

Abstract
Activators of innate immunity may have the potential to combat a broad range of infectious agents. We report that treatment with bacterial flagellin prevented rotavirus (RV) infection in mice and cured chronically RV-infected mice. Protection was independent of adaptive immunity and interferon (IFN, type I and II) and required flagellin receptors Toll-like receptor 5 (TLR5) and NOD-like receptor C4 (NLRC4). Flagellin-induced activation of TLR5 on dendritic cells elicited production of the cytokine interleukin-22 (IL-22), which induced a protective gene expression program in intestinal epithelial cells. Flagellin also induced NLRC4-dependent production of IL-18 and immediate elimination of RV-infected cells. Administration of IL-22 and IL-18 to mice fully recapitulated the capacity of flagellin to prevent or eliminate RV infection and thus holds promise as a broad-spectrum antiviral agent.
Rotavirus (RV) causes severe dehydrating diarrhea in young children and moderate intestinal distress in adults (1). RV infection of adult mice serves as a well-defined model of intestinal viral infection in which infectivity can be monitored by measuring levels of viral antigen shed in feces (2). RV predominantly infects and replicates in epithelial cells lining the small intestine (1). Bacterial flagellin, the primary component of bacterial flagella, potently activates host defense gene expression in intestinal epithelial cells (IECs) (3) and is viewed as a dominant immune activator in the intestine (4). Flagellin-induced gene expression confers both IECs and mice with resistance to a variety of challenges, including bacteria, chemicals, and radiation (5, 6). Moreover, administration of flagellin reduced susceptibility of mice to a culture-adapted strain of RV (7). Thus, we investigated the potential of flagellin to protect mice against highly contagious pathogenic mouse RV.
Oral inoculation of adult C57BL/6 mice with mouse RV (EC strain), using a dose 100,000 times that required to infect 50% of mice, resulted in uniform RV shedding that peaked 3 to 4 days after inoculation and resolved several days later, indicating viral clearance (2). Administration of a single dose of flagellin via intraperitoneal injection shortly before infection temporarily prevented infection (fig. S1A). Repeated administration of flagellin further delayed (fig. S1B) or completely prevented infection (Fig. 1A). In contrast, repeated administration of bacterial lipopolysaccharide (LPS) only transiently protected against RV infection (fig. S2).
Fig. 1. Flagellin protects mice from RV infection and diarrhea.
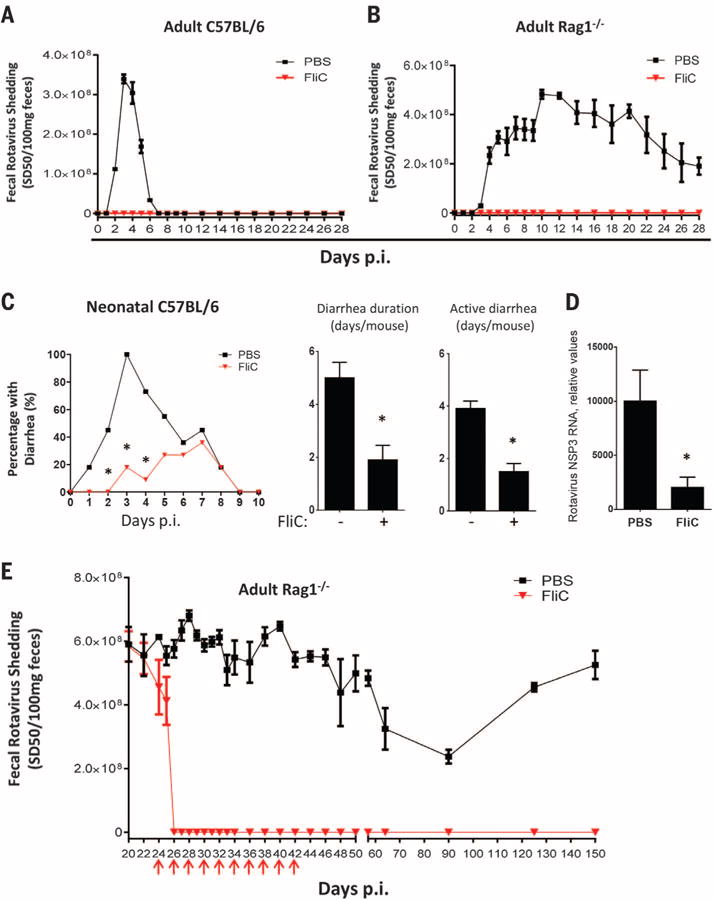
(A and B) Eight-week-old female (A) C57BL/6 mice or (B) Rag1−/− mice were orally inoculated with mouse RV, EC strain. Mice were administered 0.2 ml of phosphate-buffered saline (PBS) (vehicle) ± 20 μg of flagellin by means of intraperitoneal injection, and then every other day from 0 to 18 days after inoculation. Feces were collected daily and assayed for RV antigens by means of enzyme-linked immunosorbent assay (ELISA). Results are shown as mean ± SEM [two-way analysis of variance (ANOVA), n = 4 mice, P < 0.001) for (A) and (B). p.i., post-inoculation. (C) Seven-day-old C57BL/6 mice were orally inoculated with RV (supplementary materials). Mice were treated with PBS or flagellin (10 μg) every