

Abstract
The short average service life of traditional dental composite restorative materials and increasing occurrence of secondary caries adjacent to composite restorations and sealants are necessitating the development of new, longer lasting compositions. Novel monomers and their polymers, reinforcing fillers, and adhesive components are needed. The goal of this research is to develop resin systems for use in restorations, sealants, and other dental services that are superior in properties and endurance to currently used bisphenol A glycidyl dimethacrylate/triethylene glycol dimethacrylate (Bis-GMA/TEGDMA) and urethane–dimethacrylate products. Ether-based monomers and their polymers that were not susceptible to enzymatic or hydrolytic degradation were prepared and characterized. They showed no degradation under hydrolytic and enzymatic challenges, whereas the hydrolysis of ester links weakened contemporary resins within 16 days under these challenges. The success of the ether-based materials is promising in making durable systems that are subjected to long-term biochemical and hydrolytic challenges in oral environments.
Graphical abstract
INTRODUCTION
In the USA alone, 122.7 million dental composite restorations were placed in 2006, an increase of ≈ 40 % from 1999 (ADA Health Resources Policy Survey). The current trend in clinical dentistry indicates that this number is likely to increase. These contemporary dental composites1–4 generally contain three key components: (1) a bisphenol A glycidyl dimethacrylate/triethylene glycol dimethacrylate (Bis-GMA/TEGDMA), and/or a urethane dimethacrylate (all containing intramolecular hydrolyzable ester connecting groups) that produces the resin network, (2) reinforcing filler particles treated with coupling agents (containing intramolecular hydrolyzable ester connecting groups) to bind the resin to the particles, and (3) dentin/enamel bonding agents (also containing intramolecular hydrolyzable ester connecting groups).
These composites have been in service since Dr. Rafael Bowen first introduced them into dentistry in the early 1960s.5 However, many if not most of the currently available materials and their accompanying instructions for use do not produce satisfactory durability and esthetics over time. Composites based on Bis-GMA/TEGDMA contain undesirable ester groups [–C(═O)O–C–]. Many of these linking ester groups can eventually come apart by acidic, basic, or enzymatic-induced hydrolysis or saponification in the stressful intraoral environment, especially at or near polymer-tooth interfaces.3,4 Human saliva contains esterase, including cholesterol esterase and pseudocholine esterase, which can hydrolyze ester-containing compounds. Also, cariogenic bacteria such as Streptococcus mutans secrete esterase that can split ester groups.6–14 When subjected to thermal, mechanical, and biochemical challenges, contemporary composite restorations can lose interfacial-sealing integrity leading to staining and secondary decay. The short average service life of these systems and concerns regarding leached unreacted monomers, and possibly bisphenol A (BPA),15,16 and degradation products from these systems are evincing a need for new, long lasting composites to improve the dental and oral health globally.
New materials have been designed and new concepts proposed to enhance the performance and durability of the dental resin composites. Click chemistry17 and thiol–ene18,19 were introduced into dentistry by groups in Colorado to provide smart, reconfigurable, and responsive network.20,21 Adding thio-urethane oligomers improved the performance of resin composites.22,23 In-situ formation of antibacterial nanoparticles showed very promising results.24
The objective of the present work is to design and develop ether-based monomers that are superior in resistance to esterase and hydrolytic degradation in oral environments to the currently used Bis-GMA/TEGDMA, ester-containing monomers. The hypothesis to test is that the ether-based compounds will not be susceptible to salivary and/or other esterases, and thereby be more resistant to degradation in the oral cavity. Figure 1 illustrates a tooth restorative system, including tooth mineral, adhesive, resin network, coupling agent, and reinforcing filler, with ether-based compounds. As an example, three copolymerizable compounds, erythritol divinylbenzyl ether (E-DVBE), triethylene glycol divinylbenzyl ether (TEG-DVBE), and Glycine, N-2-hydroxy-3-(4-vinylbenzyloxy)-propyl-N-(4-methylphenyl), monosodium salt, (NTG-VBGE) (see Figure 1 for their structures) are applied as substitutes for the currently used Bis-GMA, TEGDMA, and NTG-GMA (Glycine, N-(2-hydroxy-3-(2-methyl-1-oxo-2-propenyl)propyl)-N-(4-methylphenyl), monosodium salt) [CAS No. 133736-31-9],25 respectively. The new compounds implement the good functionalities of Bis-GMA/TEGDMA. The E-DVBE and TEG-DVBE have two terminal double bonds, which can each readily copolymerize using the existing photopolymerization systems in dentistry. The TEG-DVBE will likewise be used to adjust and control the viscosity of the monomers to obtain good handling properties of dental composite restorative systems. The NTG-VBGE, incorporated in the form of the sodium, or other salt, will be the active ingredient in redesigned dentin/enamel bonding agents.26 Physical and chemical properties of restoration systems can be adjusted through subtle changes in the structure of the new compounds. E-DVBE is an amphiphilic compound with two hydrophobic vinylbenzyl groups at its ends and a flexible hydrophilic center (two hydroxyl groups from erythritol). The vicinal hydroxyl groups can more easily form clusters of hydrogen bonds with the readily accessible hydroxyl groups of other such monomers.27 Modeling suggests that such “clustering” increases monomer density relative to its polymer, which should contribute to polymerization shrinkage reduction.28 Other properties that can be improved through chemical structure changes include viscosity29 and hydrophilicity/hydrophobicity for enhancing adhesion to enamel and dentin.30,31
Figure 1.
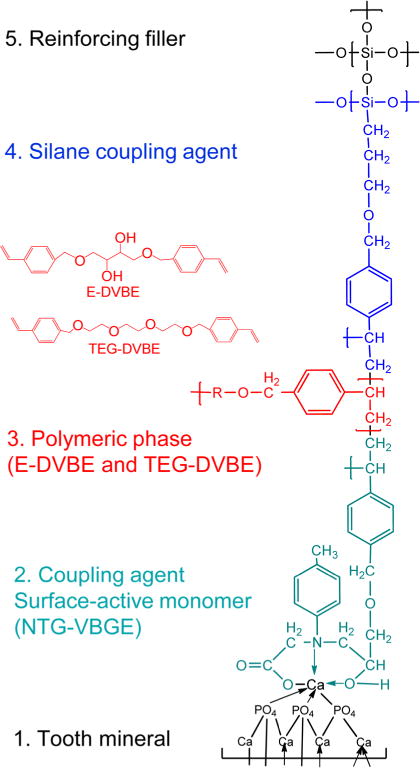
Illustration of a tooth restorative system, including tooth mineral, adhesive, resin network, coupling agent, and reinforcing filler with ether-based compounds.
