

Abstract
Nonsyndromic orofacial clefts are common birth defects with multifactorial etiology. The most common type is cleft lip, which occurs with or without cleft palate (nsCLP and nsCLO, respectively). Although genetic components play an important role in nsCLP, the genetic factors that predispose to palate involvement are largely unknown. In this study, we carried out a meta-analysis on genetic and clinical data from three large cohorts and identified strong association between a region on chromosome 15q13 and nsCLP (P = 8.13×10−14 for rs1258763; relative risk (RR): 1.46, 95% confidence interval (CI): 1.32–1.61)) but not nsCLO (P = 0.27; RR: 1.09 (0.94–1.27)). The 5 kb region of strongest association maps downstream of Gremlin-1 (GREM1), which encodes a secreted antagonist of the BMP4 pathway. We show during mouse embryogenesis, Grem1 is expressed in the developing lip and soft palate but not in the hard palate. This is consistent with genotype-phenotype correlations between rs1258763 and a specific nsCLP subphenotype, since a more than two-fold increase in risk was observed in patients displaying clefts of both the lip and soft palate but who had an intact hard palate (RR: 3.76, CI: 1.47–9.61, Pdiff<0.05). While we did not find lip or palate defects in Grem1-deficient mice, wild type embryonic palatal shelves developed divergent shapes when cultured in the presence of ectopic Grem1 protein (P = 0.0014). The present study identified a non-coding region at 15q13 as the second, genome-wide significant locus specific for nsCLP, after 13q31. Moreover, our data suggest that the closely located GREM1 gene contributes to a rare clinical nsCLP entity. This entity specifically involves abnormalities of the lip and soft palate, which develop at different time-points and in separate anatomical regions.
Author Summary
Clefts of the lip and palate are common birth defects, and require long-term multidisciplinary management. Their etiology involves genetic factors and environmental influences and/or a combination of both, however, these interactions are poorly defined. Moreover, although clefts of the lip may or may not involve the palate, the determinants predisposing to specific subphenotypes are largely unknown. Here we demonstrate that variations in the non-coding region near the GREM1 gene show a highly significant association with a particular phenotype in which cleft lip and cleft palate co-occur (nsCLP; P = 8.13×10−14). Our data suggest that the risk is even higher for patients who have a cleft lip and a cleft of the soft palate, but not of the hard palate. Interestingly, this subphenotype corresponds to the expression of the mouse Grem1 gene, which is found in the developing lip and soft palate but not in the hard palate. While Grem1-deficient mice display no lip or palate defects, we demonstrate that ectopic Grem1 protein alters palatal shelve morphogenesis. Together, our results identify a region near GREM1 as the second, genome-wide significant risk locus for nsCLP, and suggest that deregulated GREM1 expression during craniofacial development may contribute to this common birth defect.
Introduction
Nonsyndromic cleft lip with or without cleft palate (nsCL/P) is a common human birth defect with a multifactorial etiology, including a strong genetic component [1, 2]. Previous studies have identified 16 genetic risk loci for nsCL/P. These studies comprised candidate gene and linkage analyses [3–5], genome-wide association studies (GWAS) with follow-up approaches [6–11], and a meta-analysis [12]. Despite these advances in deciphering the genetic architecture of nsCL/P, a number of additional risk loci still await identification. Some of these as yet unknown susceptibility variants may be detectable in GWAS datasets but have escaped detection at a genome-wide significant level due to low statistical power, which is secondary to limited sample sizes. This suggests that further risk variants for nsCL/P might be identified via one of the following approaches: the combining of available data sets, targeted replication analyses in independent cohorts, and/or the reduction of clinical heterogeneity using detailed subphenotype information.
NsCL/P shows considerable phenotypic variability in terms of affected anatomical structures, and can be subdivided into two main forms: nonsyndromic cleft lip only (nsCLO) and clefts involving both the lip and the palate (nsCLP) [2]. This distinction is important in terms of the degree of physical handicap and treatment. Although epidemiological data indicate that these subtypes are determined at least in part by genetic predisposition [13], few data are available concerning the specific genetic factors determining the formation of nsCLP as opposed to nsCLO. To date, one locus (at 13q31) has shown a specific association with nsCLP but not with nsCLO [12, 14], while IRF6 has shown a predominant effect in nsCLO [5].
Previous research has implicated the Gremlin-1 (GREM1) locus in human orofacial clefting. This research has involved the investigation of gene networks and, more recently, the finding of association between variants at 15q13 and human nsCL/P [10, 15]. However, associations of common variants were not yet significant at the genome-wide level. Also, sequencing studies of GREM1 in nsCL/P patients and controls have been conducted in limited sample sizes only, with inconclusive results: although our group has previously generated some evidence for the role of rare variants in the GREM1 coding and untranslated region [16], the functional relevance of the identified variants remained unclear, and the results of burden analyses varied depending on the test applied. In another sequencing study, no deleterious rare variants were identified in GREM1 [15].
Analyses of Grem1-deficient mouse models have shown that during embryogenesis Grem1 function is crucial for limb development and kidney formation. However, complete loss of Grem1 function causes no obvious craniofacial defects [17, 18]. GREM1 acts as a secreted antagonist of various members of the bone morphogenetic protein (BMP) family, which has been shown to play a critical role in both lip and palate development [19, 20]. Notably, previous research has indicated a particular role for BMP4, which is involved in facial genesis [21, 22]. Moreover, rare mutations within BMP4 have been associated with human clefting [23], and it is established that soluble GREM1 binds with high affinity to BMP4 [24]. Loss-of-function and overexpression studies of the related Bmp antagonist Noggin in mice have previously demonstrated the critical role of restricted Bmp signaling during lip and palate development, as well as for midfacial morphogenesis [25–27].
The present study analyzed the putative risk locus on human chromosome 15q13 in different nsCL/P cohorts, using detailed clinical subphenotype information. The genetic analyses were complemented by functional analyses of the mouse ortholog of the GREM1 gene, which is located adjacent to the associated region. Finally, existing comprehensive genomics data sets were analyzed to annotate the associated region and to establish GREM1 as a plausible candidate gene for nsCL/P at 15q13.
Results
The 15q13 region is a genome-wide significant risk locus for nsCL/P
Regional association statistics for 219 variants at 15q13 (chr15: 32.95–33.5 Mb, hg19) were extracted from a previously published meta-analysis (referred to as Ludwig 2012 meta-analysis) [12] (Table 1, S1 Dataset). The top associated variant at 15q13 in this Ludwig 2012 meta-analysis data set was rs1258763 (PnsCL/P_meta_all = 1.81×10−06, Fig 1 and S1 Dataset), with the A-allele representing the risk allele.
Table 1. Sample overview.
| Study cohort a | Design | Ethnicity | Sample size c | Subphenotype information available? | References d | |
|---|---|---|---|---|---|---|
| Sample description | Genotypes | |||||
| Ludwig 2012 meta-analysis b | case-control | Central European e | 399 cases, 1,318 controls | yes | [29] | [10], [12] f |
| trio | European | 666 trios | yes | [6] | [12] | |
| Asian | 795 trios | |||||
| replication I | case-control | Central European e | 223 cases, 978 controls | yes | [29] | Genotyped for rs1258763 in the present study |
| Mexican | 156 cases, 337 controls | yes | [30] | |||
| Yemeni | 231 cases, 422 controls | no | [31] | |||
| replication II | trio | European (EuroCran) | 600 trios | yes | [10] | [10] g |
a—The meta-analysis of all three study cohorts in the present study is referred to as “combined analysis”.
b—The Ludwig 2012 meta-analysis contained two analyses, i.e., metaEuro (in which the Central European case-control cohort and the European trios were combined), and metaall (which additionally included the Asian trios). Full information on these analyses at 15q13 are provided in S1 Dataset.
c—Number of individuals included in each study. For replication I, pre-genotyping numbers are provided here while post-genotyping data can be found in S2 Dataset.
d—References are provided separately for description of the samples and genotype data for rs1258763, respectively.
e—All individuals are drawn from the Bonn cohort. Individuals included in the Ludwig 2012 meta-analysis have not been included in the replication I study. Therefore, both study cohorts can be considered independently.
f—In the present study, the 15q13 region was imputed using genotypes from Ludwig et al 2012.
g—In the EuroCran study that was part of Mangold et al. 2010, 65 trios from the Bonn cohort were included. To avoid overlap of individuals in the combined analysis of the present study, these individuals were excluded and data re-analyzed. For further details, see sample description in the Methods section.