day from 0 to 9 days after inoculation and monitored for incidence of diarrhea daily (χ2 test, n = 11 mice, *P < 0.01). Flagellin-treated mice exhibited significantly reduced duration of diarrhea and days of active diarrhea (Student’s t test, n = 11 mice, *P < 0.01). (D) Seven-day-old mice, treated as in (C), were euthanized 3 days after inoculation. Total RNA of small intestines from those mice were prepared and analyzed for RV NSP3 RNA level (Student’s t test, n = 6 mice, *P < 0.05). (E) Three-week-old Rag1−/− mice were inoculated with mouse RV. Three weeks after inoculation, at which point a chronic infection had been established, mice were treated with PBS ± flagellin (20 μg) every 2nd day from 22 to 40 days after inoculation as indicated with red arrows. Feces were collected at indicated days and assayed for RVantigens by means of ELISA. Asterisk indicates that the difference between mice given PBS and flagellin was statistically significant. Results are shown as mean ± SEM (two-way ANOVA, n = 5 mice, P < 0.0001).
RV infection induces an adaptive immune response, which is normally required to clear infection (8). However, we saw very little elevation in antibodies to RV in flagellin-treated mice (fig. S3), suggesting that flagellin’s protection against RV was independent of adaptive immunity. Confirming this notion, the protective effect of flagellin was fully maintained in mice lacking recombination activating gene 1 (Rag1−/−), making them deficient in mature B and T lymphocytes and susceptible to chronic RV infection (Fig. 1B) (9).
Analogous to young children, infection of neonatal mice with RV causes watery diarrhea (10). The incidence, duration, and active days of such RV-induced diarrhea and associated viral loads were reduced via flagellin treatment (Fig. 1, C and D). Flagellin treatment also diminished reovirus load after oral inoculation, suggesting broad antiviral activity (fig. S4).
We next investigated whether flagellin could treat established chronic RV infection in immune-compromised mice. Although Rag1−/− mice of all ages develop chronic RV infection, high viral loads are attained by infecting 3-week-old Rag1−/− mice (9). Flagellin treatment eliminated detectable RV shedding by 2 days after treatment. Moreover, 10 doses of flagellin treatment over a 20-day period abolished RV shedding for at least 150 days, whereas untreated mice shed virus over their lifetime (Fig. 1E). Accordingly, RV antigens, which were detectable by means of immunostaining in villus epithelial cells in untreated Rag1−/− mice, were absent in mice treated with flagellin by 24 hours (fig. S5A). Moreover, treatment with flagellin resulted in reduced levels of RV RNA within hours of administration, and RV RNA was undetectable 48 hours after treatment. Reduced levels of RV replication preceded a reduction in levels of RV genomic RNA (fig. S5B). In Rag1−/− mice, a substantial degree of replication occurs extra-intestinally, especially in the liver, resulting in hepatitis (11). Levels of RV RNA in the liver were greatly reduced within 24 hours of flagellin treatment and undetectable within 48 hours (fig. S5C). Similarly, flagellin treatment also eliminated the more modest levels of RV RNA in the spleen (fig. S5D). These results indicate that flagellin treatment cures chronic RV infection in immune-compromised mice.
