

Abstract
Purpose
Retinitis pigmentosa (RP) is a group of clinically and genetically heterogeneous hereditary retinal diseases that result in blindness due to photoreceptor degeneration. Mutations in the rhodopsin (RHO) gene are the most common cause of autosomal dominant RP (adRP) and are responsible for 16% to 35% of adRP cases in the Western population. Our purpose was to investigate the contribution of RHO to adRP in the Israeli and Palestinian populations.
Methods
Thirty-two adRP families participated in the study. Mutation detection was performed by whole exome sequencing (WES) and Sanger sequencing of RHO exons. Fluorescence PCR reactions of serially diluted samples were used to predict the percentage of mosaic cells in blood samples.
Results
Eight RHO disease-causing mutations were identified in nine families, with only one novel mutation, c.548-638dup91bp, identified in a family where WES failed to detect any causal variant. Segregation analysis revealed that the origin of the mutation is in a mosaic healthy individual carrying the mutation in approximately 13% of blood cells.
Conclusions
This is the first report of the mutation spectrum of a known adRP gene in the Israeli and Palestinian populations, leading to the identification of seven previously reported mutations and one novel mutation. Our study shows that RHO mutations are a major cause of adRP in this cohort and are responsible for 28% of adRP families. The novel mutation exhibits a unique phenomenon in which an unaffected individual is mosaic for an adRP-causing mutation.
Keywords: retinal degeneration, inherited blindness, genetic defects, rhodopsin, mosaicism
Retinitis pigmentosa (RP) is a heterogeneous group of the most common form of inherited retinal degenerations (IRDs), with a prevalence of approximately 1:4500 in the United States and Europe1–4 and 1:2100 in the vicinity of Jerusalem.5 Early clinical symptoms include loss of night and peripheral vision, leading eventually to total vision loss in most patients at later stages of the disease.6 The disorder can be inherited in different patterns including autosomal recessive (arRP), autosomal dominant (adRP), and X-linked (xlRP).7
Rhodopsin (RHO, OMIM *180380) was the first gene in which mutations were reported to cause RP.8 A single mutation (p.Pro23His) was identified in a large number of American patients with adRP and is currently known to be the most common RP mutation in North America.9 More than 150 RHO mutations have been identified so far that are responsible for 16% to 35% of adRP cases and for approximately 10% of all RP cases in Europe and the United States.10–18 In addition, a few RHO mutations are known to cause arRP19 and adCSNB20 (autosomal dominant congenital stationary night blindness).
Genetic analyses of the Israeli and the Palestinian populations in the last decade have revealed a large number of founder mutations as the cause of arRP21–24; however, no mutations associated with adRP have been reported so far in these populations. Owing to the high prevalence of RHO mutations as the cause of adRP in other populations, we selected this gene as the first to be systematically screened for mutations among our adRP cohort. Our analysis revealed previously reported mutations as well as a novel mutation. In addition, we identified the first mosaic individual to be reported with an RP-causing mutation.
Materials and Methods
Patient Recruitment and Clinical Evaluation
The tenets of the Declaration of Helsinki were followed during the course of the study. Before obtaining a blood sample for DNA analysis, informed consent was obtained from patients and family members who participated in this study.
Clinical evaluations included a detailed family history, a full ophthalmologic examination, electrooculography, full-field electroretinography, color vision testing using the Ishihara 38-plate test and Farnsworth D-15 panel, optical coherence tomography (OCT), color and infrared fundus photography, autofluorescence imaging, and fluorescein angiography, performed as previously described.25
Genetic Analysis
Thirty-two families with adRP were enrolled in the study: three Arab-Muslim families of Palestinian origin and 29 Israeli Jewish families. Genomic DNA was extracted from peripheral blood of the participants by using FlexiGene DNA kit (QIAGEN, Hilden, Germany). Nine samples underwent an in-house whole exome sequencing (WES) analysis (see Supplementary Methods).