Three ether-based monomers were synthesized and characterized. TEG-DVBE and its polymers were used to evaluate the resistance of new ether-based materials to hydrolytic and enzymatic degradation challenges in sodium phosphate buffer, cholesterol esterase, and pseudocholine esterase, respectively. The performance of TEG-DVBE and its polymer was compared with Bis-GMA and TEGDMA monomers and their copolymers. The hydrolysis sequence of the two ester groups on TEGDMA was discussed. In addition, the hydrolysis of the ester groups of the traditional resin monomers and their copolymers were assessed by high performance liquid chromatography (HPLC) in terms of monomer recovery and the production of methacrylic acid, which was further correlated with the change of mass and surface hardness of the copolymer/polymer.
MATERIALS AND METHODS
Enzyme Preparation
Cholesterol esterase from Pseudomonas sp. (CE; C9281, Sigma, Saint Louis, MO, USA) and pseudocholine esterase from horse serum (PCE, C4290, Sigma, Saint Louis, MO, USA) were reconstituted at desired concentrations in phosphate-buffered saline (D-PBS, 14190-144, Gibco, Grant Island, NY, USA). The prepared enzyme solutions used for replenishing enzyme activity in the biodegradation experiments were stored at −20 °C until needed.
Enzyme Activity Assay
The activity of CE was determined by para-nitrophenyl acetate (p-NPA) hydrolysis assay. The substrate, p-NPA (N8130, Sigma, Saint Louis, MO, USA), was prepared by dissolving p-NPA in methanol (100 mM), and diluting with a 100 mM sodium acetate buffer, pH 5.0, to give a final p-NPA concentration of 1 mM.32 In a typical CE activity assay, 50 μL of p-NPA solution, 50 μL of CE solution (1 unit/mL) and 100 μL sodium phosphate buffer (50 mM), pH 8.8, were added to a 96-well plate to give a final pH of 7.0,33 and the change of absorbance over time was measured at 410 nm at 25 °C using a SpectraMax Microplate reader (Molecular Devices, Sunnyvale, CA, USA). The absorbance was corrected by light path length correction from the absorbance of water at 975 nm and background at 900 nm from D-PBS blank samples on cuvettes with 1 cm path length compared to 200 μL filled microplate wells. One unit of CE activity was defined as a change of absorbance of 0.01 per minute.33 CE enzyme inhibition was assessed with the addition of 4 μL of phenylmethanesulfonylfluoride (PMSF, 50 mM in methanol). PCE (1 unit/mL) activity was determined with acetylcholinesterase activity assay kit (MAK119, Sigma, Saint Louis, MO, USA) by measuring a change in absorbance at 412 nm, using butylthiocholine (BTC)34 as a substrate. One unit of PCE activity was defined as the formation of 1.0 μmol of butyrate released per 1 mL of enzyme per minute at pH 7.5 and 25 °C.35,36
Synthesis and Characterization of Monomers
General Information
Commercially available materials purchased from Alfa Aesar, Sigma-Aldrich, and TCI America were used as received. Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on Bruker (600 MHz) or JEOL GSX (270 MHz) spectrometers using 5 mm tubes. Chemical shifts were recorded in parts per million (ppm, δ) relative to tetramethylsilane (δ = 0.00), dimethyl sulfoxide (δ = 2.50), or chloroform (δ = 7.26). 1H NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), quartet (q), dd (doublet of doublets), m (multiplets), etc. All first-order splitting patterns were assigned on the basis of the appearance of the multiplet. Splitting patterns that could not be easily interpreted are designated as multiplet (m) or broad (br). Melting points were measured on a METTLER FP62 melting point instrument in open capillary tubes. Hi-Res mass spectra were recorded on a JEOL AccuTOF, and 0.1 % ammonium formate in water (50 %) and MeOH (50 %) was used as the mobile phase for ESI analysis. Fourier transform infrared spectroscopy analysis (FTIR) was performed on a Thermo Nicolet NEXUS 670 FTIR spectrometer. Column chromatography was performed on silica gel (VWR, 230–400 mesh). Analytical thin-layer chromatography (TLC) was carried out on EMD Millipore 60 F254 precoated silica gel plate (0.2 mm thickness). Visualization was performed using UV radiation (254 nm).
1,12-Bis(4-vinylphenyl)-2,5,8,11-tetraoxadodecane (TEG-DVBE)
Triethylene glycol (8.02 mL, 9.01 g, 60 mmol) in DMF (30 mL) was added dropwise to a stirred suspension of NaH (95 %, 3.79 g, 150 mmol) in DMF (120 mL) at 0–4 °C under argon atmosphere over 30 min. After the reaction mixture was stirred for 2 h at room temperature, 4-vinylbenzyl chloride (90 %, 20.3 mL, 22.0 g, 120 mmol) in DMF (50 mL) was added dropwise over 30 min, and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was quenched by slow addition of a saturated NH4Cl aqueous solution (50 mL) at room temperature. The resulting solution was diluted with distilled water (600 mL) and extracted with ethyl acetate (3 × 200 mL). The combined ethyl acetate layers were washed with distilled water (2 × 200 mL). The organic layer was dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure to give crude product as a dark orange oil. Flash column chromatography (silica gel, 30 % ethyl acetate in hexane) afforded pure product as a pale yellow oil (27.5 g, 60 %). Log P: 4.36 (calculated by ChemBioDraw Ultra 14.0, CambridgeSoft Corporation, PerkinElmer, Waltham, MS, USA); 1H NMR (600 MHz, DMSO-d6) δ 7.43 (d, J = 8.1 Hz, 4 H), 7.29 (d, J = 8.1 Hz, 4 H), 6.72 (dd, J = 17.8, 11.0 Hz, 2 H), 5.81 (d, J = 17.8, 2 H), 5.24 (d, J = 11.0 Hz, 2 H), 4.47 (s, 4 H), 3.55 (m, 12 H) ppm; 13C NMR (600 MHz, DMSO-d6) δ 138.7, 136.9, 136.7, 128.2, 126.5, 114.5, 72.2, 70.4, 70.3, 69.6 ppm. Hi-Res MS (ESI): m/z calcd. for C24H30O4, 382.2144; found [M-NH4]+: C24H34NO4+, 400.2481.