Flagellin-induced remodeling of intestinal gene expression requires Toll-like receptor 5 (TLR5) (12). Flagellin can also be recognized intracellularly by the NOD-like receptor C4 (NLRC4) inflammasome, resulting in caspase 1–mediated production of the cytokines interleukin-1β (IL-1β) and IL-18 (13, 14). We expected that only TLR5 would be required for flagellin protection against RV infection. However, loss of either pathway of flagellin recognition reduced, but did not completely eliminate, the capacity of flagellin to prevent RV infection (Fig. 2, A and B). In contrast, absence of both TLR5 and NLRC4 or myeloid differentiation primary response gene 88 (MyD88), which is required for signaling by TLR5 and inflammasomeassociated cytokines, eliminated flagellin’s protection against RV infection (Fig. 2, C and D).
Fig. 2. Flagellin’s antiviral activity is mediated by TLR5/NLRC4 on dendritic cells.
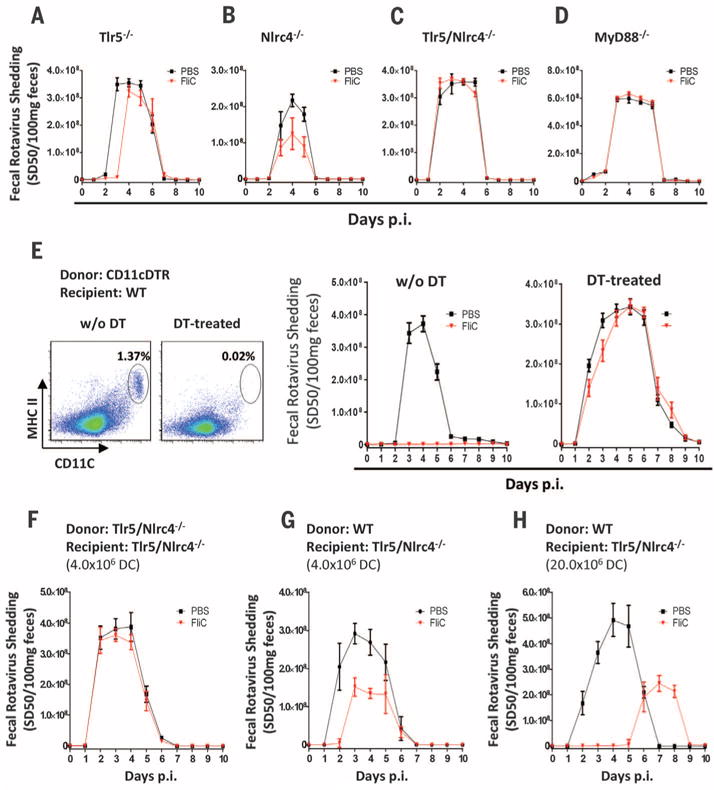
(A to D) Eight-week-old (A) Tlr5−/−, (B) Nlrc4−/−, (C) Tlr5/Nlrc4−/−, and (D) MyD88−/− mice were orally inoculated with mouse RV and administered PBS ± flagellin (20 μg) every other day from 0 to 8 days after inoculation. Feces were assayed for RV antigens by means of ELISA. Results in (A) to (D) are shown as mean ± SEM [Student’s t test, n = 5 mice, P < 0.05 on day 3 in (A) and day 5 in (B)]. Differences between PBS and flagellin groups in (C) and (D) were not significant [two-way ANOVA, n = 4 to 5 mice, P = 0.6361 for (C) and P = 0.3871 for (D)]. (E) CD11c-DTR reconstituted bone marrow chimeras were either untreated or injected with DT at 8 ng/gram body weight once a day for 2 days. Flow cytometry plots show the extent of depletion of DCs (CD45+/CD19−/MHC class II+/CD11c+ splenocytes). Mice were then studied as in (A) to (D). The difference between the PBS and flagellin groups was statistically significant in absence of DT (two-way ANOVA, n = 4 mice, P < 0.001) and nonsignificant in DT-treated group (two-way ANOVA, n = 6 to 7 mice, P = 0.3821). (F and G) Tlr5/Nlrc4−/− mice were adoptively transferred with 4 × 106 fluorescence-activated cell–sorted DCs (purity > 98.5%) from Tlr5/Nlrc4−/− (F) or WT C57BL/6 (G) mice. Twelve hours later, the mice were studied as in (A) to (D). The difference between the PBS and flagellin groups was statistically significant in (G) (Student’s t test on days 2 to 4, n = 4 mice, P < 0.05) but not (F) (Student’s t test, n = 4 mice, P > 0.05 on all days). (H) Tlr5/Nlrc4−/− mice were adoptively transferred with 20 million magnetic bead–sorted DCs (purity > 95.0%) from WT C57BL/6 mice, and 12 hours later, the mice were studied as in (A) to (D) (Student’s t test on days 2 to 5, n = 4 mice, P < 0.05).
