

Abstract
Purpose
Oxidative stress and metabolic dysregulation of the RPE have been implicated in AMD; however, the molecular regulation of RPE metabolism remains unclear. The transcriptional coactivator, peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) is a powerful mediator of mitochondrial function. This study examines the ability of PGC-1α to regulate RPE metabolic program and oxidative stress response.
Methods
Primary human fetal RPE (hfRPE) and ARPE-19 were matured in vitro using standard culture conditions. Mitochondrial mass of RPE was measured using MitoTracker staining and citrate synthase activity. Expression of PGC-1 isoforms, RPE-specific genes, oxidative metabolism proteins, and antioxidant enzymes was analyzed by quantitative PCR and Western blot. Mitochondrial respiration and fatty-acid oxidation were monitored using the Seahorse extracellular flux analyzer. Expression of PGC-1α was increased using adenoviral delivery. ARPE-19 were exposed to hydrogen peroxide to induce oxidative stress. Reactive oxygen species were measured by CM-H2DCFDA fluorescence. Cell death was analyzed by LDH release.
Results
Maturation of ARPE-19 and hfRPE was associated with significant increase in mitochondrial mass, expression of oxidative phosphorylation (OXPHOS) genes, and PGC-1α gene expression. Overexpression of PGC-1α increased expression of OXPHOS and fatty-acid β-oxidation genes, ultimately leading to the potent induction of mitochondrial respiration and fatty-acid oxidation. PGC-1α gain of function also strongly induced numerous antioxidant genes and, importantly, protected RPE from oxidant-mediated cell death without altering RPE functions.
Conclusions
This study provides important insights into the metabolic changes associated with RPE functional maturation and identifies PGC-1α as a potent driver of RPE mitochondrial function and antioxidant capacity.
Keywords: retinal pigment epithelium, PGC-1, metabolism, oxidative stress, age-related macular degeneration
Age-related macular degeneration (AMD) is the leading cause of blindness in the elderly population of industrialized countries,1 affecting more than 2 million individuals in the United States alone.2 Atrophic (“dry”) forms of AMD are the most prevalent (85%–90%) but lack any form of efficient treatment to either prevent or rescue RPE dysfunction and retinal degeneration.3 Dry AMD is characterized by RPE defect and/or atrophy leading to choriocapillaris involution and progressive photoreceptor degeneration.4 Although AMD pathogenesis remains largely unknown, a compelling set of clinical and experimental evidence has identified oxidative damage and mitochondrial dysfunction in RPE as key early events in AMD progression. One of the greatest risk factors for AMD, cigarette smoking, is known to increase RPE oxidative damage.5 Dry AMD is also associated with accumulation of iron, a pro-oxidant, in RPE and Bruch's membrane.6 Decreased expression or signaling of key RPE antioxidant components and increased mitochondrial DNA damage, as a consequence of oxidative stress, has been described in aged and AMD eyes.7–9 Finally, mouse models have confirmed that alteration of oxidative phosphorylation (OXPHOS) metabolism or increased oxidative stress in the RPE specifically can recapitulate some of the cardinal features of AMD.5,10,11
Despite the significant implication of oxidative damage in RPE dysfunction and atrophy, the molecular regulation of RPE oxidative metabolism under normal and pathological conditions remains unknown. The RPE are particularly exposed to oxidative stress due to their location between the choriocapillaris (main source of oxygen and nutrients to the retina) and photoreceptors, high levels of light irradiation, and abundance of photosensitizers such as lipofuscin.12 In addition, robust generation of ATP by OXPHOS in mitochondria is required to support RPE functions such as the phagocytosis of photoreceptor outer segments (OS). As a result of this metabolic burst, OS phagocytosis leads to a significant production of H2O2 by the RPE.13 Adaptation of RPE to such oxidative conditions depends on a high level of antioxidant enzymes and antioxidant small molecules12 regulated in part via the transcription factor NFE2L2 (commonly named NRF2), whose expression is induced by oxidative stimuli.14
The transcriptional coactivators, peroxisome proliferator-activated receptor-gamma coactivator 1α and β (PGC-1α/β) are powerful activators of mitochondrial biogenesis, oxidative metabolism, and fatty-acid β-oxidation in many tissues.15 Through their ability to coactivate numerous DNA-binding transcription factors, including nuclear respiratory factors, PPARα, and ERRα, PGC-1 isoforms activate the expression of a core genetic program needed for mitochondrial function, DNA replication, and transcription. The PGC-1s thus link the regulation of the nuclear and mitochondrial genomes. Forced expression of either PGC-1α or β dramatically increases mitochondrial biogenesis and oxidative metabolism in certain tissues in vivo,16–18 whereas deletion of both PGC-1s has the opposite effect, severely limiting respiration in skeletal muscle or heart.19–22
PGC-1s also play an important role in defending against the reactive oxygen species (ROS) generated by mitochondrial activity through their ability to induce numerous antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidases (GPX).23 In the retina, PGC-1α has been shown to regulate normal and pathological angiogenesis, regulate sensitivity of photoreceptors to pathologic levels of light, and modulate ganglion cell sensitivity to excitotoxic reagents.24–26 However, little is known about the roles of PGC-1 in RPE biology, mitochondrial homeostasis, or anti-ROS program.
Here, we investigated the function of PGC-1s in RPE maturation, oxidative metabolism, and antioxidant response. Our results demonstrate that in vitro maturation of RPE cells is associated with increased mitochondrial abundance, increased expression of PGC-1α but not PGC-1β , increased expression of OXPHOS genes, and increased expression of antioxidant genes. We show that PGC-1α induces OXPHOS and fatty-acid gene and protein expression and regulates oxidative metabolism and fatty-acid beta-oxidation in the RPE. Furthermore, we demonstrate that PGC-1α can induce an antioxidant program in RPE and protect RPE from oxidant-mediated cell death.
Materials and Methods
Cell Culture and Treatments
The human RPE cell line ARPE-19 (CLR-2302; ATCC, Manassas, VA, USA) was cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 (Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA), 2 mM L-glutamine (Lonza), and 1% penicillin-streptomycin solution (Lonza) at 37°C, 10% CO2. To perform maturation studies, passages 2 to 6 of ARPE-19 were plated on laminin (Sigma-Aldrich Corp., St. Louis, MO, USA) coated 0.4-μm Transwells (Corning Life Sciences, Corning, NY, USA) at 1.5 × 105 cells/cm2 and cultured as previously described.27 For H2O2 treatment, confluent ARPE-19 were serum-starved for 24 hours in phenol red-free DMEM/F12 (Life Technologies, Grand Island, NY, USA) supplemented with 2 mM L-glutamine and 1% penicillin-streptomycin solution (serum-free media). Solutions of H2O2 (Sigma-Aldrich Corp.) were freshly prepared in serum-free media. Human fetal RPE (hfRPE) cells were isolated from eyes obtained from Novogenix (CA) and prepared using established methods.28 Passage 1 (P1) hfRPE were cultured on laminin-coated 0.4-μm Transwells (Corning Life Sciences).