(4R,5R)-2,2-Dimethyl-4,5-bis(((4-vinylbenzyl)oxy)methyl)-1,3-dioxolane
(−)-2,3-O-Isopropylidene-D-threitol (5 g, 30.8 mmol) in DMF (20 mL) was added dropwise to a stirred suspension of NaH (95 %, 1.95 g, 77.1 mmol) in DMF (60 mL) at 0–4 °C under argon atmosphere over 30 min. After the reaction mixture was stirred for 2 h at room temperature, 4-vinylbenzyl chloride (90 %, 9.60 mL, 10.4 g, 61.2 mmol) in DMF (50 mL) was added dropwise over 30 min, and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was quenched by slow addition of a saturated NH4Cl aqueous solution (20 mL) at room temperature. The resulting solution was diluted with distilled water (300 mL) and extracted with ethyl acetate (3 × 100 mL). The combined ethyl acetate layers were washed with distilled water (2 × 200 mL). The organic layer was dried over anhydrous potassium carbonate, and the solvent was removed under reduced pressure to give crude product as a dark orange oil. The crude product was used without further purification.
(2R,3R)-1,4-Bis((4-vinylbenzyl)oxy)butane-2,3-diol (E-DVBE)
(4R,5R)-2,2-Dimethyl-4,5-bis(((4-vinylbenzyl)oxy) methyl)-1,3-dioxolane crude was added to a stirred suspension of Dowex 50W2X (10 g) in MeOH (200 mL) at room temperature. The reaction mixture was then stirred and refluxed at 70 °C for 18 h. The mixture was filtered, and the filtrate was evaporated under reduced pressure. The resulting mixture was diluted with distilled water and extracted with CH2Cl2 (3 × 150 mL), and the combined organic layers were washed with distilled water (3 × 200 mL). The organic layer was dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure to give crude product as a yellow solid. Flash column chromatography (silica, 0–5 % MeOH in CH2Cl2) afforded pure product as a pale yellow oil (7.8 g, 71 %). Log P: 3.59; 1H NMR (600 MHz, CDCl3) δ 7.37 (d, J = 8.2 Hz, 4 H), 7.26 (d, J = 8.2 Hz, 4 H), 6.70 (dd, J = 17.6, 10.9 Hz, 2 H), 5.74 (d, J = 17.6, 2 H), 5.25 (d, J = 10.9 Hz, 2 H), 4.49 (s, 4 H), 3.85 (m, 2H), 3.55 (m, 4H), 3.31 (br, 2H) ppm; 13C NMR (270 MHz, CDCl3) δ 137.6, 137.3, 136.6, 128.1, 126.4, 114.0, 73.3, 71.9, 70.7 ppm. Hi-Res MS (ESI): m/z calcd. for C22H26O4, 354.1831; found [M-NH4]+: C22H30NO4+, 372.2175.
Sodium N-(2-Hydroxy-3-(4-vinylbenzyloxy)propyl)-N-(p-tolyl)-glycinate (NTG-VBGE)
In a preliminary synthesis (by AMG) to obtain NTG-VBGE, sodium hydroxide solution (2 M) was added dropwise to a mixture of the acid and sodium salt forms of N-p-tolylglycinate in water/tetrahydrofuran (THF; 1:1, 20 mL) until the nominal pH reached 8.7. After this mixture was stirred for 30 min at room temperature, 1.0 g of 4-vinylbenzyl glycidyl ether was added dropwise over 30 min and stirred at room temperature for 48 h. The resulting reaction mixture was diluted with acetone (200 mL), prompting the formation of a needle-like crystalline precipitate, and was then kept at 4 °C for 18 h. The crystals were isolated by gravity filtration, followed by drying under reduced pressure to afford white needle-like crystals (1.13 g, 57 %). Log P: 3.35; 1H NMR (270 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.47 (d, J = 7.3 Hz, 2 H), 7.33 (d, J = 8.0 Hz, 2 H), 6.90 (d, J = 8.1 Hz, 2 H), 6.73 (dd, J = 18.9, 10.8 Hz, 1 H), 6.39 (d, J = 7.9 Hz, 2 H), 5.83 (d, J = 17.7 Hz, 1 H), 5.24 (d, J = 11.0 Hz, 1 H), 4.51 (s, 2 H), 3.82 (d, J = 15.1 Hz, 1 H), 3.62 (d, J = 16.2 Hz, 1 H), 3.42 (m, 4 H), 3.15 (d, J = 5.4 Hz, 1 H), 2.13 (s, 3 H) ppm; 13C NMR (270 MHz, DMSO-d6) δ 176.23, 145.73, 138.46, 136.47, 135.32, 129.23, 127.72, 126.12, 122.77, 114.12, 111.12, 72.51, 72.17, 66.13, 58.73, 57.12, 19.93 ppm; mp 91.0–91.2 °C; Hi-Res MS (ESI): m/z calcd. for C21H24NO4Na, 378.1760; found [M-H]+: C21H25NO4Na+, 378.2116.
Monomer Degradation Assay
TEGDMA was dissolved in DMSO (20 mM), and diluted in D-PBS with or without enzymes to give a final concentration of 0.4 mM. In a 48-well plate, monomer solutions (750 μL) were incubated with six different media for 24 h at 37 °C (n = 3) to reach a total volume of 1.5 mL in each well. The four media were, D-PBS as blanks, CE (2 units/mL), PCE (2 units/mL), and PCE (2 units/mL) + PMSF (0.5 mM). Samples (400 μL of media from each well) were taken at 1, 8, and 24 h of incubation, and their enzyme activity was quenched by adding 266 μL methanol. The mixtures were then centrifuged at 16000 rcf for 30 min to eliminate large particles and stored at 4 °C for HPLC analysis.
Monomer Recovery Assay after Enzymatic Challenges
In a 48-well plate, Bis-GMA, TEGDMA, and TEG-DVBE were each dissolved in DMSO (20 mM), and diluted in D-PBS with or without enzymes [CE (2 units/mL) or PCE (2 units/mL)] to give a final concentration of 0.4 mM. These monomer solutions (325 μL) were incubated for 24 h at 37 °C (n = 3). Then, 500 μL of methanol was added to each well and incubated for an hour to quench the enzyme activity. The mixtures were subsequently transferred to a new plate. The residue in the wells were collected by rinsing the well with methanol (500 μL). These two fractions per well were centrifuged at 16 000 rcf for 30 min to eliminate large particles and stored at 4 °C separately for HPLC analysis. Monomer concentration in these two fractions were determined separately by HPLC and combined to calculate the percentage of monomer recovery.