Next, we generated bone marrow chimeric mice using wild-type (WT) and Tlr5/Nlrc4−/− mice to determine the extent to which flagellin-mediated protection against RV infection requires recognition of flagellin by hemopoietic or nonhemopoietic cells. In contrast to our expectation that IEC recognition of flagellin would be essential, transplant of WT bone marrow to irradiated Tlr5/Nlrc4−/− mice completely restored the capacity of flagellin to prevent RV infection, whereas administration of Tlr5/Nlrc4−/− bone marrow to WT mice eliminated flagellin’s anti-RV activity (fig. S6, A and B). Subsequent studies demonstrated that TLR5-dependent protection was restricted to hemopoietic cells, whereas NLRC4 expression by either cell compartment was sufficient for flagellin-mediated protection (fig. S6, C to F). This is consistent with reports of NLRC4 expression in macrophages and IECs (15, 16). Furthermore, hemopoietic and nonhemopoietic cells produce IL-18 in response to flagellin (fig. S7).
We next examined the role of hemopoietic cell types in mediating flagellin’s antiviral effect. Although flagellin induced neutrophil recruitment to the intestine (fig. S8), neutrophils were not required for flagellin’s antiviral effect (fig. S9). Nor were natural killer (NK) cells or intestinal macrophages required (figs. S10 and S11). Dendritic cells (DCs) mediate intestinal production of antibacterial peptides in response to flagellin (17). To investigate whether these cells play a role in flagellin’s antiviral effect, we generated chimeric mice in which bone marrow–derived cells were engineered to express the diphtheria toxin receptor (DTR) under control of the CD11c promoter so that CD11c-expressing cells, primarily DCs, could be depleted by the administration of diphtheria toxin (DT) (18). Both DT-treated WT mice and untreated chimeric mice were protected against RV infection by flagellin treatment (Fig. 2E and fig. S12). However, flagellin was unable to protect DT-treated chimeric mice against RV infection (Fig. 2E). Next, we investigated whether DCs expressing TLR5 and NLRC4 are sufficient for flagellin protection against RV infection. Tlr5/Nlrc4−/− mice were intravenously administered purified DCs [CD19−/major histocompatibility complex (MHC) class II+/CD11c+/F4/80−] isolated from WT or Tlr5/Nlrc4−/− mice. Only the WT DC were capable of partial restoration of flagellin-mediated protection against RV infection, which was dependent on the number of DCs transferred (Fig. 2, F to H). Together, these results indicate that activation of TLR5/NLRC4 on DCs is necessary and sufficient for flagellin’s antiviral effect.
Interferons (IFNs) play a central role in antiviral immunity. However, flagellin’s antiviral effect was fully maintained in mice lacking type I and/or II IFN receptors or in mice lacking signal transducer and activator of transcription 1 (STAT1), which mediates IFN signaling (fig. S13, A to D). Nor did flagellin induce expression of type III IFN (fig. S13E), which is induced by RV (19), or IFN-associated gene expression (20). Thus, flagellin prevents and clears RV infection by a previously unrecognized antiviral pathway.
DC TLR5 activates the IL-12/IL-23 axis, resulting in innate lymphoid cells’ (ILCs) production of IL-17 and IL-22 (17, 21). Hence, we investigated whether ILC and this axis was involved in flagellin’s antiviral action. Mice deficient in p40, which is a component of both IL-12 and IL-23, and Rag2/Il2rg−/− mice, which lack ILCs (and mature B and T lymphocytes), were not effectively protected by flagellin treatment (Fig. 3, A and B). Flagellin protection against RV infection was not affected by neutralization of IL-17 (Fig. 3C) but was almost completely abolished by genetic or antibody-mediated blockade of IL-22 (Fig. 3, D and E). These results suggest a central role for ILC-mediated IL-22 production in mediating flagellin’s antiviral action. Accordingly, LPS does not elicit robust IL-22 production (21). Conversely, the requirement of NLRC4 suggested possible roles for inflammasome cytokines IL-1β and IL-18. Ablation of IL-1 receptor signaling modestly impaired flagellin’s protection against RV infection, whereas two different means of IL-18 ablation markedly reduced flagellin’s antiviral effect (Fig. 3, F to H). In contrast to flagellin-induced IL-22 production, which requires ILCs, and DC expression of TLR5 (fig. S7) (22), flagellin-induced IL-18 production was unimpaired in Rag2/Il2rg−/− or DC-ablated mice (fig. S7), indicating that distinct signaling events drive IL-22 and IL-18 production in distinct cell types.