DNA samples of the 32 index cases were screened for mutations in RHO (NCBI Reference Sequence NM_ 000539.3) exons (and flanking intronic sequences) with PCR followed by Sanger sequencing. Primers (Supplementary Table S1) were designed by using the PRIMER3 program (http://frodo.wi.mit.edu/; provided in the public domain by the Whitehead Institute for Biomedical Research, Cambridge, MA, USA). For fluorescent PCR of RHO–exon 3, the forward primer was 5′ labeled with Fam. Twenty-seven PCR cycles were performed in a volume of 30 μL. A genomic DNA sample of a heterozygous patient (MOL1076 III:1) was serially diluted with a sample of a wild-type family member (MOL1076 I:2), to obtain mutation ratios of 50% (no dilution), 25%, 12.5%, 6.25%, 3.13%, and 1.56%. The fluorescence was measured with ABI PRISM 3100 Genetic Analyzer (Life Technologies, Grand Island, NY, USA). Peaks were analyzed with Peak Scanner 2 program.
Results
In the last decade, we recruited 649 families with RP, 32 of whom showed a clear AD inheritance pattern. We performed WES analysis on nine cases (from seven families) and identified the cause of disease in two families: a PRPF3 mutation in MOL010826 and a previously reported RHO mutation (p.Arg135Trp) in MOL0843 (Table 1). We subsequently performed Sanger sequencing analysis of the five RHO exons in all of the remaining 30 index cases with no identified mutation. A combination of WES and Sanger sequencing revealed eight disease-causing mutations in nine families; only one of the mutations is novel (Fig. 1; Table 1). The seven mutations reported previously are missense, and the novel mutation is a tandem duplication of 91 nucleotides. Five of the seven reported missense mutations alter amino acid residues that have been previously shown to be affected by other mutations (Table 1). While seven of the mutations were family-specific, one (c.403C>T, p.Arg135Trp) was observed in two unrelated families from different origins (MOL0843 of North African Jewish origin and MOL0418 of Turkmenistan Jewish origin).
>Table 1.
A List of Families and RHO Mutation Details
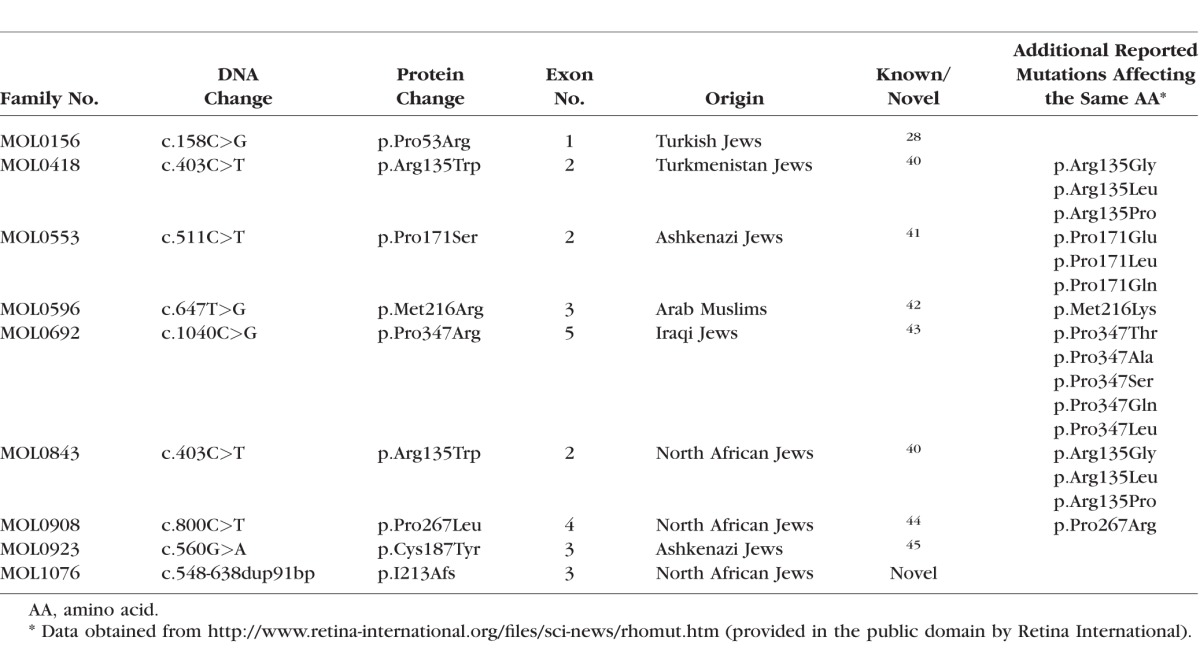
Figure 1.