MitoTracker Staining and Analysis
ARPE-19 were plated on laminin-coated glass coverslips at 150,000 cells/cm2 for maturation and at 135,000 cells/cm2 for 70% confluence. ARPE-19 was either stained with MitoTracker the following day (for 70% confluence) or maintained in low serum media as previously described.27 To perform MitoTracker staining, ARPE-19 were incubated with 100 nM MitoTracker Orange CMTMRos (Life Technologies) in serum-free media for 30 minutes at 37°C, 10% CO2. Cells were then washed with serum-free media, fixed with 4% paraformaldehyde in serum-free media, washed in PBS (Sigma-Aldrich Corp.) and permeabilized with 0.01% Triton X-100 (Sigma-Aldrich Corp.) for 5 minutes. Nuclei were stained using a 1:100 dilution of 4′,6-diamidino-2-phenylindole (DAPI) before mounting. Fluorescence photomicrographs were acquired using an Axioskop 2 mot plus (Carl Zeiss Microscopy, Thornwood, NY, USA) running AxioVision 4.8 software (Carl Zeiss Microscopy). Images were taken using a ×20/0.60 NA Plan-apochromat objective and a ×40/0.75 NA Plan-Neofluar objective. Seven fields per coverslip were imaged using the ×20 objective and were analyzed using ImageJ (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). Median fluorescence intensity (MFI) for each image was quantified and normalized to total protein content obtained from coverslips plated in parallel to those used for staining. Values for percent area covered by mitochondria were obtained from images in which Li's Minimum Cross Entropy thresholding method29 was applied.
Citrate Synthase Activity
ARPE-19 were cultured on Transwells (Corning Life Sciences) as described above. Protein lysates were prepared using 1× Cell Lysis Buffer (Cell Signaling, Danvers, MA, USA) containing 2 mM phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich Corp.) and quantified using the bicinchoninic acid (BCA) method (Thermo Scientific Pierce, Rockford, IL, USA). Protein samples were diluted to 30 μg/mL and citrate synthase activity was analyzed using the MitoCheck Citrate Synthase Activity Assay Kit (Cayman Chemicals, Ann Arbor, MI, USA) according the manufacturer's instructions.
Adenoviral Infection
ARPE-19 and hfRPE were plated at a density of 50,000 cells/cm2 and 85,000/cm2, respectively, and infected 24 hours later with control adenovirus (Ad-GFP; Vector Biolabs, Philadelphia, PA, USA) or with adenovirus containing the mouse PGC-1α sequence (Ad-mPGC-1α)30 at an multiplicity of infection of 30. The following day virus was removed and media were changed. Infected cells were collected 24 hours and 48 hours postinfection for protein or RNA isolation. For H2O2 treatments, 24 hours postinfection, the media were changed to serum-free media for another 24 hours before treatment.
Measurement of Cell Death and ROS
Lactate dehydrogenase (LDH) release from the cytoplasm of damaged cells was measured using the Cytotoxicity Detection Kit (Roche Life Sciences, Indianapolis, IN, USA). Cell-free supernatants were prepared as previously described31 and assayed according to the manufacturer's instructions. Percent LDH release was calculated with 2% Triton X-100 treatment used for maximal LDH release. In samples in which adenovirus infections were used, spontaneous, experimental, and maximal release were calculated using cells infected with the same adenovirus. Apoptosis was measured by quantification of histone-complexed DNA fragments using the Nucleosome Detection Kit (Roche Life Sciences). Reactive oxygen species were assayed using DCF fluorescence. Confluent ARPE-19 cells were incubated with 10 μM CM-H2DCFDA (Life Technologies) for 30 minutes in serum-free media, washed with PBS, and then treated with H2O2 in serum-free media as above. DCF fluorescence was measured using a 488 ex/528 em filter on a Synergy2 plate reader (Bio-Tek, Winooski, VT, USA).
Respirometry
ARPE-19 were plated at 50,000 cells per well and hfRPE were plated at 85,000 cells per well in V7-PS microplates (Seahorse Biosciences, Billerica, MA, USA). ARPE-19 were infected with adenovirus as described above and switched to assay medium (minimal DMEM [Seahorse Biosciences] containing 2 mM glutamine, 1 mM pyruvate, and 25 mM glucose) 48 hours after infection. hfRPE were infected with adenovirus as described above and switched to assay medium (minimal DMEM [Seahorse Biosciences] containing 2 mM glutamine, 1 mM pyruvate, and 5 mM glucose) 48 hours after infection. Oxygen consumption rates were obtained and analyzed using a XF-24 Extracellular Flux Analyzer (Seahorse Biosciences) following injections of mitochondrial inhibitors from the XF Cell Mito Stress Kit (Seahorse Biosciences) at the following concentrations for ARPE-19: 2.5 μM oligomycin, 500 nM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 2 μM Rotenone and 2 μM Antimycin A; and for hfRPE: 1 μM oligomycin, 500 nM FCCP, and 2 μM Rotenone and 2 μM Antimycin A. Fatty-acid oxidation experiments were conducted in substrate-limited media using 0.2 mM palmitate-BSA as previously described.22
Extraction of RNA and Quantitative PCR
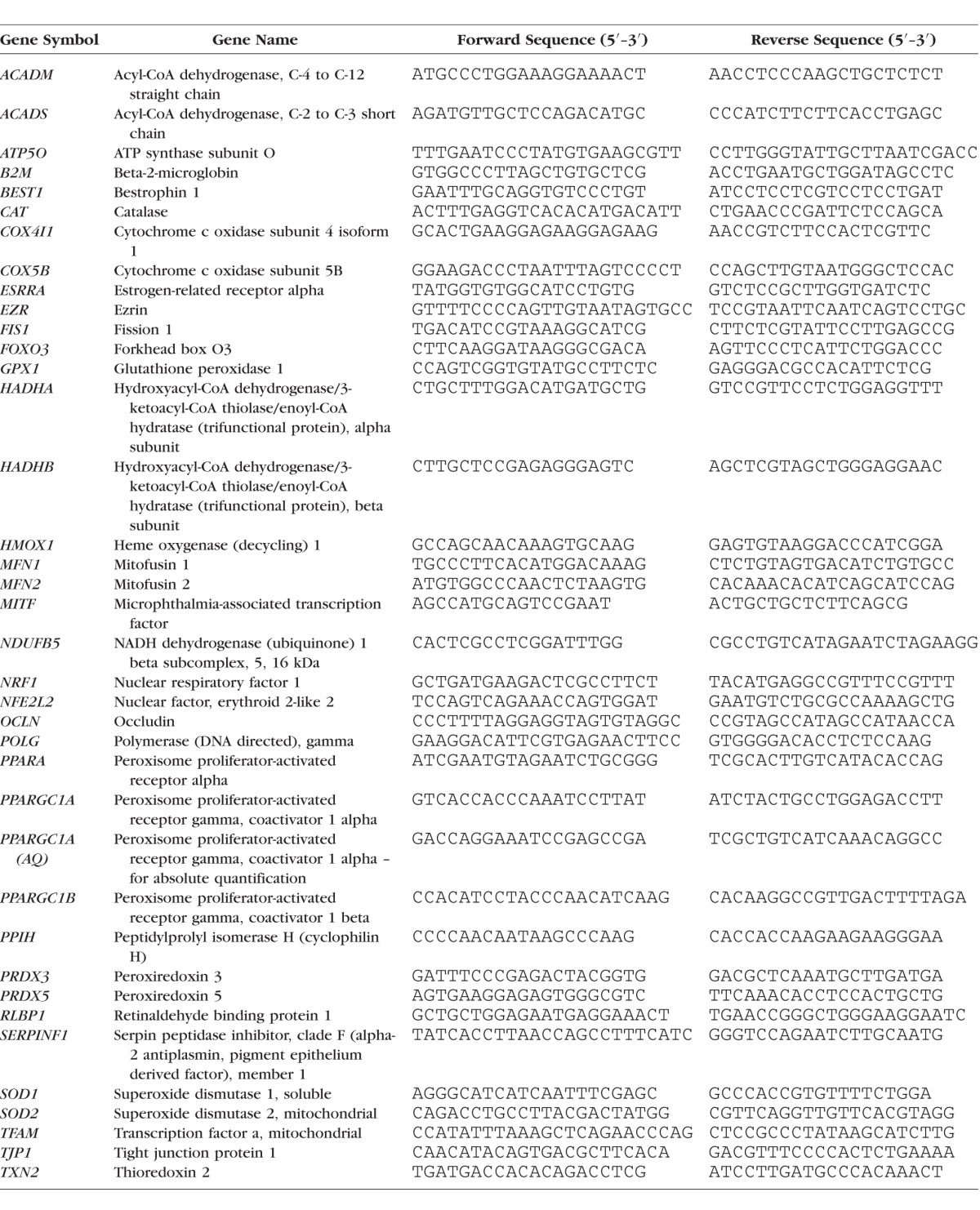