Fig. 3. Flagellin-mediated protection against RV infection requires both IL-22 and IL-18.
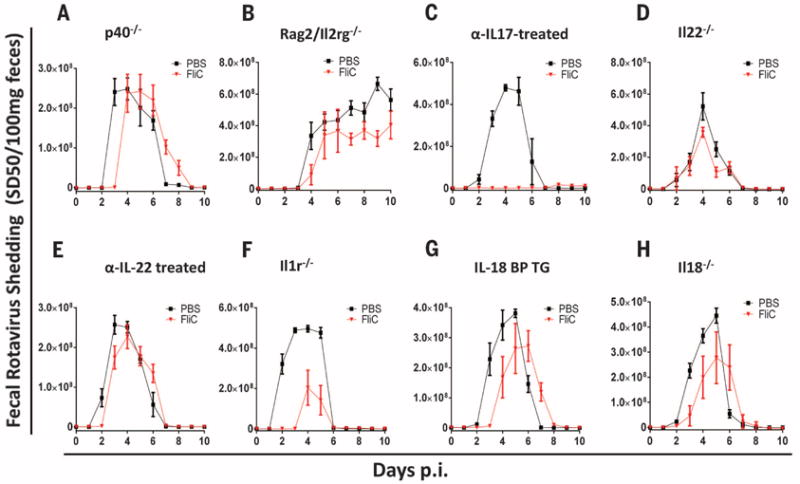
(A to H) Indicated strains of genetically modified 8-week-old mice were orally inoculated with mouse RV, EC strain. Mice were treated with PBS ± flagellin (20 μg), via intraperitoneal injection, every other day from 0 to 8 days after inoculation. Feces were collected daily and assayed for RV antigens by means of ELISA. The following strain was used in each panel: (A) p40−/−, (B) Rag2/Il2rg−/−, (C) WT C57BL/6 mice treated with IL-17–neutralizing mAb, (D) Il22−/−, (E) WT C57BL/6 mice treated with IL-22–neutralizing mAb, (F) Il1r−/−, (G) IL-18BP TG, and (H) Il18−/− mice. Results in (A) to (H) are shown as mean ± SEM (n = 4 to 6 mice). The difference between mice given PBS and flagellin was statistically significant for (C) and (F) (two-way ANOVA, P < 0.0001) and significant at individual days of (A), (B), (D), (E), (G), and (H) [Student’s t test, P < 0.05 on day 3 in (A), days 7 and 9 of (B), day 5 of (D), day 2 of (E), days 3 and 4 in (G), and days 3 and 4 in (H)].
We next investigated the extent to which recombinant IL-22 and IL-18 recapitulate flagellin’s antiviral action. When administered prophylactically, IL-18 or IL-22 had only partial protective efficacy, but together conferred complete protection against a broad range of RV inoculation (Fig. 4A and fig. S14). Moreover, co-administration of IL-18 and IL-22 eliminated viral shedding in chronically RV-infected Rag1−/− mice (Fig. 4B). These results did not reflect a strong dependence on one of these cytokines for driving the expression of the other in that flagellin-induced IL-18 expression is independent of TLR5 (14), whereas flagellin-induced IL-22 is largely independent of NLRC4 (fig. S15). Rather, our results suggest that parallel signaling pathways activated by IL-22 and IL-18 protect against RV infection and promote clearance of this virus, respectively. Consequently, combined treatment with IL-22 and IL-18 recapitulated the capacity of flagellin to cure RV infection in mice lacking mature T and B lymphocytes. Such combined IL-22/IL-18 treatment eliminated RV from Rag1−/− mice within 24 hours (versus 48 hours for flagellin) and, in contrast to flagellin, was effective in mice lacking ILCs (fig. S16). Like flagellin, prophylactic IL-22/IL-18 administration also afforded protection against the severe diarrhea seen in neonates (Fig. 4C). Furthermore, treating neonate mice with IL-22/IL-18 after diarrhea manifested moderately shortened its course (fig. S17). Thus, recapitulating flagellin’s antiviral effect with IL-22/IL-18 might offer broad antiviral therapeutic potential even in immune-compromised hosts.