Fig 1. Regional association plot for the 15q13 region.

P-values for SNPs at 15q13 that were analyzed as part of the Ludwig 2012 meta-analysis (PnsCL/P_meta) are plotted against their chromosomal position (hg19). Full data are provided in S1 Dataset. For each variant, color code denotes linkage disequilibrium to rs1258763, based on 1000genomes. After combination with data from replication I and II, the top variant rs1258763 (purple diamond; indicated by dotted line) reaches genome-wide significance. Plot was generated using LocusZoom [28].
As part of the present study, rs1258763 was then genotyped in an independent case-control cohort of mixed ethnicity (replication I, Table 1 and S2 Dataset). After quality control, 580 cases and 1,684 controls remained in the analysis. In this replication I data, rs1258763 showed strong association with nsCL/P (PnsCL/P_rep_I = 4.34×10−09, relative risk (RR) for the major allele A: 1.58, 95% confidence interval (95% CI): 1.36–1.84, S2 Dataset). Additionally, rs1258763 had been previously genotyped in an independent trio sample from the EuroCran cohort [10] (replication II, Table 1). Re-analysis of this data (to exclude individuals that were also part of replication I) revealed PnsCL/P_rep_II = 0.072 (S2 Dataset), with the A-allele being overtransmitted to affected children.
Combined analysis of the Ludwig 2012 meta-analysis and both replication I and II data sets, respectively, totaling 979 nsCL/P cases, 3,002 controls, and 2,061 trios, yielded genome-wide significance (PnsCL/P_comb = 2.23×10−13), with a strong genetic risk observed for the major allele A (1.35 (1.24–1.46), S2 Dataset).
Imputation analysis of the 15q13 region in the Central European cohort that was part of the Ludwig 2012 meta-analysis (see Methods) revealed a 5 kb region of strongest association (Chr15: 33.050–33.055 Mb), located intergenically between GREM1 and Formin-1 (FMN1), with rs2600520 being the most strongly associated variant in the imputed data (PnsCL/P_imp = 5.05×10−07, Fig 2A and S3 Dataset).
Fig 2. Regional association plots for the 15q13 region in different types of nsCL/P.

In the imputed data of the Central European cohort, the 15q13 region was analyzed in the overall phenotype nsCL/P (A) and both subphenotypes, i.e. nsCLP (B) and nsCLO (C). For each SNP, the P-value is plotted against its chromosomal position (hg19). In nsCL/P and nsCLP, a highly associated cluster of SNPs in strong linkage disequilibrium is present, located between GREM1 and FMN1. The lowest P-value was observed for rs2600520 (purple diamond). In each panel, the top genotyped variant rs1258763 is marked by an open circle. For all other variants, color code denotes linkage disequilibrium to rs2600520, based on 1000genomes. Regional association plots were generated using LocusZoom [28].
Subphenotype analysis reveals strong association with cleft lip and palate
To determine associations between variants at 15q13 and particular clefting subphenotypes, individuals from the three study cohorts were classified as nsCLP or nsCLO on the basis of available clinical information (S2 Dataset). Variant rs1258763 showed significant association with nsCLP in the genotyped data of the Ludwig 2012 meta-analysis (PnsCLP_meta = 8.0×10−08), replication I (PnsCLP_rep_I = 1.28×10−07) and replication II (PnsCLP_rep_II = 0.035 Fig 3 and S2 Dataset). No significant association was detected for nsCLO (P>0.2 in all cohorts, S2 Dataset). For each of the three cohorts, the difference in relative risk between nsCLP and nsCLO was statistically significant (P<0.05).
Fig 3. Forest plot for association of rs1258763 and nsCL/P subphenotypes.

Subphenotype analyses of cleft lip and palate (nsCLP, black) and cleft lip only (nsCLO, grey) were conducted in the Ludwig 2012 meta-analysis data, the replication I and II cohorts, and in the combined analysis of the present study. Boxes represent point estimates of the relative risk for each of the four studies, with box sizes scaled according to the number of affected individuals. Lines indicate the extent of the confidence interval. These data illustrate the consistent association between rs1258763 and nsCLP across the various studies, and the presence of a narrow effect size range in the combined cohort. Please note that confidence intervals for nsCLO are larger due to the lower number of nsCLO patients. Informed by the specific expression of Grem1 in lip and soft palate development in the mouse embryo (see below) we also analyzed the effect size of the particular soft palate subphenotype (nsCLPsoft). The arrow indicates the point estimate for rs1258763 and nsCLPsoft.
The overall association P-value for nsCLP in the combined analysis was PnsCLP_comb = 8.13×10−14 (1.46 (1.32–1.61), Fig 3), in contrast to PnsCLO_comb = 0.27 (1.09 (0.94–1.27)) for nsCLO. The difference in relative risks between nsCLO and nsCLP in the combined sample was statistically significant in both multiplicative (P = 0.0015) and general (P = 0.0033) model. In the subphenotype analysis of the imputed data generated from the Central European case-control cohort rs2600520 remained the most significantly associated variant with nsCLP (PnsCLP_imp = 3.19×10−06, Fig 2B), while only marginal associations were observed with nsCLO (Fig 2C).
In silico annotation supports GREM1 as candidate gene at 15q13
For the 5 kb region of strongest association, no compelling evidence for the presence of regulatory elements was found in ENCODE [32], except for a 200 bp region identified as DNAse hypersensitivity site in four cell types, three of which had been derived from skin tissue (chr15:33,052,666–33,052,875, hg19) [33]. In datasets of relevance to craniofacial development [34, 35], no active regulatory elements were detected.
According to HaploRegv3 [36], the top associated variant rs2600520 and a second single nucleotide polymorphism (SNP), rs2600519, which is in high linkage disequilibrium (LD) and located four base pairs away, modify a chromatin mark that involves the transcriptional repressor Sin3A. The analysis of blood-sample expression quantitative trait loci (eQTL) yielded 38 SNPs with cis-eQTL effects on GREM1 expression, six of which had false discovery rates below 0.05 (S1 Table). The strongest eQTL effect was observed for rs17816375, which showed a P-value of 0.033 in the imputed nsCLP data. Of the 38 SNPs, the SNP with strongest association to nsCLP was rs16958561 (P = 2.82×10−05). This SNP maps around 6 kb distal to rs2600520, and the two SNPs are in substantial LD (D' = 1 in the European population (1000 Genomes phase 1)). No trans- or cis-eQTL effect was identified for FMN1, despite the fact that one FMN1-targeting probe was represented on the expression array used [37]. Analysis of chromatin interaction data [38] obtained in human epidermal keratinocytes (NHEK), umbilical vein endothelial cells (HUVEC) and mammary epithelial cells (HMEC) revealed a topologically associated domain (TAD) encompassing the entire GREM1 gene, the associated region and parts of the SCG5 and FMN1 coding regions. Standard annotation of contact domains reveals a subdomain separating the 5' region of FMN1 from its 3' end and GREM1 (S1 Fig). Further analysis of the TAD data using virtual 4C reveals evidence that direct interaction loops are present between the associated region and the GREM1 transcription start site at the given resolution (S2 Fig).
Grem1 is expressed in a specific pattern and alters palatal shelve morphogenesis in vitro
X-Gal staining of the craniofacial region of heterozygous mutants of a mouse model of Grem1 deficiency [17] revealed Grem1 expression in the proximal region of both lateral and medial nasal prominences at E11.5 and E12.5 (Fig 4A and 4B). In addition, bilateral Grem1 expression was detected in the merging zones of the maxillary and medial nasal prominences during lip development at E12.5 (Fig 4B). Grem1 was also expressed in the mesenchyme of the developing secondary palate between E12.5 and E15.5, with expression being restricted to the posterior palatal shelf region in which the soft palate forms (Fig 4C–4G).
Fig 4. Grem1 expression during mouse craniofacial development.