RNA was extracted from cells using RNA-bee (Amsbio, Lake Forest, CA, USA) under RNAse-free conditions according to the manufacturer's instructions. RNA pellets were resuspended in 15 μL Tris-EDTA, pH 8.0 (Ambion, Austin, TX, USA) and RNA was quantified using a NanoDrop 2000 (Thermo Scientific, Wilmington, DE, USA) spectrophotometer. RNA was extracted from hfRPE using Qiashredders (Qiagen, Valencia, CA, USA) followed by the RNeasy kit (Qiagen). RNA was reverse-transcribed to cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Quantitative PCR (qPCR) was performed using 5 ng cDNA template, Fast Universal SYBR Green Master mix with Rox (Roche Life Sciences), and the LightCycler 480 (Roche Life Sciences). Primers used for qPCR are listed in the Table. The primers for human GA binding protein transcription factor, alpha subunit (GABPA) (PPH02387A) and human thrombospondin1 (THBS1) (PPH00762A) were obtained from Qiagen. Relative gene expression was calculated with the 2−ΔΔCT method using the mean of two housekeeping genes (B2M and PPIH) for accurate quantification.32 For the absolute quantification (AQ) of PPARGC1A copy numbers, a standard curve was derived from the serial dilution (3 × 101 to 3 × 107 copies/reaction) of a linearized plasmid coding for the human PPARGC1A gene and amplified by the SYBR Green system. The specific PPARGC1A primer pair used for AQ was selected based on their associated amplification efficiencies of both the standard curve and cDNA (PCR efficiency 97%–105%). The level of PPARGC1A expression in each sample was calculated relative to the standard curve and normalized to the housekeeping genes B2M and PPIH.
Table.
Primer Sequences for qPCR
Transepithelial Resistance Measurement and Phagocytosis Assay
Transepithelial resistance (TER) of hfRPE cultured on Transwells was measured using a two-electrode epithelial voltohmmeter (EVOM2; World Precision Instruments, Sarasota, FL, USA). We performed phagocytosis assays using hfRPE with a TER of greater than 300 Ω•cm2. Photoreceptor OSs were isolated from bovine eyes, obtained from Research 87 (Boylston, MA, USA), as previously described.33 hfRPE were fed 50 OS per cell for 2 hours and then washed extensively with PBS. Degradation of OS was analyzed by Western blot as previously described.34
Western Analysis
Proteins were extracted from ARPE-19 or hfRPE using 1× Cell Lysis Buffer (Cell Signaling) containing 2 mM PMSF (Sigma-Aldrich Corp.). Insoluble debris were removed by centrifugation at 14,000g. Protein concentration was determined using a BCA assay (Thermo Scientific Pierce). Thirty micrograms cell lysate was electrophoresed under reducing conditions on a 4% to 20% Mini-PROTEAN TGX gel (Bio-Rad) and transferred to Immobilon-FL polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were blocked in a 1:1 mixture of Odyssey Blocking Buffer (OBB) (LI-COR Biosciences, Lincoln, NE, USA): Tris-buffed Saline, 0.1% (vol/vol) Tween-20 (TBS-T) for 1 hour at room temperature. Membranes were incubated in primary antibodies diluted in 1:1 OBB: TBS-T overnight at 4°C. Primary antibodies were used at the following concentrations: mouse anti-PGC-1α (EMD Millipore) 1:1000; rabbit anti-GFP (Abcam, Cambridge, MA, USA) 1:10,000; mouse anti-rhodopsin (RET-P1, Millipore) 1:500; anti–microphthalmia-associated transcription factor (anti-MITF) (Millipore) 1:200; anti-CRALBP (Abcam) 1:500; anti-RPE65 (Abcam) 1:500; mouse anti-α-tubulin (Sigma-Aldrich Corp.) 1:1000; rabbit anti-GAPDH (Santa Cruz Biotechnology) 1:2000; and total OXPHOS Western blot cocktail (Abcam) 1:250. Following primary antibody incubation, membranes were washed three times in TBS-T. Membranes were then incubated in secondary antibodies diluted in 1:1 OBB: TBS-T at room temperature for 1 hour. Donkey anti-mouse IR800CW, donkey anti-rabbit IR680, or donkey anti-mouse IR680 (all from LI-COR) were used at 1:15,000 dilutions. Images were acquired using a LI-COR Odyssey (LI-COR) and analyzed using Image Studio 2.0 (LI-COR) or ImageJ.
Statistical Analysis
Values are expressed as mean ± SEM. Data were analyzed by Student's t-test using Prism 5 (GraphPad, La Jolla, CA, USA) unless stated otherwise. Significance is indicated in the text as follows: * P < 0.05, ** P < 0.01, *** P < 0.001.
Results
Maturation of RPE In Vitro Increases Oxidative Metabolism and Is Associated With Increased PGC-1α Expression
We first characterized the metabolic changes associated with RPE maturation in vitro. Study of this complex morphogenic process initiated by contact inhibition and by which dissociated RPE cells re-acquire a mature native-like epithelial phenotype provides valuable insights into the central factors and pathways required for the establishment and maintenance of RPE morphology and functions.35,36 In vitro maturation of RPE cells, such as ARPE-19, a spontaneously arising cell line derived from adult human RPE,37 consists of long-term culture at confluence in low serum conditions and is demonstrated by a progressive increase in transepithelial resistance (Fig. 1A) and the induction of the RPE-specific genes MITF, RLBP1, SERPINF1, and RPE65 (Fig. 1B).
Figure 1.