Fig. 4. IL-22/IL-18 treatment prevents and treats RV infection.
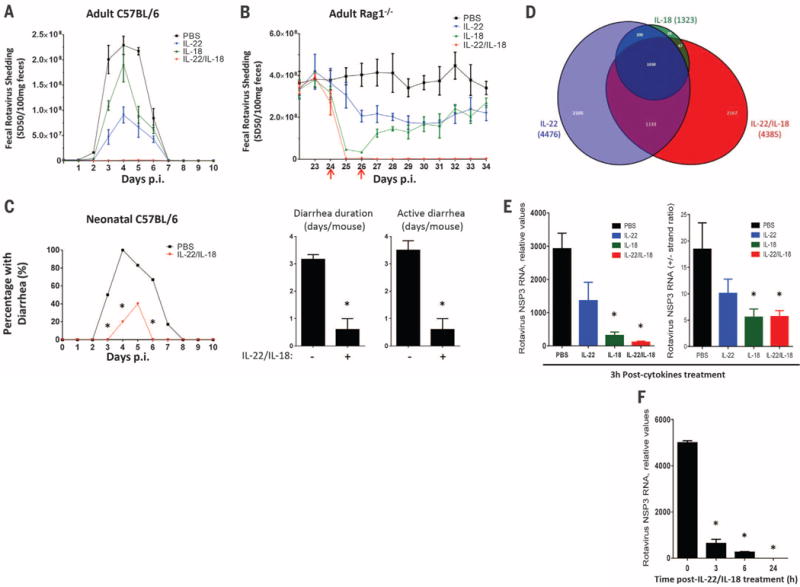
(A) Eight-week-old C57/BL6 mice were treated with 0.2 ml PBS (vehicle), 2 μg IL-22, 1 μg IL-18, or 2 μg IL-22 plus 1 μg IL-18 by means of intraperitoneal injection and subsequently inoculated with RV. Such cytokines were administered every other day from 0 to 8 days after inoculation. Feces were assayed for RV antigens by means of ELISA. Results are shown as mean ± SEM. Difference between PBS and IL-22/IL-18 groups were statistically significant (two-way ANOVA, n = 4 mice, P < 0.0001). (B) Rag1−/− mice, chronically infected with RV, were treated with PBS, 10 μg IL-22, 1 μg IL-18, or both on days 24 and 26 after inoculation (indicated with red arrows). Difference between PBS and IL-22/IL-18 groups was statistically significant (two-way ANOVA, n = 4 mice, P < 0.0001). (C) Seven-day-old C57BL/6 mice were orally inoculated with RV. Mice were administered 50 μl PBS (vehicle) or 1 μg IL-22 plus 0.2 μg IL-18 immediately before inoculation, and 1 to 9 days after inoculation, and monitored for incidence of diarrhea daily (χ2 test, n = 5 to 6 mice, *P < 0.05), duration and active days of diarrhea (Student’s t test, n = 5 to 6 mice, *P < 0.001). (D to F) Chronically RV-infected Rag1−/− mice were treated with one injection of PBS, PBS containing 10 μg IL-22, 1 μg IL-18, or 10 μg IL-22 plus 1 μg IL-18. (D) Venn diagram representation of significant changes in intestinal epithelial gene expression 3 hours after cytokine treatment. (E) Intestinal levels of RV genomes and replication rates as reflected by NSP3 RNA levels and +/− RV strand ratios at 3 hours (Student’s t test, n = 4 mice, *P < 0.001 for RV genome, *P < 0.05 for RV RNA +/− strand ratio). (F) RV genomes levels at indicated time (Student’s t test, n = 4 mice, *P < 0.0001).