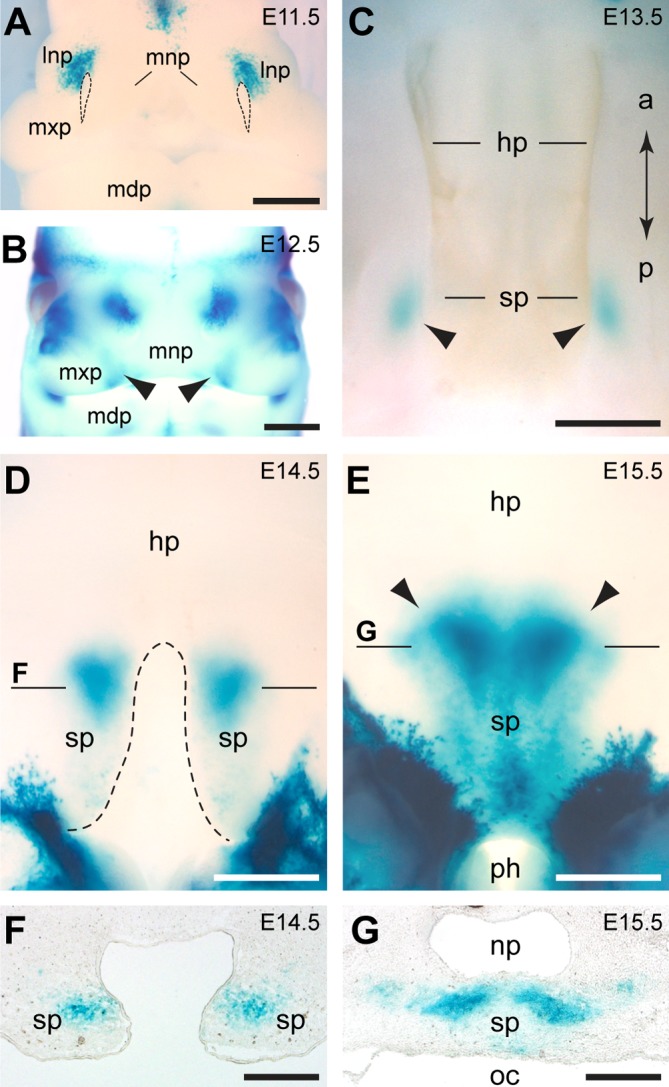
(A-E) Expression of Grem1 is visualized by X-Gal staining of heterozygous Grem1LacZ whole mount embryos. (A) At E11.5, Grem1 is expressed in the dorsal part of the lateral nasal prominence (lnp). Stippled lines demarcate the nasal pits. (B) At E12.5, Grem1-positive domains are also detectable in the merging zones (arrowheads) of medial nasal prominences (mnp) and maxillary prominences (mxp). (C-G) Secondary palate development. (C) At E13.5, Grem1-positive domains are observed in the forming soft palate (sp). (D) At E14.5, the hard palate (hp) has formed while the Grem1-expressing shelves of the soft palate are not yet fused. (E) At E15.5, the soft palate has fused and Grem1 expression extends posterior to the pharynx (ph). Note the sharp anterior boundary of Grem1 expression in the soft palate (arrowheads). (F, G) Sections of whole mount stained embryos. (F) Cross section at the level indicated in (D) showing that Grem1 expression is restricted to the mesenchyme. (G) Cross section at the level indicated in (E) showing Grem1 expression in the soft palate, which separates the nasopharynx (np) from the oral cavity (oc). Additional abbreviations: a, anterior; l, lateral; m, medial; mdp, mandibular prominence; p, posterior. Scale bars: 500μm.
Although no craniofacial defects have been reported for Grem1-deficient mice [17], we tested the hypothesis that loss of Grem1 results in alterations of lip and palate morphology without manifesting any profound craniofacial phenotype. However, a histological analysis revealed no developmental abnormalities in the embryonic secondary palate or during lip formation in Grem1-/- mouse mutants (Fig 5A–5D). Next, E13.5 secondary palate shelves were cultured in the presence of exogenous Grem1 protein to assess whether this treatment could affect their development. Significant growth (approximately 25% increase) of the palatal shelves was observed in both Grem1-treated and control groups during the 48 hour culture period, with no significant difference in the final size of the shelves observed between groups (P = 0.27, S2 Table). However, Grem1-treated palatal shelves showed a more rounded shape, with retraction of their medial edges (Fig 5E–5G). Measurement of the area between both palatal shelves at 0 and 48 hours revealed a stable area in the control group but a significant increase in the Grem1-treated group (P = 0.0014 for difference between control and Grem1-treated pairs; Fig 5H, S3 Table).
Fig 5. Analyses of the effect of Grem1 loss of function and of ectopic Grem1 protein on secondary palate development.
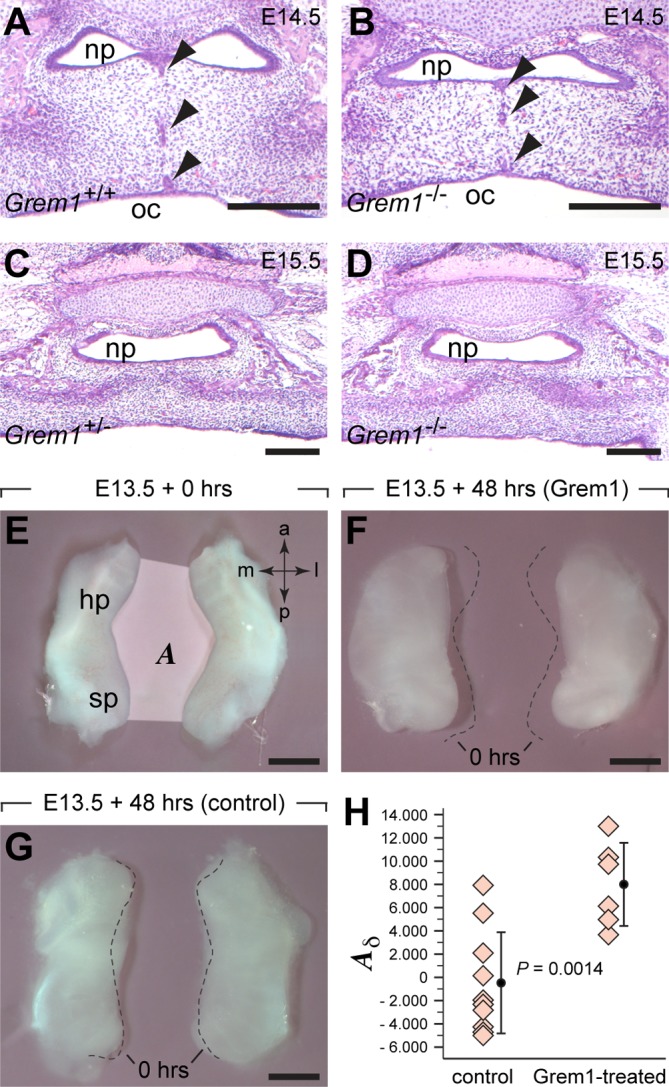
(A-D) Hematoxilin/Eosin staining of paraffin sections. (A,B) At the level of the posterior hard palate, the palatal shelves have fused and epithelial rests (arrowheads) are seen in both Grem1+/+ (A) and Grem1-/-(B) embryos at E14.5. At E15.5, the soft palate has fused in both Grem1+/- (C) and Grem1-/- embryos (D). (E-H) Organ culture experiments of secondary palatal shelves dissected at E13.5. (E) The area (A) between palatal shelves was measured at the onset and after 48 hours of culture. The presence of Grem1 protein led to an increase in the area (F), whereas the size of the area did not change in controls (G). Stippled lines demarcate the medial edges of the secondary shelves at the onset of culture. (H) The difference in area (Aδ = A48hours−A0hours) is significantly larger in the Grem1-treated palatal shelves compared to those of controls. Abbreviations: a, anterior; hp, hard palate; l, lateral; m, medial; np, nasopharynx; oc, oral cavity; p, posterior; sp, soft palate. Scale bars: 200μm in A-D, 500μm in E-G.