Pedigree structure of nine families with RHO mutations. The family number, mutation name (at the cDNA and protein levels) are noted above each pedigree. The individual number is listed below each symbol. Black shapes indicate affected family members, white shapes indicate healthy family members, gray shapes indicate subjects who are reported to have visual defect consistent with RP. Index cases are marked with black arrow.
Clinical data of 12 patients with heterozygous RHO mutations were collected and all had clinical symptoms and signs of RP (Table 2). The age of disease onset ranged from 6 to 43 years and visual acuity was from 0.3 to 1.0. The fundus appearance was compatible with typical RP except for one patient (MOL0908 II:1), who was diagnosed with sector RP. Scotopic and photopic ERG amplitudes were nondetectable in approximately half of the cases.
Table 2.
Ocular Information of Patients With RHO Mutations

Interestingly, while we were not able to identify the disease-causing mutation by WES in family MOL1076, the subsequent Sanger sequencing revealed the cause of disease (Table 1). MOL1076 includes four affected family members from two generations (Fig. 1). The index case (III:4) was diagnosed at the age of 6 years with RP accompanied by high myopia, pigmentary changes at the posterior pole and the peripheral retina, severely reduced ERG amplitudes, and normal color vision (Table 2). Samples of three family members (mother and two affected daughters) underwent WES analysis, which revealed 126,114 variants on average; of these, 77,950 were heterozygous. Filtering the WES data for minor allele frequency, pathogenicity scores, and shared genotypes revealed 27 sequence changes that were heterozygous and shared by all affected individuals. These changes were considered nonpathogenic because they were in the 5′ or 3′ untranslated region and not in the open reading frame of the main transcript, or missense changes affecting amino acids that were not evolutionarily conserved. In parallel, the index case (1076 III:4) was analyzed by Sanger sequencing for RHO mutations. The PCR analysis of exon 3 revealed three PCR products (Fig. 2A, bottom) in the affected individuals; the size of the shorter band (No. 1) was compatible with the expected size of the wild-type (WT) product (349 bp) and the two additional bands (Nos. 2, 3) were approximately 100 bp longer. Sequencing analysis of the PCR products from the affected individuals revealed a heterozygous tandem duplication of 91 bp (c.548-638dup91 bp) (Fig. 2B). The appearance of three PCR bands is likely due to the heating–cooling PCR cycles, generating two homoduplexes (WT-WT and mutant-mutant) and a heteroduplex (WT-mutant) that migrate to three different locations on the agarose gel.
Figure 2.

Analysis of the RHO c.548-638dup91bp mutation. (A) Upper panel: A condensed family tree of MOL1076. Black shapes indicate affected family members, white shapes indicate healthy family members. The individual number is listed above each individual. The haplotype surrounding the RHO gene is described below each individual symbol, and the SNPs that we used to create the haplotype are depicted at the left side (rs numbers and the location along chromosome 3). Lower panel: PCR products on a 2.5% agarose gel. The red arrow indicates the weak band of the mosaic individual. (B) Chromatograms of the duplication region in WT and heterozygous individuals. (C) Upper panel: Chromatogram of the PCR product of the mosaic individual. Middle panel: Enlargement of the duplication area. Lower panel: Chromatogram of the PCR product (red arrow in [A] of MOL1076 I:1) extracted from the agarose gel. (D) Schematic representation of the RHO gene. Light green represents the UTRs, dark green represents the exons, red represents the mutated sequence (the duplication and the frameshift). Termination codons are highlighted in exon 5.
