2014;21:443–448. doi: 10.1038/nsmb.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babon J.J., Lucet I.S., Murphy J.M., Nicola N.A., Varghese L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014;462:1–13. doi: 10.1042/BJ20140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benavente C.A., Dyer M.A. Genetics and epigenetics of human retinoblastoma. Annu. Rev. Pathol. 2015;10:547–562. doi: 10.1146/annurev-pathol-012414-040259. [DOI] [PubMed] [Google Scholar]
- 42.Sampietro J., Dahlberg C.L., Cho U.S., Hinds T.R., Kimelman D., Xu W. Crystal structure of a beta-catenin/BCL9/Tcf4 complex. Mol. Cell. 2006;24:293–300. doi: 10.1016/j.molcel.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 43.de la Roche M., Worm J., Bienz M. The function of BCL9 in Wnt/beta-catenin signaling and colorectal cancer cells. BMC Cancer. 2008;8:199–212. doi: 10.1186/1471-2407-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faust G.G., Hall I.M. YAHA: fast and flexible long-read alignment with optimal breakpoint detection. Bioinformatics. 2012;28:2417–2424. doi: 10.1093/bioinformatics/bts456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abyzov A., Gerstein M. AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics. 2011;27:595–603. doi: 10.1093/bioinformatics/btq713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.