Polymer Preparation
The composition of conventional resin was 50:50 wt % Bis-GMA:TEGDMA (Esstech, Essington, PA, USA) with 0.2 wt % Camphorquinone (CQ, 124893, Aldrich, Saint Louis, MO, USA) and 0.8 wt % ethyl 4-(diamethylamino)benzoate (4E, E24905, Aldrich, Saint Louis, MO, USA) as the photoinitiator system. TEG-DVBE was mixed with 1 wt % IRGACURE 819 (I-819) and 1 wt % bis(4-tert-butylphenyl)iodonium hexafluorophosphate (DPI) as a photoinitiation system. Photoinitiation systems for each composition were selected to achieve resins with high degree of conversion. Monomers were sandwiched into an 8 mm radius 1 mm height cylindrical Teflon mold between two Mylar films. The samples were photo cured with a Triad 2000 visible light curing unit (Dentsply, York, PA, USA) for 1 min on each side. The hardened pellets (surface area =75 mm2) were postcured overnight in a vacuum oven at 60 °C, and then incubated in D-PBS at 37 °C with stirring for 24 h to remove any unreacted monomers. Pellets were then rinsed with distilled water three times and vacuum-dried until they reached a constant mass (M1).
Polymer Mass Retention after Enzymatic Challenges
Cured polymer pellets were incubated with 500 μL 1 unit/mL CE or PCE, with media volume to polymer resin surface area ratio of 6.6 ul per mm2, for up to 16 days at 37 °C (n = 3). The incubation media was replaced every 48 h to maintain nominal enzyme activity. The enzyme in old media was quenched with addition of 400 μL methanol. The recurrent old media was pooled together for HPLC evaluations. The HPLC results on 2, 8, and 16 days of incubation periods evaluated the pooled media which was a collection of 1, 4, and 8 recurrent old media, respectively. Pooled media were centrifuged at 16000 rcf for 30 min to eliminate large particles and stored at 4 °C until analysis with HPLC. After 16 days of incubation, polymer pellets were rinsed with distilled water three times and vacuum-dried until they reach a constant mass (M2). The mass retention of the polymer pellets after enzyme degradation was (M2/M1) *100 %.
Degree of Vinyl Conversion (DC)
The DC of resin monomer and polymers was determined with the use of FTIR spectroscopy (Nexus 670 FTIR spectrophotometer (Thermo Scientific, Madison, Wisconsin, USA) equipped with attenuated total reflectance (ATR). FTIR spectra were acquired after polymer degradation. DC was calculated as the percentage change in the integrated peak area of the vinyl absorption band (1646–1616 cm−1) normalized to the peak area of aromatic C–H absorption band (1535–1496 cm−1) between the polymer (value after cure) and monomer (values before cure).37–39 The standard uncertainty associated with the DC measurement was < 1 %.
HPLC Analysis
An Agilent 1290 Infinity Binary HPLC System was used for the chromatographic separation and quantification of the degradation products. Specifically, these were the disappearance of TEGDMA, Bis-GMA, and TEG-DVBE monomers, and the appearance of methacrylic acid (MA, 155721, Aldrich, Saint Louis, MO, USA) derived from TEGDMA and Bis-GMA, and also bishydroxy propoxy phenyl propane (bisHPPP, 15137, Fluka, Saint Louis, MO, USA) derived from Bis-GMA degradation. A Zorbex Extend 5 μm C18 4.6 × 250 mm column (770450–902, Agilent Technology, Santa Clara, CA, USA) was used for the separation of products. The mobile phase consisted of 2 mM buffer solution of HPLC-grade ammonium acetate (AX1222, EMD Chemicals Inc., Billerica, MA, USA) with pH adjusted to 3.0 with 6.0 N hydrochloric acid (A144–500, Fisher Scientific, Fair Lawn, NJ, USA) and HPLC-grade methanol (MX0475, EMD Chemicals Inc., Billerica, MA, USA). The separation was achieved with 50 % to 100 % methanol in ammonium acetate buffer gradient for 30 min in order to provide comparison with previous published studies.11 Degradation products were detected by absorbance at 215 nm using a 1290 Infinity variable wavelength UV detector. Calibration curves were created by linear correlation of peak area to known concentrations of the analytes in methanol, and the amount of products formed from both monomer and polymer degradation were analyzed.
Knoop Hardness
The Knoop hardness was measured in accordance with ASTM standard E 384. A Leitz Miniload 2 microhardness machine was used with indentation loads of 0.25 N 0.5 and 1 N. Indentation sizes were measured with the same machine using a 10× objective. The loading time for an indentation was of the order of 15 s with a dwell at peak load of 15 s. Knoop hardness was the result of test force divided by the indentation projected surface area: HK = 14.229(p/d2), where P is the indenter force and d is the long diagonal length.40 The Knoop hardness test was performed on polymer pellets that had been incubated in D-PBS or with PCE. The hardness was an average of 45 measurements that were made on three specimens using three indentation loads, and five measurements per load. The standard uncertainty associated with the microindentation measurement is 5 %.
Statistical Analysis
The monomer recovery, polymer mass retention, DC, and Knoop hardness were analyzed using one-way analysis of variance (ANOVA) with a 95 % confidence interval to indicate significant differences.
RESULTS AND DISCUSSION
The ether-based monomers were synthesized to enable simultaneous, side-by-side, comparative testing of new versus traditional compounds under the same challenges. Scheme 1 is a simplified outline of the synthesis procedures for the new compounds. NTG-VBGE was synthesized by the same ring opening reaction that has been used to prepare NTG-GMA.25 To make E-DVBE, an acetonide-protected threitol was reacted with 4-vinylbenzyl chloride by SN2 reaction using NaH as base, followed by deprotection of the diol to form E-DVBE. The TEG-DVBE was synthesized using the same SN2 procedures. The chemical structures of these compounds were confirmed by 1H NMR, 13C NMR, and high-resolution mass spectroscopy.
Scheme 1.
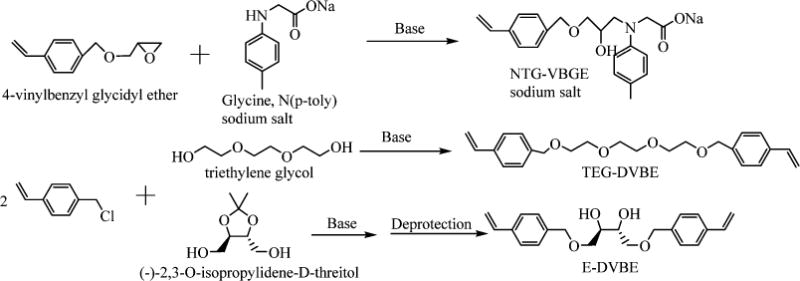
Synthetic Approaches to Make the Ether-Based Monomers
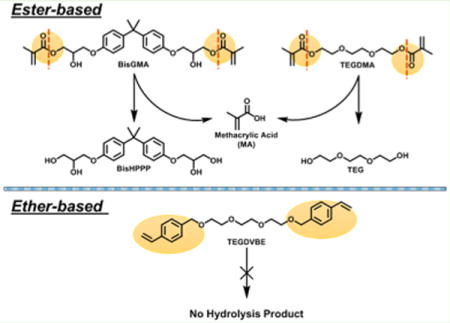
Ester function groups are subjected to hydrolyisis by acid, base, and esterases. An esterase is a hydrolase enzyme that splits esters into an alcohol and a carboxyl moiety in a chemical reaction with water, which is called hydrolysis. Both Bis-GMA and TEGDMA contain two hydrolyzable ester groups. The breakdown of ester groups in Bis-GMA and TEGDMA produces methacrylic acid (MA) and two other products of each monomer (Scheme 2). For TEGDMA, 2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl methacrylate (TEGMA) is produced when the first ester group is cleaved; and TEG is generated when both of the ester groups are hydrolyzed.