Genotype-phenotype correlation is strongest in a rare clinical entity
Informed by the murine expression pattern of Grem1, we tested for involvement of the soft palate in human data sets. Twenty-one patients with a cleft lip and a cleft of the soft palate in the presence of an intact hard palate (nsCLPsoft) were selected (see Methods). In this group, analysis of rs1258763 revealed P = 0.03 and an RR of 3.76 (95% CI: 1.47–9.61) in nsCLPsoft, representing a two-fold increase of effect size in comparison to 320 patients with complete cleft lip and palate (nsCLPhard+soft, Fig 3 and Table 2). While only a limited number of patients with this rare nsCLPsoft phenotype could be recruited this association P-value passed the statistical significance threshold after permutation-based correction (P = 0.04). No association was found in a group of 45 patients with cleft of the soft palate only and an intact lip (PnsCPO_soft = 0.94, Table 2), or in 115 patients with a submucous cleft of the soft palate (PnsCPO_submuc = 0.85, Table 2).
Table 2. Analysis of rs1258763-subphenotype effect on the soft palate.
| Phenotype | Genotypes cases | MAF cases | Genotypes controls | MAF controls | P-value | RR allelic (95% CI) |
|---|---|---|---|---|---|---|
| nsCLP_hard+soft | 13/130/167 | 0.252 | 149/590/578 | 0.337 | 3.53×10−05 | 1.51 (1.24–1.85) |
| nsCLP_soft | 1/3/17 | 0.119 | 2.99×10−03 | 3.76 (1.47–9.61) | ||
| nsCPO_soft | 1/28/16 | 0.333 | 0.94 | 1.02 (0.65–1.59) | ||
| nsCPO_submuc | 12/55/48 | 0.343 | 0.85 | 0.97 (0.73–1.29) |
Abbreviations: MAF—minor allele frequency, RR—allelic odds ratio (provided for the risk allele A), CI—confidence interval, ns—non-syndromic, CLP—cleft lip and palate, CPO—cleft palate only, submuc—submucous. Genotypes are presented with the minor allele first, i.e. GG/AG/AA.
Discussion
Previous studies have reported suggestive associations between markers in the 15q13 region and nsCL/P [10, 15]. In the present study, we now conclusively confirm 15q13 as risk locus for nsCL/P by reaching genome-wide significance. In addition, the present study is the first to demonstrate a specific subphenotype effect in patients with nsCLP. While 15q13 is not associated with nsCLO in any cohort analyzed in this study, a strong association with nsCLP was observed in different ethnicities, suggesting the 15q13 risk locus generally acts on various population backgrounds. The strength of association, however, varied between ethnicities (1.28 to 1.87 in the present study), which might be due to heterogeneity at the population or allelic level, respectively. Notably, we observed the lowest effects size in the replication II cohort which is a trio data set of European ethnicity. As heterogeneity can be considered rather unlikely here, an additional explanation would be differences in the composition of the samples, e.g., in the frequency of individuals with a positive family history. This should be investigated in further studies.
The region of strongest association, which was identified by imputation of common variants in Central Europeans, encompasses several variants in high LD located within a 5 kb region, about 40 kb downstream of the GREM1 transcription start site (TSS). Two hypotheses regarding the nature of the functional causative variant(s) arise: First, the common variant(s) might be functionally relevant themselves. This has been previously observed in nsCL/P for common variants at the high-risk locus IRF6, where a common variant in the IRF6 enhancer mediates craniofacial disturbances by altering an Ap-2alpha binding site [5] and, recently, for rs227727 at the 17q22-locus [39]. Alternatively, rare but highly penetrant sequence variants might confer functional effects in some patients. Those variants are missed by imputation, and their detection would require re-sequencing of the entire associated region in large cohorts of either multiply affected families (to infer co-segregation) or trios with sporadic cases (to detect de novo occurrences). The successful outcome of such an approach has recently been demonstrated in a large trio re-sequencing study of nsCL/P risk loci. In that study, functionally relevant rare variants were identified, including a non-coding variant in FGFR2 [39].
The formation of lip and palate is completed by the 10th week of human embryonic development. In the absence of functionally annotated material from human embryonic craniofacial tissue we here used different approaches to assess a potential regulatory effect of the common variant rs2600520 or any other highly associated variants. First, we checked the functional regulatory landscape at 15q13 using previously published Hi-C data [38]. This analysis showed that GREM1 locates within a topologically associated domain (TAD) that includes the associated region as well as parts of the adjacent genes SCG5 and FMN1. Notably, in silico annotation of contact domains suggests that the 3' FMN1 region, together with GREM1, is located in one functional unit which is different from that containing the FMN1 5' region and FMN1 promoter. While closer analysis of the region reveals evidence for an interaction of the GREM1-TSS with the GREM1/FMN1 intergenic region, little evidence is provided for interaction between the associated region and any of the SCG5 or FMN1 promoters.
We also accessed data from a large and systematic analysis of gene expression from blood samples [37], using FMN1 and GREM1 as query genes. While no result was returned for FMN1, numerous eQTLs for GREM1 were identified. This suggests a regulatory effect of SNPs in this intergenic region on GREM1 in general, however, none of the SNPs identified at an FDR < 5% are associated with nsCLP in our imputed data. Notably, two of the variants with suggestive evidence (rs16958561, rs16958734) are significantly associated in our imputed case-control data and are in high LD with rs2600520. This suggests that common functional variant(s) located on haplotypes tagged by rs2600520 could have an effect on GREM1 expression and might be stronger eQTLs in data sets of relevant craniofacial tissue. Next, the 5 kb risk locus was analyzed in comprehensive data from the ENCODE project [32] and published datasets of relevance to craniofacial development [34, 35]. No compelling evidence for the presence of regulatory elements was found in these data, with the exception of a 200 bp DNA hypersensitivity site [33] in ENCODE, which indicates accessible chromatin in this region. Notably, the DNA region around rs2600520 and rs2600519 has previously been identified as chromatin mark involving the transcriptional repressor Sin3a [36]. Together with histone deacetylases (HDAC1 and HDAC2) Sin3a can form part of a repressor complex that is targeted to specific DNA regions by sequence-specific transcription factors [40]. Interestingly, at E10.5 of mouse development Sin3a is expressed in the developing limb buds and in craniofacial prominences in a pattern overlapping with that of Grem1 (Emage: #2906, [41]). It is conceivable that a failure to recruit the transcriptional repressor Sin3a to this putative regulatory region could result in up-regulated GREM1 expression, which in turn would affect lip and palate development by disrupting Bmp signaling. However, the identity of the DNA-binding transcription factors that recruit Sin3a and recognize target sites near or at rs2600520 and rs2600519 in craniofacial prominences remain to be identified.
The functional annotation approach used in this study has two limitations. First, most of the analyses in functional data sets are based on tissue types with no direct relevance for nsCL/P. However, given the lack of appropriate nsCL/P-relevant material at the moment, these data provide the opportunity to understand basic regulatory mechanisms that might be present at the GREM1 locus. Second, methodological issues such as under-investigation of particular epigenetic marks or activity states (such as silencers) in the queried datasets might have been confounding factors in our analysis. To identify regulatory non-coding regions and to decipher how the intergenic GREM1/FMN1 region interferes with normal craniofacial development, the generation of comprehensive data sets in relevant tissues is warranted which might be accomplished by consortia projects such as Facebase [42].
Our data suggest that intergenic variants located close to FMN1 affect regulatory elements which are targeting the adjacent GREM1 gene. In nsCL/P, such long-range effects of genetic variants on distally located genes have been previously suggested, e.g. at the 1p22 locus: Here, associated variants map intronically within the ABCA4 gene while expression and mutation analyses suggest the adjacent ARHGAP29 gene as causative gene [43]. However, experimental proof of this regulation has not yet been obtained. For the 15q13 locus, evidence for long-distance regulatory effects on GREM1 is provided by studies in other mammals. Research in mice has shown that the 3’-region of the Fmn1 gene is necessary for cis-regulation of Grem1 transcription [44]. Remarkably, the developmental limb and kidney defects observed in Grem1-/- mice are similar to those of ld (limb deformity) mutant mice [17, 44], which carry mutations in the Fmn1 3’-region. These findings suggest that the Grem1-loss of function mutation is allelic with ld, and that Grem1 expression might be regulated by a global control region (GCR) located near or at the Grem1/Fmn1 intergenic region [17, 44]. The activity and function of specific regions within the GCR, however, might be different, depending on the developmental processes or tissues [45].