Maturation of RPE in vitro is associated with increased mitochondrial mass. (A) Transepithelial resistance of ARPE-19 matured on Transwells for up to 4 weeks (n = 3 for each time point). (B) Gene expression of RPE-specific genes measured by qPCR during ARPE-19 maturation (n = 3 for each time point). (C) MitoTracker CMTMRos staining demonstrates increased mitochondrial network with maturation time. Images were acquired with variable exposure times using a ×40 objective. Scale bar: 20 μm. (D) Quantification of image area covered by MitoTracker fluorescence as a measure of mitochondrial network size. Area covered is significantly greater at 4 weeks compared with 70% confluent and 1 week confluent (n = 9 for 70% and 1 week and n = 6 for 4 weeks). (E) MitoTracker staining images acquired with identical exposure times using a ×20 objective demonstrates increased fluorescence at longer maturation times. Values shown indicate MFI per microgram of protein. Scale bar: 50 μm. (F) Quantification of MFI per microgram of protein and (G) MFI per cell nucleus. Significantly greater MFI per microgram and MFI/nucleus is observed with longer maturation times (n = 9 for 70% and 1 week and n = 6 for 4 weeks). (H) Measure of maximal citrate synthase activity showed a significant increase at 4 weeks (n = 4 per time point). (I) Relative expression of genes involved in mitochondrial dynamics (MFN1, MFN2, FIS1) and mitochondrial DNA replication and transcription (POLG, TFAM) during RPE maturation in vitro. Significant increases in FIS1 and MFN1 occur with RPE maturation (n = 3 per time point). Data were analyzed by ANOVA followed by Tukey's multiple comparison test.
Changes in oxidative metabolism associated with ARPE-19 maturation were evaluated by measuring mitochondrial abundance using MitoTracker CMTMRos38 and citrate synthase activity, a marker of mitochondrial mass.39 Compared with ARPE-19 cultured at 70% confluence, representative of an immature mesenchymal-like phenotype, or ARPE-19 confluent for 1 week, fully matured ARPE-19 (i.e., confluent for 4 weeks) had significantly greater mitochondrial signal (Figs. 1C, 1D), 2.0-fold greater MFI per microgram of protein, 2.0-fold greater MFI per nucleus (Figs. 1E–G) and 2.5-fold greater citrate synthase activity (Fig. 1H). These data indicate that in vitro maturation of RPE is associated with increased mitochondrial mass. This increased mitochondrial content was associated with changed expression of genes involved in mitochondrial fission and fusion (Fig. 1I). Both fission 1 (FIS1) and mitofusin 1 (MFN1) were significantly increased in ARPE-19 confluent for 1 week compared with 70% confluent cells and MFN1 was significantly increased in ARPE-19 confluent cells for 4 weeks compared with 70% confluent cells. Although ARPE-19 cell line is a commonly used culture model for studying RPE biology, recent transcriptome analysis has identified significant deviations of the ARPE-19 gene signature when compared with primary cells and native tissue.40 Of particular interest, some genes involved in fatty-acid metabolism were underexpressed in ARPE-19. To test if increased mitochondrial biogenesis during RPE maturation is a general phenomenon, we conducted similar analysis on primary hfRPE, which closely resembles native cells both morphologically and functionally.28,41 P1 hfRPE were matured on Transwells for up to 14 days, at which time cells acquired a TER comparable to the values recorded on native tissue (Supplementary Fig. S1A).42 Citrate synthase activity was much higher in unpolarized hfRPE than ARPE-19 and increased as early as day 3 (Supplementary Fig. S1B) concomitantly with the induction of the mitochondrial dynamic genes, FIS1 and MFN1 (Supplementary Fig. S1C).
Next we characterized the effect of RPE maturation on the protein levels of major OXPHOS subunits: ATP5A, a component of the F0-F1-ATPase; UQCR2, a component of complex III; MTCO-1, a component of complex IV; SDHB, a component II; and NDUFB8, a component of complex I. In ARPE-19, MTCO-1 and SDHB were significant increased during maturation (Fig. 2A). NDUFB8 was under the level of detection. The induction of mitochondrial proteins was more robust in primary hfRPE with UQCR2, MTCO-1, SDHB, and NDUFB8 being all significantly increased (Fig. 2B). We next analyzed gene expression of OXPHOS subunits: ATP5O, a component of the F0-F1-ATPase; COX4I1 and COX5B, components of complex IV; and NDUFB5, a component of complex I. A robust induction of ATP5O, COX5B, and NDUFB5 was observed in ARPE-19 cells after 1 week of maturation (Fig. 2C). At 4 weeks of confluence, gene expression of all subunits studied returned to a level similar or even lower in the case of NDUFB5 to subconfluent cells (Fig. 2C). This set of genes was also studied during hfRPE maturation. In primary RPE cells, expression of ATP5O was significantly higher after 3 days on Transwells, and continued to increase at 7 and 14 days (Fig. 2D). Gene expression of COX5B was significantly upregulated only at 3 days of culture on Transwells, and COX4I1 expression was not significantly increased compared with 1 day at any time point. Gene expression of NDUF5B also significantly increased with duration of maturation peaking at 7 days of culture (Fig. 2D).
Figure 2.

Increased OXPHOS protein and gene expression during RPE maturation. Levels of OXPHOS proteins during (A) ARPE-19 maturation and (B) hfRPE maturation and normalized to glyceraldehyde-3-phosphate dehydrogenase. For (A), n = 4 per time point; *P < 0.05, **P < 0.01 vs. 70% confluent: for (B), n = 3 per time point; *P < 0.05 vs. 1 day. ANOVA followed by Dunnett's multiple comparison test. (C) Gene expression of OXPHOS components measured by qPCR during ARPE-19 maturation and (D) hfRPE maturation. For (C), n = 3 per time point; ***P < 0.001 vs. 70% confluent; ##P < 0.01, ###P < 0.001 vs. 4 week confluent. ANOVA followed by Tukey's multiple comparison test. For (D), n = 3 per time point; *P < 0.05; **P < 0.01; ***P < 0.001 vs. day 1; ###P < 0.001 day 3 vs. day 7 or day 14; $P < 0.05 day 7 vs. 14. ANOVA followed by Tukey's multiple comparison test). (E, F) Gene expression of PGC-1α (PPARGC1A) and PGC-1β (PPARGC1B) measured by qPCR during (E) ARPE-19 maturation or (F) hfRPE maturation. Expression for (E), n = 3 for each time point; *P < 0.05, **P < 0.01 vs. 70% confluent. For (F), n = 3 for each time point; **P < 0.01, ***P < 0.001 versus day 1; #P < 0.05 day 3 vs. day 7. Data were analyzed by ANOVA followed by Tukey's multiple comparison test.
Based on these findings, we examined the expression the PGC-1 family of transcriptional coactivators, known regulators of OXPHOS gene expression. Maturation of ARPE-19 leads to a 5- to 6-fold increase in PPARGC1A (PGC-1α) expression at 1 and 4 weeks compared with PPARGC1A expression in subconfluent cells (Fig. 2E). In contrast to PPARGC1A genes, PPARGC1B (PGC1β) expression was significantly decreased (Fig. 2E) in matured ARPE-19. Maturation of hfRPE also was associated with increased expression of PPARGC1A but not PPARGC1B (Fig. 2F), suggesting that the increase in OXPHOS subunits observed during RPE maturation may be due to the concomitant induction of PPARGC1A. Absolute quantification of the PGC-1α transcripts indicated that although the expression level of PGC-1α is overall higher in hfRPE during the early stages of in vitro maturation, there is no statistical significance in transcript numbers between the two cell types once post-mitotic maturation is completed (Supplementary Fig. S2).