Scheme 2.
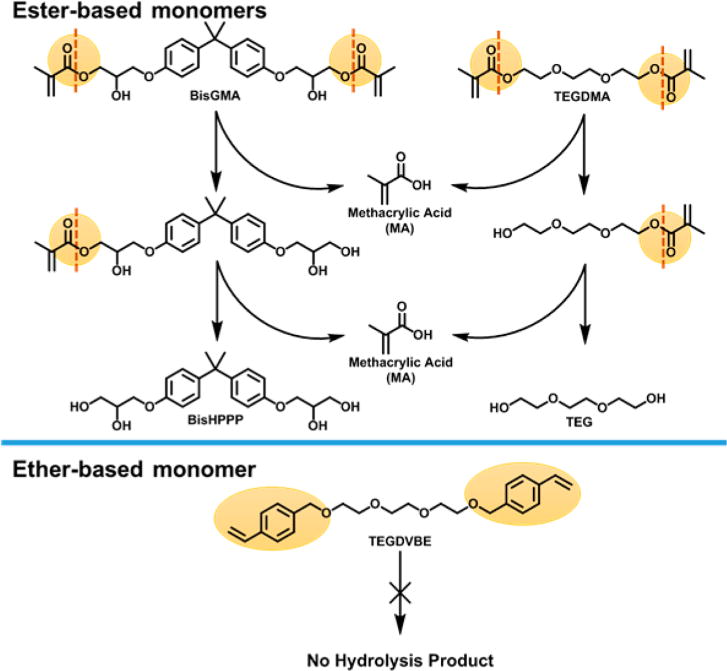
Sketch of Bis-GMA and TEGDMA Hydrolysis and the Resistance of Ether-Based Monomer to Hydrolysis
The enzymatic activity (Figure 2A,2B) of Pseudomona CE was 0.15 ± 0.02 units/μg protein using p-NPA as a substrate, approximately half of the reported enzymatic activity of human serum CE on p-NPA, and was within the range of the enzymatic activity found in human saliva: 0.09–0.26 unit/μg.11 The addition of PMSF, with a final concentration of 1 mM, as esterase inhibitor decreased the CE activity to 0.10 ± 0.01 units/μg protein, which was 33 % lower than that without inhibitor. The enzymatic activity of PCE (Figure 2C,D) from horse serum was 0.055 ± 0.01 units/μg protein when BTC was used as a substrate, which was much higher than activities of human saliva on BTC in literature (0.004–0.018 units/μg protein).11 The addition of PMSF with a final concentration of 1 mM reduced the PCE activity to 0.001 ± 0.001 μg protein, which was a 98 % reduction on enzymatic activity.11 The change of enzyme sources lead to substantial variances in enzymatic activities. Using enzymes with stronger activities provided opportunities to correlate the hydrolysis of compounds with material performance in a short period of time.
Figure 2.
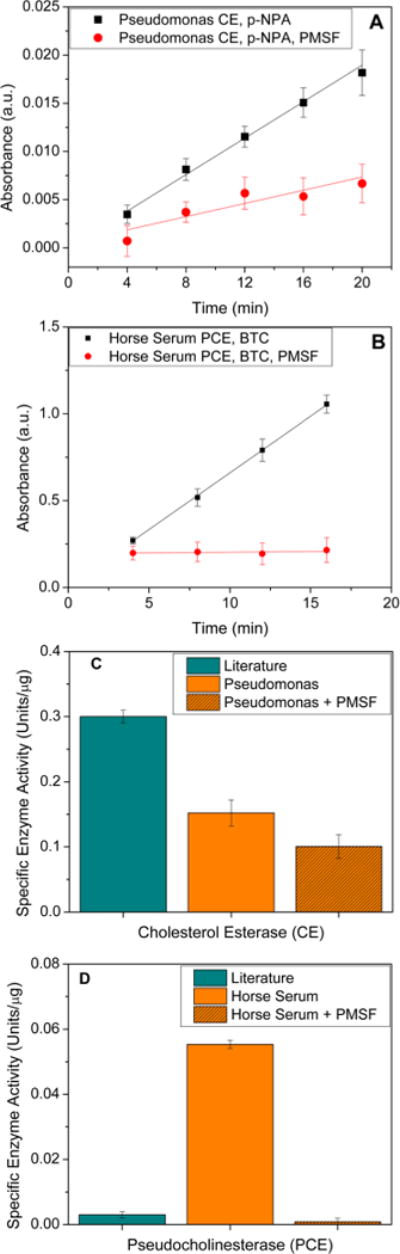
CE and PCE activity assay. (A) Absorbance from the degradation of p-NPA substrate by CE with/without PMSF measured at 410 nm; (B) absorbance from the degradation of BTC substrate by PCE with/without PMSF measured at 415 nm; (C) enzymatic activity of CE with/without PMSF measured spectroscopically; and (D) enzymatic activity of PCE with/without PMSF measured spectroscopically. All data is reported as mean ± standard error (N = 3).
The TEGDMA degraded and produced MA in different challenges. D-PBS was the medium and was also used as a negative control for esterase CE and PCE. PMSF was used as an inhibitor for PCE. The mass change of monomer TEGDMA (retention time: 11.6 min), TEGMA (retention time: 4.6 min), and MA (retention time: 4.3 min) over time (1, 8, and 24 h in D-PBS, CE, and PCE and 24 h in the presence of PMSF) are shown in Figure 3. Only a small amount (4 % and 10 %) of TEGDMA was lost at 24 h in D-PBS and CE, respectively. Significant loss of TEGDMA was seen under PCE challenges. No TEGMDA was detected after 8 h incubation. Approximately 25 % by mass of TEGDMA was decomposed in the first hour of incubation. We believe that the extraordinary high PCE activity contributed to the complete disappearing of TEGDMA within a short time. The addition of PMSF significantly reduced the degradation rate and approximately 35 % by mass of the TEGDMA survived after 24 h of incubation.