Our study also provides first evidence for genetic components underlying a rare clinical entity, i.e., a cleft lip and a cleft soft palate in the presence of an intact hard palate. Cuddapah et al (2015) coined the phrase “interrupted clefting” to describe this phenotype [46] and challenged the classical view that cleft lip with or without cleft palate are always manifestations of a single etiological continuum. During mouse embryogenesis, highly localized expression of Grem1 was observed in the soft palate and in the processes forming the lip, while no expression was detectable in the developing hard palate. These observations correspond well with the considerably increased risk we observed in patients presenting with "interrupted clefting". Thus, our data support a more complex situation as they suggest that a cleft palate may form independently of a cleft lip and that nsCLP can be caused by disruptions of genetic pathways that are required in both lip and palate development. Although intriguing, this observation was based on a low number of individuals only and, therefore, has to be confirmed in further independent samples for which detailed clinical information is available. Within this context, it is interesting to note that genotype-phenotype correlations in six previously reported nsCLP multiplex families with rare mutations in GREM1 [16] revealed a correlation between the presence of these mutations and cleft soft palate status in 11 out of 13 affected individuals (S3 Fig). All variants were located in the 3' UTR, and the functional impact of the identified mutations has not been established. Importantly, it has been recently shown that rare variants in the 3' and 5' UTR regions might have a strong effect on disease risk, which might even outweigh the effect of rare coding mutations [47]. We hypothesize that rare GREM1 mutations might have modifier effects, thereby influencing which of the palatal structures are affected in patients predisposed to nsCLP. This would contribute to the explanation why unaffected individuals in these families are mutation carriers. Although not fully conclusive yet, this observation should be followed up in the future, including experimental validation of the functional role of the non-coding mutations.
Given the critical requirement of Grem1 in kidney and limb formation during mouse embryogenesis [17, 18] it is unlikely that a ubiquitous downregulation of GREM1 transcription during human embryogenesis would exclusively affect lip and palate development. Moreover, our mouse data indicate that the presence of Grem1 is not essential for the development of the lip and palate. However, Grem1 expression is required for limb bud development, where it maintains a positive Shh-Fgf feedback loop through restriction of Bmp signaling, which in turn is a negative regulator of Shh expression in the limb mesenchyme [17, 48]. In marked contrast, a previous study identified Bmp signaling as a positive regulator of both Shh expression in the palatal shelf epithelium and cell proliferation in the anterior palatal shelf mesenchyme [22]. These findings suggest that increased or ectopic expression of Grem1 might interfere with normal secondary palate development through: (i) inhibition of Bmp4 or Bmp2, both of which are expressed in the anterior palate at E12.5 [22]; or (ii) inhibition of Bmp4 and Bmp7, both of which are strongly expressed in the posterior region of the palatal shelves at E13.5 in the mouse [49]. The results of our organ culture experiments suggest that a pathogenic effect can be caused by increased levels of Grem1, as these data demonstrated a marked effect of ectopic Grem1 protein on the morphogenesis of the embryonic palatal shelves during a critical phase of secondary palate development.
Furthermore, the nsCL/P risk allele for rs1258763 has been associated with increased nasal width [50, 51], which is consistent with results from a previous meta-analysis showing an increased nasal width in unaffected parents of nsCL/P patients [52]. This provides additional support for the hypothesis that regulatory regions near the GREM1 locus plays an active role in modulating morphogenesis during craniofacial development.
Although our results support the hypothesis that GREM1 is a candidate gene at 15q13 and expression analyses of various isoforms of murine Fmn1 transcripts [53] failed to implicate Fmn1 in lip and palate development, our data do not entirely exclude the possibility that Fmn1 could play some role in craniofacial development. Fmn1 has been shown to regulate aspects of mouse limb and kidney organogenesis similar to those controlled by Grem1 [54]. However, contrary results in terms of expression regulation have been obtained. Whereas Grem1 expression was upregulated in a Fmn1 null mouse mutant in which the Grem1/Fmn1 regulatory region was intact [54], earlier observations demonstrated reduced Grem1 expression as a result of the ld mutation, which affects the 3’-region of Fmn1 [44]. Thus, the Grem1/Fmn1 regulatory landscape exhibits a complex architecture, which, at present, cannot be delineated more precisely due to the limited resolution of chromatin interaction data and the close proximity of the GREM1 and FMN1 genes. However, our results suggest strong, and functionally distinct regulatory activities in the region, which warrants investigation in future studies that might include higher chromatin resolution and targeted conformation assays.
Concluding remarks
The present study established that the 15q13 region contains a genetic risk factor for nsCL/P. This is the second locus after 13q31 to show a particularly strong association with nsCLP and not with nsCLO. Moreover, our results suggest this risk factor to be involved in nsCLPsoft, and our genetic and functional analyses provide strong support for the hypothesis that GREM1 is an effector gene that contributes to this rare clinical subphenotype. In aggregate, our data suggest that GREM1 plays a specific role not only in the development of the lip, but also during formation of the soft palate. These findings provide a framework for further functional analyses of GREM1 in human cell systems and for inducible modulation of Grem1 expression during the development of specific craniofacial regions in model organisms. These analyses may broaden our understanding of the processes that regulate facial morphogenesis, and help to decipher the molecular mechanisms underlying the manifestation of specific nsCL/P subphenotypes in humans.
Materials and Methods
Ethics statement
Human genetic studies
The study was approved by the ethics committees of the respective medical faculties, and informed consent was obtained from all participants.
Animal studies
Breeding and mouse embryo production were approved by the local veterinary authorities (permit 98/2011, Veterinäramt Zürich) in accordance with Swiss federal law (TSchG, TSchV) and cantonal by-laws in full compliance with European Guideline 86/609/EC.
Sample description
Genome-wide cohorts
Association statistics for variants at 15q13 were obtained from a large nsCL/P meta-analysis performed by our group, which is described elsewhere [12] and referred to as “Ludwig 2012 meta-analysis” throughout this manuscript. This study included analyses of (i) European individuals (399 cases, 1.318 controls and 666 case-parent trios, referred to as metaEuro), and (ii) Asian and European individuals (inclusion of additional 795 Asian trios, referred to as metaall). Please note that the Central European cases from the case-control cohort were drawn from the same Bonn cohort as was part of the replication I sample. However, individuals were included either in the GWAS or in the replication; none of the individuals was included in both studies.
Replication cohorts
Replication I (case-control cohort). The first replication cohort was a case-control cohort that comprised nsCL/P samples from three different populations (Bonn, Mexico, Yemen). The Bonn sample comprised 223 independent nsCL/P patients. A total of 978 volunteer blood donors were included as controls, which should not result in any appreciable reduction in power given the low prevalence of nsCL/P in the general population [55]. The Mexican case-control sample was recruited as described elsewhere, and subphenotype information was available for these subjects [30]. The Yemeni case-control sample was recruited as described elsewhere [31]. No information on subphenotypes was available for the Yemeni sample.
Replication II (EuroCran trio cohort). Genotypes for individuals from the EuroCran cohort for rs1258763 were retrieved from a previously published study [10]. In this study from 2010, the EuroCran sample contained 65 trios that were from the Bonn cohort and therefore, some of the affected index patients overlapped with individuals from the replication I cohort. To avoid any overlap in the statistical analysis, we excluded these 65 Bonn trios from the EuroCran data and re-analyzed the nsCL/P association (replication II). This explains differences in T/NT ratios and P-values between the present study and Mangold et al 2010. Notably, in Mangold et al. 2010, no subphenotype analysis for rs1258763 was performed.