PGC-1α Drives Oxidative Phosphorylation and Fatty-Acid Oxidation in RPE
We next investigated the modulation of oxidative metabolism by PGC-1α in RPE by using adenovirus to overexpress murine PGC-1α (Ad-mPGC-1α) (Fig. 3A). Infection of ARPE-19 with Ad-mPGC-1α did not significantly affect endogenous human PPARGC1A; however, endogenous human PPARGC1B gene expression was significantly decreased (Fig. 3B), paralleling what we observed during ARPE-19 maturation (Fig. 2E). The effect of PGC-1α on downstream metabolic transcription factors including PPARA (which regulates genes of fatty-acid utilization); ESRRA, NRF1, and GABPA (which regulate OXPHOS genes); and NRE2L2 and FOXO3 (which regulate antioxidant genes) was analyzed. In ARPE-19, expression of mPGC-1α induced robust expression of PPARA, ESRRA, and GABPA, whereas expression of NRE2L2 was significantly reduced (Fig. 3B). Consistent with these results, multiple OXPHOS and fatty-acid β-oxidation genes were also significantly induced in Ad-mPGC-1α–infected ARPE-19 (Figs. 3C, 3D), suggesting that PGC-1α could regulate oxidative metabolism in the RPE. Measurement of the oxygen consumption rate (OCR) in ARPE-19 by extracellular flux analysis showed that PGC-1α overexpression led to a robust and significant increase in multiple phases of respiration measured, including basal respiratory rate, ATP-dependent, maximal respiratory rate, and spare respiratory capacity (Figs. 3E, 3F). We then analyzed the ability of substrate-limited ARPE-19 to use exogenously added palmitate for oxidative respiration. In ARPE-19 expressing mPGC-1α, a significant increase in OCR was observed following palmitate addition (Figs. 3F, 3G). Similar to the results obtained with ARPE-19, infection of confluent hfRPE monolayers with Ad-mPGC-1α led to the induction of the transcription factors PPARA, ESRRA, GABPA, and FOXO3 (Fig. 4A) and numerous OXPHOS protein subunits, both at the protein and gene levels (Figs. 4B–D). Measurement of hfRPE respiration showed a comparable increase in maximal respiratory rate and spare respiratory capacity, whereas basal respiration, which in the control cells was much higher than ARPE-19, was unaffected (Figs. 4E, 4F). All together, these data demonstrate that PGC-1α strongly induces mitochondrial respiration and promotes fatty-acid β-oxidation in RPE cells.
Figure 3.

Increased oxidative metabolism in ARPE-19 overexpressing PGC-1α. (A) Western blot of ARPE-19 infected with Ad-GFP or Ad-mPGC-1α for 48 hours. The Ad-mPGC-1α construct contains an enhanced green fluorescent protein tag fused to the N-terminal region of the PGC-1 sequence. Detection of mPGC-1α (113 kDa), GFP (26 kDa), and α-tubulin as a loading control in protein lysates. (B–D) Quantitative PCR analysis of endogenous PGC-1s, transcription factor genes (B), OXPHOS (C), and fatty-acid oxidation genes (D) in ARPE-19 48 hours following infection with Ad-GFP or Ad-mPGC-1α (n = 3 each group). (E) Oxygen consumption rates in GFP-infected (black line) and mPGC-1α–infected (gray line) ARPE-19. Oligomycin (2.5 μM), FCCP (500 nM), and rotenone and antimycin A (2 μM) were injected following the third, sixth , and ninth measurement, respectively. (F) Analysis of OCR parameters. PGC-1α significantly increased multiple phases of respiration in ARPE-19 (n = 3 each group). (G) Oxygen consumption rate following fatty-acid addition. Vertical bar indicates substrate addition. (H) Area under the curve analysis of OCR in the presence of BSA or palmitate (0.2 mM). PGC-1α significantly increased fatty-acid substrate oxidation in ARPE-19 (n = 3 each group; *P < 0.05 versus Ad-mPGC-1α plus BSA; #P < 0.05 versus Ad-GFP plus palmitate). Data analyzed by ANOVA followed by Tukey's multiple comparison test.
Figure 4.

Overexpression of mPGC-1α in hfRPE increases transcription factor and OXPHOS gene expression, and oxidative metabolism. (A) Gene expression analysis by qPCR of PPARGC1A and PPARGC1B and selected transcription factors 48 hours following ad-mPGC-1α infection of hfRPE. (B, C) Representative Western-blot (B) and quantification of OXPHOS proteins (C) shows increased expression of UQCR2 and MTCO1 in hfRPE 48 hours after adenovirus infection (n = 3 each group). (D) Gene expression analysis confirmed the induction of multiple OXPHOS genes in hfRPE (n = 3 each group). (E) Oxygen consumption rates in GFP-infected (black line) and mPGC-1α–infected (gray line) hfRPE. Oligomycin (1 mM), FCCP (500 nM), and rotenone and antimycin A (2 mM) were injected following the third, sixth, and ninth measurement, respectively. (F) Analysis of OCR parameters. PGC-1α significantly increased multiple phases of respiration in hfRPE (n = 5 each group).
Overexpression of PGC-1α Does Not Alter RPE Function
Gene expression analysis revealed that PGC-1α also induced the expression of some RPE-signature genes, such as BEST1, MITF, and TFEB in ARPE-19 (Fig. 5A); however, this effect was not observed in hfRPE (Fig. 5B), which expresses basal high levels of RPE-signature genes compared with ARPE-19.40 Western blot analysis confirmed the lack of effect on of PGC-1α overexpression on the critical RPE proteins MITF, cellular retinaldehyde binding protein (CRALBP), and RPE65 in hfRPE (Figs. 5B, 5C). Roggia and Ueta43 recently described that PGC-1α silencing alters ARPE-19 phagocytic activity. Using hfRPE, we found no difference in the degradation of rhodopsin following OS phagocytosis in Ad-mPGC-1α–infected cells compared with Ad-GFP–infected cells (Figs. 5D, 5E), indicating that although PGC-1α loss of function may be detrimental to RPE phagocytic function, PGC-1α gain of function has no enhancing effect. A significant decrease in TER was observed in Ad-GFP–infected hfRPE (Fig. 5F), but not in Ad-mPGC-1α, suggesting that whereas adenovirus infection may alter RPE barrier function, this deleterious effect is rescued by PGC-1α. Thus, overexpression of PGC-1α does not adversely affect RPE maturation or phagocytic function.
Figure 5.

Overexpression of PGC-1α does not alter hfRPE signature gene expression and functions. (A) Gene expression analysis by qPCR of ARPE-19 and hfRPE infected with Ad-GFP or Ad-mPGC-1α (n = 3 per group). (B) Western blot of RPE proteins in hfRPE infected with Ad-GFP or Ad-mPGC-1α. (C) Quantification of Western blots (n = 4 per group). (D) Western blot of rhodopsin following outer segment phagocytosis in hfRPE 0, 4, and 12 hours after phagocytosis. (E) Quantification of Western blot (n = 3 per time point). (F) Transepithelial resistance of hfRPE pre- and postinfection with Ad-GFP and Ad-mPGC-1α (n = 26 per group, paired t-test, Ad-GFP P < 0.0001; Ad-PGC-1α P = 0.1600).