Following the identification of the duplication by Sanger sequencing, we took a closer look at the WES data of RHO exon 3. A base coverage analysis along exon 3 of three patients and three controls did not reveal any significant differences (Supplementary Fig. S1). In addition, no variants in the duplication region were identified in the WES data.
The duplication of 91 nucleotides is predicted to result in a frameshift; such frameshift mutations usually create a premature stop codon. However, the nucleotide sequence of the new mutated frame did not contain a premature termination codon, and the mutation therefore is expected to result in a 360-amino acid (aa) long protein (12 aa longer than the WT protein) with a termination codon in the terminal exon 5 (Fig. 2D).
A bioinformatics analysis using OCTOPUS showed that the last two transmembrane domains (Nos. 6, 7) of the rhodopsin protein are eliminated by the frameshift mutation, and the C-terminal mutated protein sequence is expected to be located intracellularly, forming a globular loop (Supplementary Fig. S2A). This topology modification is also supported by a Phyre2 analysis (Supplementary Fig. S2B). The affected C-terminus region includes the retinal-binding site, sites of interaction with cytoplasmic proteins, and phosphorylation sites.27 In addition, the mutation is expected to cause a conformational change of the protein, not only in the secondary structure level, as predicted by OCTOPUS and Phyre2, but also in the tertiary structure (Supplementary Figs. S2C, S2D), as predicted by Phyre2 and PDBsum. Although a new alpha helix structure is formed, the structure at the protein level is disorganized and the new alpha helix is not aligned to other protein components. The predicted conformational shift is also expected (by using PDBsum analysis) to affect protein regions upstream of the mutation site (Supplementary Fig. S2D).
To follow the segregation of the mutation along generations, Sanger sequencing of exon 3 was performed in individuals 1076 I:1 and 1076 I:2, and only the WT allele was evident by initial inspection of the chromatogram (Fig. 2C, top panel). We subsequently performed a haplotype analysis (Fig. 2A) by using six single nucleotide polymorphism (SNP) markers that flank the mutation site (three markers on each side of the mutation). The analysis revealed that the three affected individuals (1076 II:5, III:1, III:4) share the same haplotype that was inherited from the unaffected grandfather (1076 I:1; Fig. 2A, top panel). Interestingly, the PCR analysis of 1076 I:1 showed a faint band on the agarose gel (Fig. 2A, red arrow in the lower panel) at a location that is expected to represent the mutated allele. The fact that the haplotype carrying the mutation was inherited from 1076 I:1, who also produced this faint band, prompted us to check the original chromatogram at a higher resolution. Although the chromatogram was dominated by peaks representing the WT nucleotides, starting from the duplication site, a low but steady appearance of peaks representing the mutated allele was evident (Fig. 2C, middle panel). To obtain a better quality sequence, we performed multiple PCR reactions (resulting in a high amount of PCR product) and ran it at a slow pace on a 2.5% agarose gel. A gel slice containing this faint band was cut and the DNA was sequenced, resulting in a clear representation of the mutated allele (Fig. 2C, bottom). Our result indicated that blood cells of individual 1076 I:1 either contained two WT RHO copies or were heterozygous for the mutation; in other words, 1076 I:1 is mosaic for the analyzed mutation. To quantify the percentage of cells carrying RHO mutation in 1076 I:1, we performed a fluorescence-based PCR assay with a limited number of PCR cycles (n = 27) aiming to avoid the saturation phase. We then calculated the area below peaks representing the WT versus the mutated alleles (Fig. 3A) in a series of samples representing different concentrations of the mutated allele to generate the calibration curve (Fig. 3B), which showed a high coefficient of determination (r2 = 0.998). A comparison of the mutated allele peak area in 1076 I:1 to the serial dilutions of the mutated allele (Fig. 3B) demonstrated that the mutant allele accounted for approximately 6.5% of the PCR product (based on the following equation: y = 1.0877X − 0.0023, x = 0.062209), representing approximately 13% of heterozygous blood cells. We performed visual acuity and ERG testing on individual 1076 I:1 who was found to be mosaic for the c.548-638dup91bp mutation with normal visual acuity and scotopic and photopic ERG amplitudes that were within the normal range.