Analysis of clinical subtypes
To characterize the cleft lip and palate phenotype in more detail, all available clinical information was accessed. For individuals of the Central European case-control cohort from Mangold et al. 2010, phenotype information was retrieved from our in-house clinical database. A total number of 310 patients with a complete cleft of the lip and clefts of the hard and soft palates were identified (CLPhard+soft). Thirteen patients from the Bonn cohort (8 from the GWAS, 5 from the replication I cohort) displayed a cleft of the lip and the soft palate in the presence of an intact hard palate (CLPsoft), indicating the rarity of this clinical subphenotype. To increase the size of this CLPsoft group we reached out to other clinical nsCL/P cohorts and identified eight patients meeting the CLPsoft criteria in a Dutch nsCL/P sample [56]: Six of these patients were part of the EuroCran cohort while two patients were drawn from an independent Dutch sample [57]. In addition, rs1258763 was genotyped in: (i) 45 patients with a cleft of the soft palate only (CPOsoft), which is an orofacial clefting subform with a different genetic background; and (ii) 115 patients from a cohort of submucous cleft palate patients (CPOsubmuc) [58].
Genotyping and statistical analysis
Genotyping of rs1258763 in the nsCL/P replication sample and nsCPOsoft individuals was performed using the Sequenom MassArray system (Agena Bioscience, San Diego, USA). nsCLPsoft-patients from the Dutch cohort and the nsCPOsubmuc patients were genotyped using ABI3130XL sequencing and BigDye v3.1.
The replication II sample was analyzed using the transmission disequilibrium test. In the replication I cohort, genotype frequencies in the cases and controls of each of the three subsamples (Bonn, Mexico, Yemen) were compared using the Cochran-Armitage trend test. To combine all results for rs1258763, effect estimates for the different studies were combined in an inverse variance-weighted fixed-effects meta-analysis. To test for heterogeneity of the genotypic RRs between the nsCLO and nsCLP phenotypes, a heterogeneity likelihood-ratio test was applied using a general and a multiplicative model. For each of these analyses, this resulted in an asymptotic Chi2 null distribution with two degrees of freedom.
For the analysis of rs1258763 in the nsCL/P subphenotypes nsCLPhard+soft, nsCLPsoft, nsCPOsoft, and nsCPOsubmuc, a common set of 1,318 controls from the Central European case-control cohort was used. To determine whether a statistically significant difference was present between the results for nsCLPhard+soft and nsCLPsoft and to account for the limited number of individuals in the nsCLPsoft group, an empirical P-value was determined using 100,000 permutations (larger test statistics were assigned a value of 1, equal test statistics were assigned a value of 0.5).
Imputation analysis
Genotypes from the Central European case-control cohort that was part of the meta-analysis [10] were imputed using IMPUTE2 [59] and 2,184 alleles of the 1000genomes project. Dosage values were included in a logistic regression model (using SNPTEST and—method expected) in which we included as covariates the first five components eik (which were obtained from MDS analysis [60], to account for population stratification. Only variants showing a SNPTEST info-score (detailed description: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.v2.pdf) of at least 0.4 and a minor allele frequency of at least 1% were retained, as accuracy of imputation is compromised for low frequency variants. The resolution of the 15q13 region (250 kb, chr15:32.95 Mb—33.2 Mb) increased from 106 variants in the genotyped data to 1,042 variants in the imputed data. Relative risks were calculated using the beta values, representing the logarithm of the RR. Please note that inclusion of the genome-wide data from Beaty et al. 2010 in the imputation analysis was not possible at the time of the present study, due to insufficient coverage of that approach in the dbGaP approval used for the Ludwig 2012 meta-analysis.
In silico annotation
Potential regulatory effects of the top associated region on either GREM1 or FMN1 were assessed using previously published eQTL and HiC-datasets. Since there is a complete absence of eQTL studies of fetal craniofacial samples, eQTLs obtained in large datasets from other tissues were used. Here we used a recently published and comprehensive study of blood samples by Westra et al. [37]. Both FMN1 and GREM1 probes are present on the Illumina arrays used in the Westra et al. study. To identify altered transcription factor binding we used the v3 version of the HaploReg tool, which was developed to annotate disease-associated genetic variants located in non-coding regions [36]. In particular, information on transcription factor binding altered by nsCL/P risk alleles was used. These analyses were complemented by recently published HiC-data illustrating chromatin formation and loops at 15q13 using the Juicebox tool [38] and virtual 4C as provided at http://promoter.bx.psu.edu/hi-c/virtual4c.php using the same data.
Mouse husbandry and Grem1 expression analysis
Grem1LacZ mutant mice [17] were kept on a C57Bl/6 genetic background and embryos were staged using mid-day on the day of vaginal plug detection as embryonic day 0.5 (E0.5). Genotyping of embryos was carried out using allele-specific PCR. The wild type allele was detected as a 435bp product using primers 5’-TGCAATTGTGTCAGGAGCCA-3’ and 5’-ACTGGGTCTGCTCAGAGTCA-3’. The lacZ allele was detected as an approximately 550bp product using primers 5’-TGCAATTGTGTCAGGAGCCA-3’ and 5’-GGGAACAAACGGATTGACCG-3’. Following embryo dissection, mandibles and tongues were removed to facilitate the penetration of fixatives and staining solutions. Expression of Grem1 was visualized by X-Gal staining of heterozygous Grem1LacZ whole mount embryos using standard procedures, as described elsewhere [61]. To assess tissue-specific domains of Grem1 expression, 15μm cryosections of whole mount stained embryos were prepared and mounted onto glass slides. Images were obtained using an Axiocam color camera mounted on a Zeiss Stemi SV11. Images were processed using the softwares Axiovision AC (release 4.4) and Photoshop C4 (version 11).
Organ culture experiments and histology
Organ culture experiments of secondary palatal shelves were carried out using conditions essentially similar to those described elsewhere [61, 62]. Briefly, palatal shelves were dissected at E13.5 and cultured for 48 hours on sterilized filter papers (Millipore) placed on a metal grid located on the central well of a culture dish. With the aboral side facing downwards, the palatal shelves were cultured in Dulbecco’s modified eagles medium (DMEM), supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin (5000 units/ml, 5000ug/ml), 1% glutamine (200mg/ml) and 1% (v/v) ascorbic acid (50mg/ml). Grem1 recombinant protein (Peprotech (USA)) was used at a final concentration of 10μg/ml medium. The organ rudiments were cultured at 37°C using 5% CO2 for 48 hours and culture medium was replaced after 24 hours. Images were obtained using a Axiocam mounted onto a Zeiss Stemi SV6 stereomicroscope. Areas between palatal shelves were measured using imagej (http://imagej.nih.gov/ij/). To calculate P-values, arbitrary units quantifying the area between the palatal shelves were analyzed using a one-tailed t-test. For the craniofacial phenotype analysis of Grem1-deficient mouse embryos, processing of heads for embedding in paraffin and generation of Hematoxilin/Eosin stained sections were performed as described elsewhere [61].
URLs
For the purposes of the present study, the following databases were accessed: https://www.broadinstitute.org/mpg/snap/ [63], http://genenetwork.nl/bloodeqtlbrowser/ [37], http://www.emouseatlas.org/gxdb/dbImage/segment1/2906/2906.html [41], http://www.aidenlab.org/juicebox/ [38].
Supporting Information
Chromatin interaction data for three different celltypes were drawn from [38]. RefSeq genes are plotted above the interaction map, together with CTCF binding sites identified in the respective cell lines. GREM1 position is highlighted in red, adjacent genes SCG5 and FMN1 are also labeled. + /—below the RefSeq annotation denotes strand orientation of the gene. Yellow lines indicate contact regions, blue squares show regions of loop interactions, both as defined by the original study. The dotted line indicates co-localization of the GREM1 / 3‘FMN1 region within one topologically associated domain (TAD) which is separate from the 5‘ FMN1 region. TAD structure is stable between each of the three cell types. (A) NHEK–normal human epidermal keratinocytes, in situ combined, 5kb resolution, (B) HUVEC–human umbilical vein endothelial cells, in situ combined dataset, 5kb resolution(C) HMEC–human mammary epithelial cells.