PGC-1α Modulates Antioxidant Gene Expression and Reduces Oxidant-Mediated Cell Death in RPE
Consistent with our observations of increased oxidative metabolism and PGC-1α expression, RPE maturation was also characterized by the induction of several antioxidant enzymes. Specifically, CAT and SOD2 were both induced after 1 and 4 weeks of ARPE-19 maturation, respectively (Fig. 6A). Maturation of hfRPE was associated with the additional induction of SOD1 and GPX1 (Supplementary Fig. S3A). Overexpression of mPGC-1α in ARPE-19 upregulated a wide range of antioxidant enzymes, including CAT, GPX1, PRDX3, SOD1, SOD2, and TXN2 (Fig. 6B). Similar results were obtained in mPGC-1α–infected hfRPE (Supplementary Fig. S3B).
Figure 6.

PGC-1α induces antioxidant gene expression in RPE and protects RPE from oxidant-mediated cell death. (A) Gene expression of antioxidant genes measured by qPCR during ARPE-19 maturation (n = 3 per group, data analyzed by ANOVA followed by Tukey's multiple comparison test). (B) Expression of antioxidant genes analyzed by qPCR 48 hours after Ad-GFP or Ad-mPGC1α infection in ARPE-19. (C) Three-hour H2O2 treatment of ARPE-19 increases DCF fluorescence in a dose-dependent manner (n = 4 per group). (D) Eighteen-hour H2O2 treatment of ARPE-19 increases LDH release into cell culture supernatants in a dose-dependent manner (n = 4 per group). (E) Lactate dehydrogenase release measured in cell culture supernatants after 18 hours of H2O2 treatment was significantly reduced in PGC-1α–infected cells compared with GFP-infected cells (n = 4 per group; *P < 0.05; **P < 0.01; ***P < 0.001). (F) Phase-contrast and fluorescent photomicrographs of Ad-GFP– and Ad-mPGC-1a–infected ARPE-19 before and after H2O2 treatment. ARPE-19 were infected with Ad-GFP or Ad-mPGC1a for 48 hours, serum-starved, and then treated with H2O2 for 18 hours. (G) Treatment of serum-starved ARPE-19 with 250 μM hydroquinone for 12 hours strongly induces apoptosis quantified using the Nucleosome ELISA kit. Compared with ad-GFP controls, PGC-1α overexpressing cells were significantly protected against HQ-induced apoptosis (n = 3–4, ANOVA followed by Dunnett's multiple comparison test). Scale bar: 200 μm.
The protective role of PGC-1α against RPE oxidative damage was evaluated by exposing RPE cells to clinically relevant oxidants. Treatment of serum-starved, confluent ARPE-19 with 1 mM H2O2, a strong endogenous oxidant produced during mitochondrial respiration and OS phagocytosis,13 induced ROS accumulation after 3 hours and caused 35% cell death measured by quantification of LDH release after 18 hours (Figs. 6C, 6D). Ad-mPGC1α significantly reduced H2O2-induced cytotoxicity from 25% in ARPE-19 infected with Ad-GFP to 8% (Fig. 6E). Increased viability of Ad-mPGC1-α overexpressing ARPE-19 exposed to H2O2 was further confirmed by their improved cellular morphology in contrast to control cells that appear rounded, shrunk, and detached from the cell culture plate (Fig. 6F). A similar rescue effect of Ad-mPGC1-α was observed in ARPE-19 cells exposed to a cytotoxic dose of the cigarette-smoke oxidant hydroquinone (Fig. 6G). These results demonstrate that PGC-1α is protective against oxidant-mediated RPE cell death.
Discussion
Increased Mitochondrial Mass, OXPHOS Gene Expression, and PGC-1α Expression During RPE Maturation
Functional maturation depends on the induction of a specific metabolic program able to support the energetic requirements of specialized tissues. By controlling the rate of cellular growth and division, a metabolic switch from glycolysis to oxidative metabolism is generally observed during terminal differentiation of cells.44 The present study showed that, like several other cell types, RPE maturation is associated with mitochondrial biogenesis. Consistent with an increase in mitochondrial mass and expression of OXPHOS genes, PPARGC1A was found to be robustly and rapidly induced during RPE maturation. Increased PGC-1α expression has been associated with the differentiation of many cell types, including myotubes, mesenchymal stem cells differentiated into hepatocytes, dendritic cells, Schwann cells, and intestinal epithelium.45–50 In addition, the increased expression of genes involved in both mitochondrial fission (FIS1) and fusion (MFN1) indicates that increased mitochondrial mass observed during RPE maturation is likely a dynamic process, with both mitochondrial fission and fusion occurring. Similar to other cell types, such as mytotubes, dendritic cells, and hepatic differentiation of mesenchymal stem cells, we found that RPE maturation was also associated with increased OXPHOS proteins. All together, our data suggest that induction of mitochondrial biogenesis and OXPHOS may be an important driver of RPE maturation. This concept is supported by the recent report that blockade of the tricarboxylic acid cycle and electron transport chain blunt the induction of RPE-specific genes during maturation51; however, definitive evidence of a critical role for PGC-1α in RPE functional differentiation will required the generation of RPE cells genetically deficient for PGC-1α.
Differential Expression of PGC-1 Isoforms in RPE
Conversely to PPARGC1A, PPARGC1B expression remained low or even decreased during in vitro RPE maturation. Isoforms of PGC-1 are generally considered to share many transcription factor targets and therefore to function similarly; however, recent evidence indicates that regulation and function of PGC-1 isoforms differ in a number of cellular systems. In the liver, PGC-1α expression is specifically induced in response to starvation to promote gluconeogenesis.52 On the other hand, dietary fat intake induces PGC-1β expression in the liver, where it regulates lipid synthesis and trafficking.53 Other examples of differential regulation were identified in adipose tissues. PGC-1α is induced in brown fat after cold exposure to promote thermogenesis,54,55 whereas PGC-1β is specifically upregulated during adipocyte differentiation in order to control energy expenditure.55 Clearly, there is strong precedence for the nonredundant, although nonexclusive roles of the PGC1 isoforms. Surprisingly, in RPE, we also observed that overexpression of PGC-1α repressed even further the expression of PPARGC1B. While an interaction between PGC-1α and PGC-1β has been demonstrated, as PGC-1β can drive PGC-1α expression in 10T1/2 cells,17 this is to our knowledge the first example of the transcriptional repression of PGC-1β by PGC-1α. The molecular regulation of PPARGC1B expression in RPE is unclear and will be explored in future studies.