Figure 3.

(A) Electropherograms of fluorescence-based PCR of WT, heterozygous, and mosaic individuals visualized by the Peak Scanner software. The x-axis shows the size of the fragment and the y-axis shows the fluorescence intensity. The WT product is 292-bp long and the mutated product is 383-bp long. The area underneath the peaks is presented in a box next to the peak. (B) Standard curve of the c.548-638dup91bp allele quantity, derived from serial dilutions of DNA from a heterozygous patient (MOL1076 III:1) and a control (MOL1076 I:2), in which 50%, 25%, 12.5%, 6.25%, 3.13%, and 1.56% of the DNA has a mutation. For calculations, the area under the peaks was used. The mutated allele in the grandfather MOL1076 I:1 exhibits approximately 6.5% of the PCR product, which means that he carries approximately 13% heterozygous mutated blood cells.
Discussion
We report here the first genetic analysis of an adRP gene in general and the rhodopsin gene in particular, in the Israeli and Palestinian populations. Our data showed that RHO mutations are a major cause (and are probably the major cause) of adRP in these populations and are responsible for 28% (9/32 families) of adRP families (including 36 RP patients) in this cohort. Our data are consistent with those obtained from other countries: Italy: 28%,14 United Kingdom: 30%,28 United States: 24%29 and 30%,17 and France: 10.3%.10 A review analysis has reported that 25% of adRP cases worldwide are caused by RHO mutations.7
Of the eight mutations we identified, only one (c.548-638dup91bp) was novel and the remaining are likely to represent mutational hotspots. The fact that most mutations identified here were already reported in other populations is surprising since marked differences in mutation spectrum have been reported in autosomal recessive RP-causing genes among the Israeli/Palestinian populations versus the European/US populations.5,7 The similarity in adRP mutation spectrum among different populations might justify a relatively simple mutation analysis protocol for RHO in populations that were not studied earlier, while such an analysis for autosomal recessive genes is unlikely to be efficient.
At least 20 amino acids in the rhodopsin protein sequence are affected by different multiple mutations (http://www.retina-international.org/files/sci-news/rhomut.htm; provided in the public domain by Retina International); it is therefore not surprising that five of the mutations identified here are located in such positions. Only one of the mutations (c.403C>T, p.Arg135Trp) was shared by patients from unrelated families, and since the two families are from different origins (Turkmenistan Jews and North African Jews), this is also likely to be due to a mutational hotspot.
Patient MOL0908 II:1 was found to be heterozygous for the p.Pro267Leu mutation and was diagnosed with a relatively mild form of RP that is compatible with sector RP, by fundus appearance and ERG analysis. Interestingly, the same mutation has been reported elsewhere30,31 in four patients with a mild phenotype of retinitis pigmentosa. Another mutation affecting the same amino acid (p.Pro267Arg) is reported to cause RP32; however, no clinical data are reported.
The c.548-638dup91bp mutation we identified uncovers a unique and novel phenomenon in the genetics of RP and can potentially aid in our understanding of the required amount of normal rhodopsin protein level that is sufficient to prevent retinal degeneration and hence the clinical features of RP. Affected individuals from two generations in family MOL1076 who are heterozygous for the duplication were diagnosed with RP at a relatively early age. The mutant transcript does not include a premature stop codon and is likely to produce a mutant protein that will lead to adRP. It was therefore intriguing that none of the grandparents reported any visual problems at 75 years of age. Indeed, a detailed ocular evaluation, including ERG, did not reveal any visual abnormalities in the grandparents. However, a detailed genetic analysis of the grandfather revealed that a small proportion of his blood cells carry the mutation. In addition, two of his five children are affected with RP, one of whom was available for the study and carried the duplication heterozygously. It is therefore reasonable to assume that individual 1076 I:1 carries a de novo pathogenic RP-causing mutation, appearing both in somatic cells and in the germline, making him the first individual, to the best of our knowledge, to be reported as a mosaic for an RP-causing mutation. Identification of mosaics for disease-causing mutations is highly important, as it might shed light on the fraction of WT cells needed to protect a person from developing an inherited disease. It is obvious that one cannot measure the fraction of mutated alleles in the relevant tissue, the retina; however, by using a sensitive PCR assay, we were able to accurately determine the fraction of mutated alleles in blood cells (6.5%), resulting in 13% cells that are heterozygous for the mutation. If indeed this value reflects other organs as well as the retina, such allelic fraction is not sufficient to cause retinal degeneration. In addition to the mosaic mutation we reported here, three cases of de novo RHO mutations have been reported thus far (c.538C>T; p.P180S,33 c.641T>A; p.I214N,33 and c.158C>G; p.P53R34,35). Such mutations are likely to result in inheritance patterns that will not be interpreted as autosomal dominant and therefore the wrong sets of genes might be screened for mutations. One should therefore bear in mind that isolated RP cases or nonconclusive inheritance patterns might be due to de novo and/or mosaic mutations in affected as well as unaffected family members.