Figure 3.
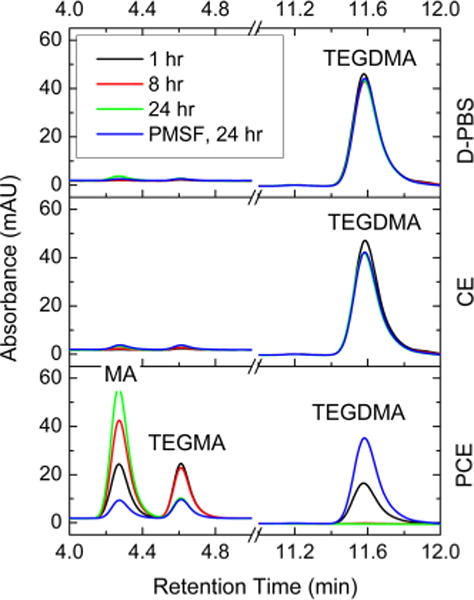
HPLC analysis of hydrolysis sequence of the two ester groups on TEGDMA.
The relative molar ratio of MA and TEGMA for samples in PCE at 1 h, 8 h, 24 h, and 24 h with PMSF were analyzed according to peak area, which was approximately 1, after 1 h incubation with PCE and 24 h incubation with PMSF. In both cases, large amounts of TEGDMA were still present. However, after 8 and 24 h incubation with PCE peak-area ratios of MA/TEGMA were 1.8 and 5.8, respectively, and TEGDMA was not detected. In-depth stoichiometric correlation analysis indicated that one ester group of TEGDMA was hydrolyzed first, and the majority of the second ester groups on the same molecule were split after TEGDMA monomers were all consumed.
A monomer recovery assay was carried out to evaluate the degradation of Bis-GMA and TEG-DVBE and cross-check the finding in TEGDMA. Figure 4 shows the percentage of recovered monomers under challenges of D-PBS, CE and PCE, respectively. The results from monomer recovery of TEGDMA agreed well with the findings by the monomer degradation assay: A small amount of TEGDMA degraded under challenges of D-PBS and CE, and TEGDMA had completely disappeared after 24 h incubation with PCE. Bis-GMA had no degradation in D-PBS, whereas enzyme CE was more effective on Bis-GMA than PCE. This selectivity of CE and PCE on Bis-GMA and TEGDMA was also reported in the literature.41,42 In contrast to the traditional monomers, no mass loss of TEG-DVBE was detected in any of the culture conditions.
Figure 4.
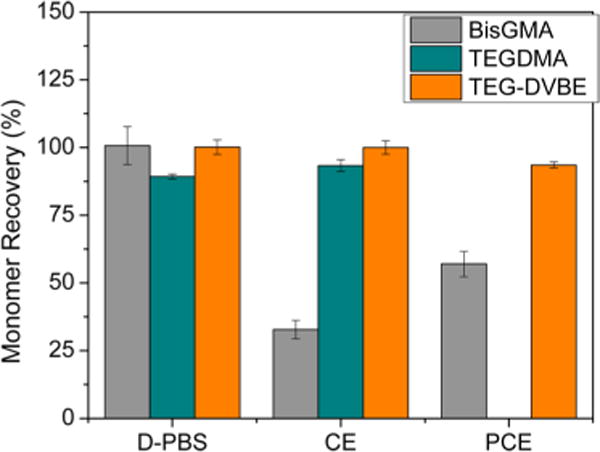
Monomer recovery after enzymatic degradation evaluated by HPLC. Cumulative amounts of monomer recovered from aliquots and methanol wash from wells.
We maximized the degradation rate without using filler. The degradation rate of traditional resins increased as the amount of filler decreased.12 No new peaks were detected by HPLC when TEG-DVBE pellets were subjected to all of the challenges. The MA production of the traditional polymer was evaluated by HPLC to indicate the progress of polymer degradation (Figure 5). After 2 and 8 d of incubation, no significant difference in terms of MA production was detected when the polymer pellets under challenges of CE or PCE; significantly more MA was produced under PCE challenges after 16 d of incubation. The MA production can only come from unpolymerized methacrylate groups because polymerized methacrylate groups do not produce MA. Table 1 lists the degree of conversion of pellets under different challenges. Although the DC of Bis-GMA/TEGDMA pellets after PCE challenges is the lowest (P < 0.05), the difference is too small to be correlated with the amount of MA released.
Figure 5.
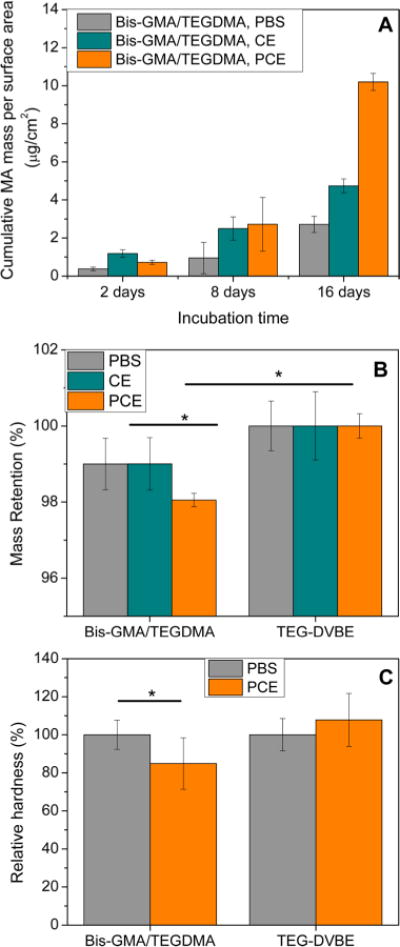
Polymer biodegradation. (A) MA released from degradation of Bis-GMA:TEGDMA (1:1) resins with CE and PCE esterase. (B) Mass retention of Bis-GMA:TEGDMA and TEG-DVBE resins after 16 days incubation with and without CE or PCE enzyme. (C) Relative hardness of Bis-GMA:TEGDMA and TEGDVBE resins with and without the presence of PCE. (N = 45, P < 0.05).
Table 1.
Degree of Conversion of Samples under Different Challenges
| Bis-GMA/TEGDMA
|
TEG-DVBE
|
|||
|---|---|---|---|---|
| DC | STDEV | DC | STDEV | |
| average DC before degradationa | 84.7 | 0.5 | 97.2 | 2.1 |
| PBS only | 83.9 | 0.3 | 95.6 | 0.8 |
| CE | 84.0 | 0.3 | 94.2 | 1.5 |
| PCE | 82.5 | 0.6 | 95.5 | 1.9 |
Note: These pellets were prepared at the same time as those being challenged.