PGC-1α Protects RPE Against Oxidant-Mediated Cell Death
Age- and/or disease-dependent imbalance between oxidants and antioxidant capacity has been proposed as an important contributor to the progressive dysfunction and atrophy of RPE during AMD. Exposure to environmental oxidants such as cigarette smoke56 combined with the lifelong accumulation of pro-oxidative degradation products from OS phagocytosis57 can lead to deleterious ROS levels and increased mitochondrial DNA damage. Susceptibility of RPE to oxidative stress is further enhanced by the dysregulation and/or decreased activity of key components of the cell's antioxidant mechanism observed during aging and/or AMD.8,58 For example, CAT protein levels and activity were both found to be decreased in the aging eye.9,59,60 Additionally, aged RPE failed to increase the expression of the transcription factor NFE2L2, a critical regulator of antioxidant enzymes, in response to oxidative insult, whereas such induction was observed in the RPE of younger animals.61 We show here that overexpression of PGC-1α in RPE is able to powerfully increase the expression of multiple antioxidant enzymes including CAT, GPX1, PRDXs, and SOD2. Moreover, PGC-1α effectively reduced RPE cell death induced by H2O2 and hydroquinone, two potent oxidants that have been implicated in AMD pathogenesis.62,63 CAT and GPX1 have been previously shown to protect RPE from H2O2-mediated damage and cell death,64,65 and therefore, may be contributing to the cytoprotective effect of PGC-1α. Retinal pigment epithelium dysfunction and progressive atrophy of the outer retina induced by genetic deletion of Sod1 or Sod2 in mice suggest also a role for impaired superoxide clearance in AMD.10,66 Recently, the ARMS/HTRA1 AMD-associated risk haplotype has been associated with an altered SOD2 stress response in patient-derived iPS-RPE,67 suggesting that RPE in patients carrying the ARMS/HTRA1 risk alleles have increased susceptibility to oxidative damage and that gene therapy approaches to increase SOD2 expression could serve as potential treatment for AMD. Surprisingly, overexpression of SOD2 alone in ARPE-19 increases protein oxidative damage likely due to cytoplasmic H2O2 accumulation.65 However, when GPX1 was also overexpressed, protein carbonyls were reduced.65 Therefore, the ability of PGC-1α to simultaneously induce the expression of both cytoplasmic and mitochondrial antioxidant enzymes in RPE would most likely counteract the effect of increased protein carbonyls secondary to SOD2 overexpression alone. This observation underscores the potential benefit of targeting PGC-1α to activate multiple and properly coordinated antioxidant responses.
PGC-1α as a Potential Gene Therapy for Dry AMD
Our results demonstrate that increased expression of PGC-1α in RPE increases oxidative metabolism and gene expression of OXPHOS subunits. Concomitantly, numerous antioxidant genes were also induced, potentially limiting the increase of ROS due to increased oxidative metabolism. Thus, as a powerful transcriptional coactivator, PGC-1α gain of function is likely to alleviate the increased oxidative stress and decreased oxidative metabolism associated with AMD.8,9,12 Similar approaches aimed at increasing PGC-1α level and/or function are currently under significant investigations for the treatment of neuronal degenerative diseases, such as Huntington's and Parkinson's.68–70 Manipulation of PGC-1α expression for therapeutic purposes may be limited due to potential detrimental effects of overexpression of PGC-1α in, for example, dopaminergic neurons.71 However, we showed that overexpression of PGC-1α in primary cells did not affect RPE survival, expression of RPE-signature genes, or the cell's phagocytic and barrier functions. Through its ability to control RPE response to oxidative stress and increase RPE survival in the face of an oxidative insult, PGC-1α appears as a promising therapeutic target for preventing vision loss associated with dry AMD.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health Grant 1R01EY023682 (ZA, MSG), a Research to Prevent Blindness Unrestricted Grant (MSG), and donations to the Macular Degeneration Research, a program of the BrightFocus Foundation (MSG).
Disclosure: J. Iacovelli, None; G.C. Rowe, None; A. Khadka, None; D. Diaz-Aguilar, None; C. Spencer, None; Z. Arany, None; M. Saint-Geniez, None
References
- 1. Jager RD,, Mieler WF,, Miller JW. Age-related macular degeneration. N Engl J Med. 2008; 358: 2606–2617. [DOI] [PubMed] [Google Scholar]
- 2. Friedman DSD,, O'Colmain BJB,, Muñoz BB,, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004; 122: 564–572. [DOI] [PubMed] [Google Scholar]
- 3. Rickman CB,, Farsiu S,, Toth CA,, Klingeborn M. Dry age-related macular degeneration: mechanisms therapeutic targets, and imaging. Invest Ophthalmol Vis Sci. 2013; 54:ORSF68–ORSF80. [DOI] [PMC free article] [PubMed]
- 4. Bhutto I,, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med. 2012; 33: 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cano M,, Thimmalappula R,, Fujihara M,, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010; 50: 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hahn P,, Milam AH,, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol. 2003; 121: 1099–1105. [DOI] [PubMed] [Google Scholar]
- 7. Lin H,, Xu H,, Liang FQ,, et al. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011; 52: 3521–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karunadharma PP,, Nordgaard CL,, Olsen TW,, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010; 51: 5470–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frank RN,, Amin RH,, Puklin JE. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am J Ophthalmol. 1999; 127: 694–709. [DOI] [PubMed] [Google Scholar]
- 10. Mao H,, Seo S-J,, Biswal MR,, et al. Mitochondrial oxidative stress in the retinal pigment epithelium leads to localized retinal degeneration. Invest Ophthalmol Vis Sci. 2014; 55: 4613–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao Z,, Chen Y,, Wang J,, et al. Age-related retinopathy in NRF2-deficient mice. PLoS One. 2011; 6: e19456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012; 33: 418–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miceli MVM,, Liles MRM,, Newsome DAD. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994; 214: 242–249. [DOI] [PubMed] [Google Scholar]
- 14. Nguyen T,, Nioi P,, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009; 284: 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Finck BN,, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006; 116: 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Handschin C,, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006; 27: 728–735. [DOI] [PubMed] [Google Scholar]
- 17. Arany Z,, Lebrasseur N,, Morris C,, et al. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007; 5: 35–46. [DOI] [PubMed] [Google Scholar]
- 18. Lin J,, Wu H,, Tarr PT,, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002; 418: 797–801. [DOI] [PubMed] [Google Scholar]
- 19. Lai L,, Leone TC,, Zechner C,, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008; 22: 1948–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zechner C,, Lai L,, Zechner JF,, et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010; 12: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uldry M,, Yang W,, St-Pierre J,, Lin J,, Seale P,, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006; 3: 333–341. [DOI] [PubMed] [Google Scholar]
- 22. Rowe GC,, Patten IS,, Zsengeller ZK,, et al. Disconnecting mitochondrial content from respiratory chain capacity in PGC-1-deficient skeletal muscle. Cell Rep. 2013; 3: 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. St-Pierre J,, Drori S,, Uldry M,, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006; 127: 397–408. [DOI] [PubMed] [Google Scholar]
- 24. Saint-Geniez M,, Jiang A,, Abend S,, et al. PGC-1α regulates normal and pathological angiogenesis in the retina. Am J Pathol. 2013; 182: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Egger A,, Samardzija M,, Sothilingam V,, et al. PGC-1α determines light damage susceptibility of the murine retina. PLoS One. 2012; 7: e31272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo X,, Dason ES,, Zanon-Moreno V,, et al. PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am J Pathol. 2014; 184: 1017–1029. [DOI] [PubMed] [Google Scholar]