The genetic analysis of family MOL1076 raises yet another important issue. Although WES analysis was performed on three affected family members, the disease-causing mutation could not be identified. This can be explained by the methodology used during analysis and alignment of WES reads. In the WES protocol, DNA fragments of 110 bp are sequenced and then mapped to a reference human sequence. The analysis includes quality inspection of the sequence reads, and a quality score is obtained for each base along the sequence reads. The first and last 10 bases usually obtained low-quality values and were therefore deleted and excluded from the analysis, resulting in reads with an average length of 90 nucleotides. In addition, reads that could not reliably map to the reference sequence were also excluded from the analysis. The RHO duplication we identified is 91 bases long and can therefore cover a whole read or a substantial fraction of a read (Supplementary Fig. S1). This duplication is not expected to affect the coverage of RHO exon 3, since reads encompassing the duplication site will not be able to align the reference sequence and will be excluded from the final alignment. Such mutations will therefore not be identified in the traditional WES analysis. Recent analyses of WES data reported by others and by our group have shown that in only 50% to 60% of IRD cases analyzed by WES, a disease-causing mutation can be identified.26,36–39 One should therefore bear in mind that duplications, similar to the one we report here, might be present, requiring a specialized WES analysis that is based on base coverage along each exon in the human genome.
In conclusion, we described here for the first time the spectrum of RHO mutations in the Israeli and Palestinian populations and showed that (1) most mutations are family-specific; (2) RHO is a major adRP-causing gene in the Israeli and Palestinian populations and is responsible for the disease in 28% of families; and (3) RHO mutations may appear in mosaicism in unaffected individuals, followed by autosomal dominant inheritance in successive generations.
Supplementary Material
Acknowledgments
The authors thank all patients and family members for their participation in this study. The authors also thank Inbar Erdinest for ERG analysis, and Michelle Grunin for bioinformatic assistance.
Supported by the Foundation Fighting Blindness USA (BR-GE-0214-0639, DS and EB), the Bi-National USA-Israel Foundation (BSF- Grant No. 2011202, DS and AS), intramural research program of the National Eye Institute (AS), an ERA-RARE RHORCOD grant and the Yedidut Research grant (EB).