(TIF)
Zoom-in into the GREM1/FMN1 region is provided for the HUVEC cells. (A) Local TAD structure (chr15: 33.000.000–33.100.000) with arrow indicating potential interaction loops between the GREM1 transcription start site (TSS, 33,010,204) and the intergenic region (33,040,000–33,044,999). The exact location of the loop cannot be further narrowed down due to the given resolution (5 kb). Data were drawn from [38]. Color code denotes number of observed reads. (B) Same data in virtual 4C visualisation. Using the GREM1-TSS as anchor point, regions of interactions are provided as peaks, with number of observed read counts as quantitative measure. Again, arrow highlights a potential interaction candidate at 33,040,000–33,044,999 bp.
(TIF)
Phenotype: empty symbol–unaffected, half-filled symbol–cleft of the lip and hard palate only (soft palate intact), full symbol–cleft of the lip, hard and soft palate. Carrier status: + carrier,—non-carrier. Phenotype-genotype correlation: red symbols—individuals with concordant genotype-phenotype correlation, black symbols–individuals with discordant genotype-phenotype correlation. a—Family BN-45 has four variants that are transmitted together: rs2280738, rs117317622, rs137899769, rs151194761. Please note that four more index patients with rare mutations but without any additional affected family members have a complete cleft of lip, hard and soft palate: BN-139 (rs201006159), BN-241 (rs201134502), BN-251 (g.33023715), BN-317 (rs147141645). All positions hg19.
(TIF)
(XLS)
(XLS)
(XLS)
(PDF)
(PDF)
(PDF)
Acknowledgments
We thank R. Harland for providing Grem1 mutant mice, and A. Zuniga, P. Mossey, Julian Hecker, and Markus M. Nöthen for helpful discussions. We are grateful to all patients and their families for their participation, as well as the German support group for individuals with cleft lip and/or palate (Deutsche Selbsthilfevereinigung für Lippen-Gaumen-Fehlbildungen e.V.). We also thank the CBC staff at the Institute of Genetic Medicine at Newcastle University for their technical assistance.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The work was supported by the Medical Research Council (UK) URL: http://www.mrc.ac.uk/ Grant Number: G0400679 to HP; the Newlife Foundation for disabled children (UK) URL: http://www.newlifecharity.co.uk/ Grant number: SG/13-14/05 to HP; Deutsche Forschungsgemeinschaft (DE) URL: http://www.dfg.de/ Grant number: LU 1944/2-1 to KUL; Deutsche Forschungsgemeinschaft (DE) URL: http://www.dfg.de/ Grant numbers: MA 2546/5-1 and MA2546/3-1 to EM; BONFOR program of the Medical Faculty; and by the University of Bonn (DE) URL: https://www.ukb.uni-bonn.de/ Grant Number: O-149.0097 to KUL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12(3):167–78. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mangold E, Ludwig KU, Nöthen MM. Breakthroughs in the genetics of orofacial clefting. Trends Mol Med. 2011;17(12):725–33. 10.1016/j.molmed.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 3.Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Goldstein T, et al. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32-35. Am J Hum Genet. 2004;75(2):161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, et al. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet. 2009;18(24):4879–96. 10.1093/hmg/ddp444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, et al. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 2008;40(11):1341–7. 10.1038/ng.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42(6):525–9. 10.1038/ng.580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beaty TH, Taub MA, Scott AF, Murray JC, Marazita ML, Schwender H, et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum Genet. 2013;132(7):771–81. 10.1007/s00439-013-1283-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet. 2009;41(4):473–7. 10.1038/ng.333 [DOI] [PubMed] [Google Scholar]
- 9.Grant SF, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, et al. A genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J Pediatr. 2009;155(6):909–13. 10.1016/j.jpeds.2009.06.020 [DOI] [PubMed] [Google Scholar]
- 10.Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet. 2010;42(1):24–6. 10.1038/ng.506 [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Huang Y, Yin A, Pan Y, Wang Y, Wang C, et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat Commun. 2015;6:6414 10.1038/ncomms7414 [DOI] [PubMed] [Google Scholar]
- 12.Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44(9):968–71. 10.1038/ng.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosen D, Chevrier C, Skytthe A, Bille C, Molsted K, Sivertsen A, et al. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J Med Genet. 2010;47(3):162–8. 10.1136/jmg.2009.069385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia ZL, Leslie EJ, Cooper ME, Butali A, Standley J, Rigdon J, et al. Replication of 13q31.1 Association in Nonsyndromic Cleft Lip with Cleft Palate in Europeans. Am J Med Genet A. 2015;167A(5):1054–60. 10.1002/ajmg.a.36912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mostowska A, Hozyasz KK, Wojcicki P, Zukowski K, Dabrowska A, Lasota A, et al. Association between polymorphisms at the GREM1 locus and the risk of nonsyndromic cleft lip with or without cleft palate in the Polish population. Birth Defects Res A Clin Mol Teratol. 2015. [DOI] [PubMed] [Google Scholar]
- 16.Al Chawa T, Ludwig KU, Fier H, Pötzsch B, Reich RH, Schmidt G, et al. Nonsyndromic cleft lip with or without cleft palate: Increased burden of rare variants within Gremlin-1, a component of the bone morphogenetic protein 4 pathway. Birth Defects Res A Clin Mol Teratol. 2014;100(6):493–8. 10.1002/bdra.23244 [DOI] [PubMed] [Google Scholar]
- 17.Khokha MK, Hsu D, Brunet LJ, Dionne MS, Harland RM. Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat Genet. 2003;34(3):303–7. [DOI] [PubMed] [Google Scholar]
- 18.Michos O, Panman L, Vintersten K, Beier K, Zeller R, Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development. 2004;131(14):3401–10. [DOI] [PubMed] [Google Scholar]
- 19.Brazil DP, Church RH, Surae S, Godson C, Martin F. BMP signalling: agony and antagony in the family. Trends Cell Biol. 2015;25(5):249–64. 10.1016/j.tcb.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 20.Parada C, Chai Y. Roles of BMP signaling pathway in lip and palate development. Front Oral Biol. 2012;16:60–70. 10.1159/000337617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomason HA, Dixon MJ, Dixon J. Facial clefting in Tp63 deficient mice results from altered Bmp4, Fgf8 and Shh signaling. Dev Biol. 2008;321(1):273–82. 10.1016/j.ydbio.2008.06.030 [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129(17):4135–46. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki S, Marazita ML, Cooper ME, Miwa N, Hing A, Jugessur A, et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet. 2009;84(3):406–11. 10.1016/j.ajhg.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Church RH, Krishnakumar A, Urbanek A, Geschwindner S, Meneely J, Bianchi A, et al. Gremlin1 preferentially binds to bone morphogenetic protein-2 (BMP-2) and BMP-4 over BMP-7. Biochem J. 2015;466:55–68. 10.1042/BJ20140771 [DOI] [PubMed] [Google Scholar]
- 25.Ashique AM, Fu K, Richman JM. Endogenous bone morphogenetic proteins regulate outgrowth and epithelial survival during avian lip fusion. Development. 2002;129(19):4647–60. [DOI] [PubMed] [Google Scholar]
- 26.He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, et al. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol. 2010;347(1):109–21. 10.1016/j.ydbio.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lana-Elola E, Tylzanowski P, Takatalo M, Alakurtti K, Veistinen L, Mitsiadis TA, et al. Noggin null allele mice exhibit a microform of holoprosencephaly. Hum Mol Genet. 2011;20(20):4005–15. 10.1093/hmg/ddr329 [DOI] [PubMed] [Google Scholar]
- 28.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7. 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangold E, Reutter H, Birnbaum S, Walier M, Mattheisen M, Henschke H, et al. Genome-wide linkage scan of nonsyndromic orofacial clefting in 91 families of central European origin. Am J Med Genet A. 2009;149A(12):2680–94. 10.1002/ajmg.a.33136 [DOI] [PubMed] [Google Scholar]
- 30.Rojas-Martinez A, Reutter H, Chacon-Camacho O, Leon-Cachon RB, Munoz-Jimenez SG, Nowak S, et al. Genetic risk factors for nonsyndromic cleft lip with or without cleft palate in a Mesoamerican population: Evidence for IRF6 and variants at 8q24 and 10q25. Birth Defects Res A Clin Mol Teratol. 2010;88(7):535–7. 10.1002/bdra.20689 [DOI] [PubMed] [Google Scholar]