The sudden increase of the MA production at 16 days under PCE challenges suggested that a large amount of ester groups were hydrolyzed between 8 to 16 days, which should associate with a mass change. The mass retention of the polymer pellets are showed in Figure 4B. After 16 days of incubation, the mass retention of Bis-GMA/TEGDMA pellets under the PCE challenge was significantly smaller than those under the other two challenges, while there was no mass loss in TEG-DVBE pellets under all of the challenges.
In addition, the Knoop hardness of the Bis-GMA/TEGDMA pellets was statistically lower (p < 0.05) after PCE challenge than that of the pellets after D-PBS challenges, while the Knoop hardness of the TEGDVBE pellets showed no significant change (Figure 5C). The ANOVA analysis on Knoop hardness was based on 45 measurements for each composition per challenge. Three specimen were evaluated per composition, and each composition was evaluated under three loads (0.25 N, 0.5 and 1 N), with five measurement per load. Overall, the hydrolysis of ester groups on Bis-GMA/TEGDMA pellets caused mass loss of the polymer and softening of the resin.
CONCLUSIONS
Three ether-based monomers were synthesized and characterized. TEG-DVBE monomer and its polymer showed no degradation under the challenges from D-PBS and esterase including CE and PCE, while the traditional dental resins were decomposing to different levels under these challenges. By using a highly active esterase enzyme from horse serum, the two ester groups on one TEGDMA compound were completely split in less than 8 h. The two ester groups on TEGDMA were hydrolyzed sequentially, and the majority of the second ester group was decomposed after no TEGDMA was detected. The ester hydrolysis results were consistent with mass loss of monomers and polymers. Furthermore, the hydrolysis of ester caused significant damage, in terms of mechanical performance, to the polymer within 16 days. The resistance of ether-based materials to esterase degradation made them good candidates for making durable compositions that are subjected to long-term biochemical and hydrolytic challenges in oral environments.
Supplementary Material
Acknowledgments
This work was funded by the National Institute of Dental and Craniofacial Research (U01DE023752). Financial support was also provided through the ADA Foundation. The authors would like to thank Mr. Anthony Giuseppetti, Mr. George Quinn, and Dr. Joseph Antonucci for their technical recommendations. We would like to thank Drs. Kenneth Cole and Brian Lang at Biosystems and Biomaterials Division, National Institute of Standards and Technology (NIST), for their help on HPLC. We also would like to thank the Center for Nanoscale Science and Technology (CNST) at NIST for their technical support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.5b01069.
The 1H NMR and 13C NMR spectra of the ether-based compounds (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. A.G.M. and G.K. contributed equally to this project.
Notes
The authors declare no competing financial interest.
References
- 1.Bowen RL. Properties of a silica-reinforced polymer for dental restorations. J Am Dent Assoc, JADA. 1963;66(1):57–64. doi: 10.14219/jada.archive.1963.0010. [DOI] [PubMed] [Google Scholar]
- 2.Bowen RL, Marjenhoff WA. Dental composites glass ionomers - the materials. Adv Dent Res. 1992;6:44–49. doi: 10.1177/08959374920060011601. [DOI] [PubMed] [Google Scholar]
- 3.Ferracane JL. Resin composite-State of the art. Dent Mater. 2011;27(1):29–38. doi: 10.1016/j.dental.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 4.Ferracane JL. Resin-based composite performance: Are there some things we can’t predict? Dent Mater. 2013;29(1):51–58. doi: 10.1016/j.dental.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowen RL. US Patent US3066112. Dental filling material. 1962
- 6.Lee YK, Powers JM. Influence of salivary organic substances on the discoloration of esthetic dental materials - A review. J Biomed Mater Res, Part B. 2006;76B(2):397–402. doi: 10.1002/jbm.b.30380. [DOI] [PubMed] [Google Scholar]
- 7.Lin BA, Jaffer F, Duff MD, Tang YW, Santerre JP. Identifying enzyme activities within human saliva which are relevant to dental resin composite biodegradation. Biomaterials. 2005;26(20):4259–4264. doi: 10.1016/j.biomaterials.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Munksgaard EC, Freund M. Enzymatic-hydrolysis of (di)methacrylates and their polymers. Eur J Oral Sci. 1990;98(3):261–267. doi: 10.1111/j.1600-0722.1990.tb00971.x. [DOI] [PubMed] [Google Scholar]
- 9.Santerre JP, Shajii L, Leung BW. Relation of dental composite formulations to their degradation and the release of hydrolyzed polymeric-resin-derived products. Crit Rev Oral Biol Med. 2001;12(2):136–151. doi: 10.1177/10454411010120020401. [DOI] [PubMed] [Google Scholar]
- 10.Santerre JP, Shajii L, Tsang H. Biodegradation of commercial dental composites by cholesterol esterase. J Dent Res. 1999;78(8):1459–1468. doi: 10.1177/00220345990780081201. [DOI] [PubMed] [Google Scholar]
- 11.Finer Y, Santerre JP. Salivary esterase activity and its association with the biodegradation of dental composites. J Dent Res. 2004;83(1):22–26. doi: 10.1177/154405910408300105. [DOI] [PubMed] [Google Scholar]
- 12.Finer Y, Santerre JP. Influence of silanated filler content on the biodegradation of bisGMA/TEGDMA dental composite resins. J Biomed Mater Res, Part A. 2007;81A(1):75–84. doi: 10.1002/jbm.a.31004. [DOI] [PubMed] [Google Scholar]
- 13.Bourbia M, Ma D, Cvitkovitch DG, Santerre JP, Finer Y. Cariogenic bacteria degrade dental resin composites and adhesives. J Dent Res. 2013;92(11):989–994. doi: 10.1177/0022034513504436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delaviz Y, Finer Y, Santerre JP. Biodegradation of resin composites and adhesives by oral bacteria and saliva: A rationale for new material designs that consider the clinical environment and treatment challenges. Dent Mater. 2014;30(1):16–32. doi: 10.1016/j.dental.2013.08.201. [DOI] [PubMed] [Google Scholar]