Disclosure: A. Beryozkin, None; G. Levy, None; A. Blumenfeld, None; S. Meyer, None; P. Namburi, None; Y. Morad, None; L. Gradstein, None; A. Swaroop, None; E. Banin, None; D. Sharon, None
References
- 1. Bundey S,, Crews SJ. A study of retinitis pigmentosa in the City of Birmingham I: prevalence. J Med Genet. 1984; 21: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bunker CH,, Berson EL,, Bromley WC,, Hayes RP,, Roderick TH. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984; 97: 357–365. [DOI] [PubMed] [Google Scholar]
- 3. Peterlin B,, Canki-Klain N,, Morela V,, et al. Prevalence of retinitis pigmentosa in Slovenia. Clin Genet. 1992; 42: 122–123. [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg T. Epidemiology of hereditary ocular disorders. Dev Ophthalmol. 2003; 37: 16–33. [DOI] [PubMed] [Google Scholar]
- 5. Sharon D,, Banin E. Nonsyndromic retinitis pigmentosa is highly prevalent in the Jerusalem region with a high frequency of founder mutations. Mol Vis. 2015; 21: 783–792. [PMC free article] [PubMed] [Google Scholar]
- 6. Berson EL. Retinitis pigmentosa: the Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993; 34: 1659–1676. [PubMed] [Google Scholar]
- 7. Hartong DT,, Berson EL,, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 8. Dryja TP,, Mukai S,, Petersen R,, et al. Parental origin of mutations of the retinoblastoma gene. Nature. 1989; 339: 556–558. [DOI] [PubMed] [Google Scholar]
- 9. Dryja TP,, McGee TL,, Reichel E,, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990; 343: 364–366. [DOI] [PubMed] [Google Scholar]
- 10. Bareil C,, Hamel C,, Pallares-Ruiz N,, et al. Molecular analysis of the rhodopsin gene in southern France: identification of the first duplication responsible for retinitis pigmentosa, c.998999ins4. Ophthalmic Genet. 1999; 20: 173–182. [DOI] [PubMed] [Google Scholar]
- 11. Fernandez-SanJose P,, Blanco-Kelly F,, Corton M, et al. Prevalence of Rhodopsin mutations in autosomal dominant retinitis pigmentosa in Spain: clinical and analytical review in 200 families. Acta Ophthalmol. 2015; 93: e38–e44. [DOI] [PubMed] [Google Scholar]
- 12. Ayuso C,, Garcia-Sandoval B,, Najera C,, et al. Retinitis pigmentosa in Spain: the Spanish Multicentric and Multidisciplinary Group for Research into Retinitis Pigmentosa. Clin Genet. 1995; 48: 120–122. [PubMed] [Google Scholar]
- 13. Milla E,, Maseras M,, Martinez-Gimeno M,, et al. Genetic and molecular characterization of 148 patients with autosomal dominant retinitis pigmentosa (ADRP) [in Spanish]. Arch Soc Esp Oftalmol. 2002; 77: 481–484. [PubMed] [Google Scholar]
- 14. Ziviello C,, Simonelli F,, Testa F,, et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): a comprehensive study of 43 Italian families. J Med Genet. 2005; 42: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sullivan LS,, Bowne SJ,, Birch DG,, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006; 47: 3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gal A,, Apfelstedt-Sylla E,, Janecke AR,, Zrennert E. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Retin Eye Res. 1997; 16: 51–79. [Google Scholar]
- 17. Shastry BS. Retinitis pigmentosa and related disorders: phenotypes of rhodopsin and peripherin/RDS mutations. Am J Med Genet. 1994; 52: 467–474. [DOI] [PubMed] [Google Scholar]
- 18. Wang DY,, Chan WM,, Tam PO,, et al. Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta. 2005; 351: 5–16. [DOI] [PubMed] [Google Scholar]
- 19. Rosenfeld PJ,, Cowley GS,, McGee TL,, et al. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992; 1: 209–213. [DOI] [PubMed] [Google Scholar]
- 20. Gal A,, Orth U,, Baehr W,, Schwinger E,, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet. 1994; 7: 551. [DOI] [PubMed] [Google Scholar]
- 21. Auslender N,, Sharon D,, Abbasi AH,, et al. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Invest Ophthalmol Vis Sci. 2007; 48: 5431–5438. [DOI] [PubMed] [Google Scholar]
- 22. Bandah-Rozenfeld D,, Mizrahi-Meissonnier L,, Farhy C,, et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010; 87: 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zelinger L,, Banin E,, Obolensky A,, et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011; 88: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nevet MJ,, Shalev SA,, Zlotogora J,, Mazzawi N,, Ben-Yosef T. Identification of a prevalent founder mutation in an Israeli Muslim Arab village confirms the role of PRCD in the aetiology of retinitis pigmentosa in humans. J Med Genet. 2010; 47: 533–537. [DOI] [PubMed] [Google Scholar]