- 31.Aldhorae KA, Böhmer AC, Ludwig KU, Esmail AH, Al-Hebshi NN, Lippke B, et al. Nonsyndromic cleft lip with or without cleft palate in arab populations: genetic analysis of 15 risk loci in a novel case-control sample recruited in Yemen. Birth Defects Res A Clin Mol Teratol. 2014;100(4):307–13. 10.1002/bdra.23221 [DOI] [PubMed] [Google Scholar]
- 32.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. 10.1038/nature11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Attanasio C, Nord AS, Zhu Y, Blow MJ, Li Z, Liberton DK, et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science. 2013;342(6157):1241006 10.1126/science.1241006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rada-Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell. 2012;11(5):633–48. 10.1016/j.stem.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–4. 10.1093/nar/gkr917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–43. 10.1038/ng.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leslie EJ, Taub MA, Liu H, Steinberg KM, Koboldt DC, Zhang Q, et al. Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am J Hum Genet. 2015;96(3):397–411. 10.1016/j.ajhg.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9(3):206–18. 10.1038/nrm2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richardson L, Venkataraman S, Stevenson P, Yang Y, Moss J, Graham L, et al. EMAGE mouse embryo spatial gene expression database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D835–44. 10.1093/nar/gkt1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hochheiser H, Aronow BJ, Artinger K, Beaty TH, Brinkley JF, Chai Y, et al. The FaceBase Consortium: a comprehensive program to facilitate craniofacial research. Dev Biol. 2011;355(2):175–82. 10.1016/j.ydbio.2011.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, et al. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A Clin Mol Teratol. 2012;94(11):934–42. 10.1002/bdra.23076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuniga A, Michos O, Spitz F, Haramis AP, Panman L, Galli A, et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes Dev. 2004;18(13):1553–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spitz F, Gonzalez F, Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell. 2003;113(3):405–17. [DOI] [PubMed] [Google Scholar]
- 46.Cuddapah SR, Kominek S, Grant JH, 3rd, Robin NH. IRF6 Sequencing in Interrupted Clefting. Cleft Palate Craniofac J. 2015. [DOI] [PubMed] [Google Scholar]
- 47.Ament SA, Szelinger S, Glusman G, Ashworth J, Hou L, Akula N, et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc Natl Acad Sci U S A. 2015;112(11):3576–81. 10.1073/pnas.1424958112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuniga A, Haramis APG, McMahon AP, Zeller R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature. 1999;401(6753):598–602. [DOI] [PubMed] [Google Scholar]
- 49.Levi G, Mantero S, Barbieri O, Cantatore D, Paleari L, Beverdam A, et al. Msx1 and Dlx5 act independently in development of craniofacial skeleton, but converge on the regulation of Bmp signaling in palate formation. Mech Dev. 2006;123(1):3–16. [DOI] [PubMed] [Google Scholar]
- 50.Boehringer S, van der Lijn F, Liu F, Gunther M, Sinigerova S, Nowak S, et al. Genetic determination of human facial morphology: links between cleft-lips and normal variation. Eur J Hum Genet. 2011;19(11):1192–7. 10.1038/ejhg.2011.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F, van der Lijn F, Schurmann C, Zhu G, Chakravarty MM, Hysi PG, et al. A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet. 2012;8(9):e1002932 10.1371/journal.pgen.1002932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinberg SM, Maher BS, Marazita ML. Parental craniofacial morphology in cleft lip with or without cleft palate as determined by cephalometry: a meta-analysis. Orthod Craniofac Res. 2006;9(1):18–30. [DOI] [PubMed] [Google Scholar]
- 53.Chan DC, Wynshaw-Boris A, Leder P. Formin isoforms are differentially expressed in the mouse embryo and are required for normal expression of fgf-4 and shh in the limb bud. Development. 1995;121(10):3151–62. [DOI] [PubMed] [Google Scholar]
- 54.Zhou F, Leder P, Zuniga A, Dettenhofer M. Formin1 disruption confers oligodactylism and alters Bmp signaling. Hum Mol Genet. 2009;18(13):2472–82. 10.1093/hmg/ddp185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moskvina V, Holmans P, Schmidt KM, Craddock N. Design of case-controls studies with unscreened controls. Ann Hum Genet. 2005;69(Pt 5):566–76. [DOI] [PubMed] [Google Scholar]
- 56.Ludwig KU, Böhmer AC, Rubini M, Mossey PA, Herms S, Nowak S, et al. Strong association of variants around FOXE1 and orofacial clefting. J Dent Res. 2014;93(4):376–81. 10.1177/0022034514523987 [DOI] [PubMed] [Google Scholar]
- 57.van Rooij IA, Vermeij-Keers C, Kluijtmans LA, Ocke MC, Zielhuis GA, Goorhuis-Brouwer SM, et al. Does the interaction between maternal folate intake and the methylenetetrahydrofolate reductase polymorphisms affect the risk of cleft lip with or without cleft palate? Am J Epidemiol. 2003;157(7):583–91. [DOI] [PubMed] [Google Scholar]
- 58.Reiter R, Brosch S, Goebel I, Ludwig KU, Pickhard A, Högel J, et al. A post GWAS association study of SNPs associated with cleft lip with or without cleft palate in submucous cleft palate. Am J Med Genet A. 2015;167A(3):670–3. 10.1002/ajmg.a.36891 [DOI] [PubMed] [Google Scholar]
- 59.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kouskoura T, Kozlova A, Alexiou M, Blumer S, Zouvelou V, Katsaros C, et al. The etiology of cleft palate formation in BMP7-deficient mice. PLoS One. 2013;8(3):e59463 10.1371/journal.pone.0059463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michos O, Goncalves A, Lopez-Rios J, Tiecke E, Naillat F, Beier K, et al. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development. 2007;134(13):2397–405. [DOI] [PubMed] [Google Scholar]
- 63.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–9. 10.1093/bioinformatics/btn564 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chromatin interaction data for three different celltypes were drawn from [38]. RefSeq genes are plotted above the interaction map, together with CTCF binding sites identified in the respective cell lines. GREM1 position is highlighted in red, adjacent genes SCG5 and FMN1 are also labeled. + /—below the RefSeq annotation denotes strand orientation of the gene. Yellow lines indicate contact regions, blue squares show regions of loop interactions, both as defined by the original study. The dotted line indicates co-localization of the GREM1 / 3‘FMN1 region within one topologically associated domain (TAD) which is separate from the 5‘ FMN1 region. TAD structure is stable between each of the three cell types. (A) NHEK–normal human epidermal keratinocytes, in situ combined, 5kb resolution, (B) HUVEC–human umbilical vein endothelial cells, in situ combined dataset, 5kb resolution(C) HMEC–human mammary epithelial cells.
(TIF)
Zoom-in into the GREM1/FMN1 region is provided for the HUVEC cells. (A) Local TAD structure (chr15: 33.000.000–33.100.000) with arrow indicating potential interaction loops between the GREM1 transcription start site (TSS, 33,010,204) and the intergenic region (33,040,000–33,044,999). The exact location of the loop cannot be further narrowed down due to the given resolution (5 kb). Data were drawn from [38]. Color code denotes number of observed reads. (B) Same data in virtual 4C visualisation. Using the GREM1-TSS as anchor point, regions of interactions are provided as peaks, with number of observed read counts as quantitative measure. Again, arrow highlights a potential interaction candidate at 33,040,000–33,044,999 bp.
(TIF)
Phenotype: empty symbol–unaffected, half-filled symbol–cleft of the lip and hard palate only (soft palate intact), full symbol–cleft of the lip, hard and soft palate. Carrier status: + carrier,—non-carrier. Phenotype-genotype correlation: red symbols—individuals with concordant genotype-phenotype correlation, black symbols–individuals with discordant genotype-phenotype correlation. a—Family BN-45 has four variants that are transmitted together: rs2280738, rs117317622, rs137899769, rs151194761. Please note that four more index patients with rare mutations but without any additional affected family members have a complete cleft of lip, hard and soft palate: BN-139 (rs201006159), BN-241 (rs201134502), BN-251 (g.33023715), BN-317 (rs147141645). All positions hg19.
(TIF)
(XLS)
(XLS)
(XLS)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.