- 15.Calafat AM, Ye XY, Wong LY, Reidy JA, Needham LL. Exposure of the US population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116(1):39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh S, Li SSL. Bisphenol A and phthalates exhibit similar toxicogenomics and health effects. Gene. 2012;494(1):85–91. doi: 10.1016/j.gene.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 17.Xi WX, Scott TF, Kloxin CJ, Bowman CN. Click Chemistry in Materials Science. Adv Funct Mater. 2014;24(18):2572–2590. [Google Scholar]
- 18.Boulden JE, Cramer NB, Schreck KM, Couch CL, Bracho-Troconis C, Stansbury JW, Bowman CN. Thiol-ene-methacrylate composites as dental restorative materials. Dent Mater. 2011;27(3):267–272. doi: 10.1016/j.dental.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cramer NB, Couch CL, Schreck KM, Carioscia JA, Boulden JE, Stansbury JW, Bowman CN. Investigation of thiol-ene and thiol-ene-methacrylate based resins as dental restorative materials. Dent Mater. 2010;26(1):21–28. doi: 10.1016/j.dental.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cramer NB, Stansbury JW, Bowman CN. Recent Advances and Developments in Composite Dental Restorative Materials. J Dent Res. 2011;90(4):402–416. doi: 10.1177/0022034510381263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kloxin CJ, Bowman CN. Covalent adaptable networks: smart, reconfigurable and responsive network systems. Chem Soc Rev. 2013;42(17):7161–7173. doi: 10.1039/c3cs60046g. [DOI] [PubMed] [Google Scholar]
- 22.Bacchi A, Consani RL, Martim GC, Pfeifer CS. Thio-urethane oligomers improve the properties of light-cured resin cements. Dent Mater. 2015;31(5):565–574. doi: 10.1016/j.dental.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacchi A, Dobson A, Ferracane JL, Consani R, Pfeifer CS. Thio-urethanes Improve Properties of Dual-cured Composite Cements. J Dent Res. 2014;93(12):1320–1325. doi: 10.1177/0022034514551768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan C, Chu LR, Rawls HR, Norling BK, Cardenas HL, Whang K. Development of an antimicrobial resin-A pilot study. Dent Mater. 2011;27(4):322–328. doi: 10.1016/j.dental.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 25.Bowen RL. US Patent US4588756A. Multistep method for obtaining strong adhesive bonding of composites to dentin, enamel and other substrates. 1986
- 26.Bowen RL, Cobb EN, Rapson JE. Adheisve bonding of various materials to hard tooth tissues - improvement in bond strength to dentin. J Dent Res. 1982;61(9):1070–1076. doi: 10.1177/00220345820610090901. [DOI] [PubMed] [Google Scholar]
- 27.Sun J, Yu KH, Russo PS, Pople J, Henry A, Lyles B, McCarley RS, Baker GR, Newkome GR. Some structural observations of self-assembled, fibrillar gels composed of two-directional bolaform arborols. Polymeric Nanofibers. 2006;918:370–383. [Google Scholar]
- 28.Moraes RR, Garcia JW, Barros MD, Lewis SH, Pfeifer CS, Liu JC, Stansbury JW. Control of polymerization shrinkage and stress in nanogel-modified monomer and composite materials. Dent Mater. 2011;27(6):509–519. doi: 10.1016/j.dental.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun JR, Fang R, Lin N, Eidelman N, Lin-Gibson S. Nondestructive quantification of leakage at the tooth-composite interface and its correlation with material performance parameters. Biomaterials. 2009;30(27):4457–4462. doi: 10.1016/j.biomaterials.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishitani Y, Yoshiyama M, Donnelly AM, Agee KA, Sword J, Tay FR, Pashley DH. Effects of resin hydrophilicity on dentin bond strength. J Dent Res. 2006;85(11):1016–1021. doi: 10.1177/154405910608501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun JR, Forster AM, Johnson PM, Eidelman N, Quinn G, Schumacher G, Zhang XR, Wu WL. Improving performance of dental resins by adding titanium dioxide nanoparticles. Dent Mater. 2011;27(10):972–982. doi: 10.1016/j.dental.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Labow RS, Adams KAH, Lynn KR. Porcine cholesterol esterase, a multiform enzyme. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1983;749(1):32–41. doi: 10.1016/0167-4838(83)90147-4. [DOI] [PubMed] [Google Scholar]
- 33.Labow RS, Duguay DG, Santerre JP. The enzymatic-hydrolysis of a synthetic biomembrane - a new substrate for cholesterol and carboxyl esterases. J Biomater Sci, Polym Ed. 1995;6(2):169–179. doi: 10.1163/156856294x00293. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Chen SG, Ralph EC, Dwyer M, Cashman JR. Identification of Human Butyrylcholinesterase Organophosphate-Resistant Variants through a Novel Mammalian Enzyme Functional Screen. J Pharmacol Exp Ther. 2012;343(3):673–682. doi: 10.1124/jpet.112.198499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khalichi P, Cvitkovitch DG, Santerre JP. Effect of composite resin biodegradation products on oral streptococcal growth. Biomaterials. 2004;25(24):5467–5472. doi: 10.1016/j.biomaterials.2003.12.056. [DOI] [PubMed] [Google Scholar]
- 36.Finer Y, Santerre JP. Biodegradation of a dental composite by esterases: dependence on enzyme concentration and specificity. J Biomater Sci, Polym Ed. 2003;14(8):837–849. doi: 10.1163/156856203768366558. [DOI] [PubMed] [Google Scholar]
- 37.Stansbury JW, Dickens SH. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent Mater. 2001;17(1):71–79. doi: 10.1016/s0109-5641(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 38.Eidelman N, Raghavan D, Forster AM, Amis EJ, Karim A. Combinatorial approach to characterizing epoxy curing. Macromol Rapid Commun. 2004;25(1):259–263. [Google Scholar]
- 39.Antonucci JM, Fowler BO, Weir MD, Skrtic D, Stansbury JW. Effect of ethyl-alpha-hydroxymethylacrylate on selected properties of copolymers and ACP resin composites. J Mater Sci: Mater Med. 2008;19(10):3263–3271. doi: 10.1007/s10856-008-3463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nunes RAX, Costa VC, Calado VMD, Branco JRT. Wear, friction, and microhardness of a thermal sprayed PET - poly (ethylene terephthalate) coating. Mater Res. 2009;12(2):121–125. [Google Scholar]
- 41.Finer Y, Jaffer F, Santerre JP. Mutual influence of cholesterol esterase and pseudocholinesterase on the biodegradation of dental composites. Biomaterials. 2004;25(10):1787–1793. doi: 10.1016/j.biomaterials.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 42.Jaffer F, Finer Y, Santerre JP. Interactions between resin monomers and commercial composite resins with human saliva derived esterases. Biomaterials. 2002;23(7):1707–1719. doi: 10.1016/s0142-9612(01)00298-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.