- 25. Beryozkin A,, Zelinger L,, Bandah-Rozenfeld D,, et al. Identification of mutations causing inherited retinal degenerations in the Israeli and Palestinian populations using homozygosity mapping. Invest Ophthalmol Vis Sci. 2014; 55: 1149–1160. [DOI] [PubMed] [Google Scholar]
- 26. Beryozkin A,, Shevah E,, Mizrahi-Meissonnier L,, et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci Rep. In press. [DOI] [PMC free article] [PubMed]
- 27. Hargrave PA,, McDowell JH,, Feldmann RJ,, et al. Rhodopsin's protein and carbohydrate structure: selected aspects. Vision Res. 1984; 24: 1487–1499. [DOI] [PubMed] [Google Scholar]
- 28. Inglehearn CF,, Keen TJ,, Bashir R,, et al. A completed screen for mutations of the rhodopsin gene in a panel of patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1992; 1: 41–45. [DOI] [PubMed] [Google Scholar]
- 29. Sung CH,, Davenport CM,, Hennessey JC,, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 6481–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ponjavic V,, Abrahamson M,, Andreasson S,, et al. A mild phenotype of autosomal dominant retinitis pigmentosa is associated with the rhodopsin mutation Pro-267-Leu. Ophthalmic Genet. 1997; 18: 63–70. [PubMed] [Google Scholar]
- 31. Fishman GA,, Vandenburgh K,, Stone EM,, et al. Ocular findings associated with rhodopsin gene codon 267 and codon 190 mutations in dominant retinitis pigmentosa. Arch Ophthalmol. 1992; 110: 1582–1588. [DOI] [PubMed] [Google Scholar]
- 32. Souied E,, Gerber S,, Rozet JM,, et al. Five novel missense mutations of the rhodopsin gene in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1994; 3: 1433–1434. [DOI] [PubMed] [Google Scholar]
- 33. Neveling K,, Collin RW,, Gilissen C,, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012; 33: 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shanks ME,, Downes SM,, Copley RR,, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013; 21: 274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies WI,, Downes SM,, Fu JK,, et al. Next-generation sequencing in health-care delivery: lessons from the functional analysis of rhodopsin. Genet Med. 2012; 14: 891–899. [DOI] [PubMed] [Google Scholar]
- 36. Abu-Safieh L,, Alrashed M,, Anazi S,, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013; 23: 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Y,, Guan L,, Shen T,, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet. 2014; 133: 1255–1271. [DOI] [PubMed] [Google Scholar]
- 38. Jinda W,, Taylor TD,, Suzuki Y,, et al. Whole exome sequencing in Thai patients with retinitis pigmentosa reveals novel mutations in six genes. Invest Ophthalmol Vis Sci. 2014; 55: 2259–2268. [DOI] [PubMed] [Google Scholar]
- 39. Zhao L,, Wang F,, Wang H,, et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet. 2015; 134: 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sung CH,, Schneider BG,, Agarwal N,, Papermaster DS,, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vaithinathan R,, Berson EL,, Dryja TP. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics. 1994; 21: 461–463. [DOI] [PubMed] [Google Scholar]
- 42. Haim M,, Grundmann K,, Gal A,, Rosenberg T. Novel rhodopsin mutation (M216R) in a Danish family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 1996; 17: 193–197. [DOI] [PubMed] [Google Scholar]
- 43. Gal A,, Artlich A,, Ludwig M,, et al. Pro-347-Arg mutation of the rhodopsin gene in autosomal dominant retinitis pigmentosa. Genomics. 1991; 11: 468–470. [DOI] [PubMed] [Google Scholar]
- 44. Sheffield VC,, Fishman GA,, Beck JS,, Kimura AE,, Stone EM. Identification of novel rhodopsin mutations associated with retinitis pigmentosa by GC-clamped denaturing gradient gel electrophoresis. Am J Hum Genet. 1991; 49: 699–706. [PMC free article] [PubMed] [Google Scholar]
- 45. Kaushal S,, Khorana HG. Structure and function in rhodopsin, 7: point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994; 33: 6121–6128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.