- 27. Ford KM,, D'Amore PA. Molecular regulation of vascular endothelial growth factor expression in the retinal pigment epithelium. Mol Vis. 2012; 18: 519–527. [PMC free article] [PubMed] [Google Scholar]
- 28. Maminishkis A,, Chen S,, Jalickee S,, et al. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest Ophthalmol Vis Sci. 2006; 47: 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li CH,, Tam PKS. An iterative algorithm for minimum cross entropy thresholding. Pattern Recognit Lett. 1998; 19: 771–776. [Google Scholar]
- 30. Rhee J,, Inoue Y,, Yoon JC,, et al. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A. 2003; 100: 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J,, Iacovelli J,, Spencer C,, Saint-Geniez M. Direct effect of sodium iodate on neurosensory retina. Invest Ophthalmol Vis Sci. 2014; 55: 1941–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vandesompele J,, De Preter K,, Pattyn F,, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3:RESEARCH0034. [DOI] [PMC free article] [PubMed]
- 33. Tsang SH,, Burns ME,, Calvert PD,, et al. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998; 282: 117–121. [DOI] [PubMed] [Google Scholar]
- 34. Singh R,, Phillips MJ,, Wright LS,, et al. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013; 22: 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burke JM. Epithelial phenotype and the RPE: is the answer blowing in the Wnt? Prog Retin Eye Res. 2008; 27: 579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeffer BA,, Philp NJ. Cell culture of retinal pigment epithelium: special issue. Exp Eye Res. 2014; 126: 1–4. [DOI] [PubMed] [Google Scholar]
- 37. Dunn KC,, Aotaki-Keen AE,, Putkey FR,, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996; 62: 155–169. [DOI] [PubMed] [Google Scholar]
- 38. Pendergrass W,, Wolf N,, Poot M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A. 2004; 61: 162–169. [DOI] [PubMed] [Google Scholar]
- 39. Larsen S,, Nielsen J,, Hansen CN,, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012; 590: 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strunnikova NV,, Maminishkis A,, Barb JJ,, et al. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum Mol Genet. 2010; 19: 2468–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maminishkis A,, Miller SS. Experimental models for study of retinal pigment epithelial physiology and pathophysiology. J Vis Exp. 2010; (45). doi:http://dx.doi.org/10.3791/2032. [DOI] [PMC free article] [PubMed]
- 42. Quinn RH,, Miller SS. Ion transport mechanisms in native human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1992; 33: 3513–3527. [PubMed] [Google Scholar]
- 43. Roggia MF,, Ueta T. αvβ5 Integrin/FAK/PGC-1α pathway confers protective effects on retinal pigment epithelium. PLoS One. 2015; 10: e0134870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Agathocleous M,, Harris WA. Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol. 2013; 23: 484–492. [DOI] [PubMed] [Google Scholar]
- 45. Wanet A,, Remacle N,, Najar M,, et al. Mitochondrial remodeling in hepatic differentiation and dedifferentiation. Int J Biochem Cell Biol. 2014; 54: 174–185. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y,, Marsboom G,, Toth PT,, Rehman J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS One. 2013; 8: e77077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zaccagnino P,, Saltarella M,, Maiorano S,, et al. An active mitochondrial biogenesis occurs during dendritic cell differentiation. Int J Biochem Cell Biol. 2012; 44: 1962–1969. [DOI] [PubMed] [Google Scholar]
- 48. Cowell RM,, Blake KR,, Inoue T,, Russell JW. Regulation of PGC-1alpha and PGC-1alpha-responsive genes with forskolin-induced Schwann cell differentiation. Neurosci Lett. 2008; 439: 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Remels AHV,, Langen RCJ,, Schrauwen P,, Schaart G,, Schols AMWJ,, Gosker HR. Regulation of mitochondrial biogenesis during myogenesis. Mol Cell Endocrinol. 2010; 315: 113–120. [DOI] [PubMed] [Google Scholar]
- 50. D'Errico I,, Salvatore L,, Murzilli S,, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc Natl Acad Sci U S A. 2011; 108: 6603–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adijanto J,, Philp NJ. Cultured primary human fetal retinal pigment epithelium (hfRPE) as a model for evaluating RPE metabolism. Exp Eye Res. 2014; 126: 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoon JC,, Puigserver P,, Chen G,, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001; 413: 131–138. [DOI] [PubMed] [Google Scholar]
- 53. Lin J,, Yang R,, Tarr PT,, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005; 120: 261–273. [DOI] [PubMed] [Google Scholar]
- 54. Puigserver P,, Wu Z,, Park CW,, Graves R,, Wright M,, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998; 92: 829–839. [DOI] [PubMed] [Google Scholar]
- 55. Kamei Y,, Ohizumi H,, Fujitani Y,, et al. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci U S A. 2003; 100: 12378–12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Espinosa-Heidmann DG,, Suner IJ,, Catanuto P,, Hernandez EP,, Marin-Castano ME,, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006; 47: 729–737. [DOI] [PubMed] [Google Scholar]
- 57. Sparrow J. Bisretinoids of RPE lipofuscin: trigger for complement activation in age-related macular degeneration. In: Lambris JD Adamis AP, eds. Inflammation and Retinal Disease: Complement Biology and Pathology. New York, NY: Springer; 2010; 63–74. [DOI] [PubMed]
- 58. Arnouk H,, Lee H,, Zhang R,, Chung H,, Hunt RC,, Jahng WJ. Early biosignature of oxidative stress in the retinal pigment epithelium. J Proteomics. 2011; 74: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liles MR,, Newsome DA,, Oliver PD. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch Ophthalmol. 1991; 109: 1285–1288. [DOI] [PubMed] [Google Scholar]
- 60. La Paz De MA Zhang J, Fridovich I. Antioxidant enzymes of the human retina: effect of age on enzyme activity of macula and periphery. Curr Eye Res. 1996; 15: 273–278. [DOI] [PubMed] [Google Scholar]
- 61. Sachdeva MM,, Cano M,, Handa JT. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp Eye Res. 2014; 119: 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beatty S,, Koh H,, Phil M,, Henson D,, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000; 45: 115–134. [DOI] [PubMed] [Google Scholar]
- 63. Bertram KM,, Baglole CJ,, Phipps RP,, Libby RT. Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. Am J Physiol Cell Physiol. 2009; 297: C1200–C1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rex TS,, Tsui I,, Hahn P,, et al. Adenovirus-mediated delivery of catalase to retinal pigment epithelial cells protects neighboring photoreceptors from photo-oxidative stress. Hum Gene Ther. 2004; 15: 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu L,, Oveson BC,, Jo Y-J,, et al. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid Redox Signal. 2009; 11: 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Imamura Y,, Noda S,, Hashizume K,, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2006; 103: 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang J,, Li Y,, Chan L,, et al. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum Mol Genet. 2014; 23: 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tsunemi T,, Ashe TD,, Morrison BE,, et al. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012; 4:142ra97. [DOI] [PMC free article] [PubMed]
- 69. Shin J-H,, Ko HS,, Kang H,, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell. 2011; 144: 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsunemi T,, La Spada AR. PGC-1α at the intersection of bioenergetics regulation and neuron function: from Huntington's disease to Parkinson's disease and beyond. Prog Neurobiol. 2012; 97: 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Clark J,, Silvaggi JM,, Kiselak T,, et al. Pgc-1α overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased Bdnf. PLoS One. 2012; 7: e48925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.