Neither flagellin nor IL-22/IL-18 altered RV infection in cultured IECs (fig. S18). Hence, to investigate mechanisms by which IL-22 and IL-18 treatment cleared RV infection, we examined signaling events in gut epithelial cells isolated from chronically RV-infected mice treated with IL-22, IL-18, or both cytokines. RNA sequencing revealed that IL-22 treatment induced rapid reprogramming of epithelial cell gene expression, implicating genes involved in a broad array of cellular processes (Fig. 4D and figs. S19 and S20). A more modest effect was observed in response to IL-18, whereas the combination of IL-22 and IL-18 induced a number of changes in gene expression not seen with either cytokine alone. In contrast, administration of IL-18 but not IL-22 to RV-infected mice resulted in rapid activation of caspase 3, suggesting involvement of apoptosis (fig. S21). Thus, IL-22 and IL-18 induced changes in gene expression and caspase 3 activation that correlated with rapid blockade of RV replication and elimination of RV genomes within 24 hours of cytokine treatment (Fig. 4, E and F). Together, these data suggest that IL-18 induces signaling events that lead to rapid reduction in RV levels, whereas IL-22 reprograms epithelial cell gene expression, resulting in resistance to RV infection.
We demonstrate that innate immunity can be harnessed to prevent and treat viral infection. Given the health burden caused by RV, including 600,000 annual deaths in children (23) and chronic infections in immune-compromised hosts, this strategy presents a therapeutic opportunity, differences between human and mouse RV strains (24) notwithstanding. Moreover, the complex antiviral action of flagellin was fully recapitulated by IL-22 and IL-18, whose use could circumvent differences in NLRC4 function between mice and humans (25). The action of these cytokines would likely be synergistic with therapies that directly target viruses and/or those that promote adaptive immunity. Thus, we propose activation of innate immunity with flagellin, or IL-22 and IL-18, as a potential strategy to combat emerging and recalcitrant viral pathogens.
Supplementary Material
Acknowledgments
We thank H. Virgin, T. Nice, A.-U. Rasheed, R. Ahmed, G. Silvestri, B. Rouse, B. Pulendran, A. Lukacher, S-M. Kang, and D. Kalman for helpful discussions. We thank D. Moore and K. Yu for technical support. This work was supported by NIH grants DK061417—and an accompanying award from the American Reinvestment and Recovery Act—AI107943, DK064730, DK56338, AI038296, and AI080656. B.C. is supported by a fellowship from the Crohn’s and Colitis Foundation of America. T.S.D. is supported by Elizabeth B. Lamb Center for Pediatric Research. Recombinant IL-22, antibodies to IL-17 and IL-22, and Il22−/− mice were provided under a materials transfer agreement by Genentech (South San Francisco, CA). Patents for use of flagellin and IL-22/IL-18 to treat or prevent viral infection have been applied for under application nos. PCT/US2011/052301 (Emory University) and 61/888/439 (Georgia State University, provisional). RNA-sequencing data are deposited in Gene Expression Omnibus with accession no. GSE62479.
Footnotes
REFERENCES AND NOTES
- 1.Greenberg HB, Estes MK. Gastroenterology. 2009;136:1939–1951. doi: 10.1053/j.gastro.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng N, Franco MA, Greenberg HB. Adv Exp Med Biol. 1997;412:233–240. doi: 10.1007/978-1-4899-1828-4_35. [DOI] [PubMed] [Google Scholar]
- 3.Zeng H, et al. J Immunol. 2003;171:3668–3674. doi: 10.4049/jimmunol.171.7.3668. [DOI] [PubMed] [Google Scholar]
- 4.Vijay-Kumar M, Gewirtz AT. Mucosal Immunol. 2009;2:197–205. doi: 10.1038/mi.2009.9. [DOI] [PubMed] [Google Scholar]
- 5.Jones RM, et al. Gut. 2011;60:648–657. doi: 10.1136/gut.2010.223891. [DOI] [PubMed] [Google Scholar]
- 6.Burdelya LG, et al. Science. 2008;320:226–230. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vijay-Kumar M, et al. J Immunol. 2008;180:8280–8285. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 8.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. J Virol. 2009;83:10322–10335. doi: 10.1128/JVI.01186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang JQ, He XS, Feng N, Greenberg HB. J Virol. 2008;82:6812–6819. doi: 10.1128/JVI.00450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ball JM, Tian P, Zeng CQ, Morris AP, Estes MK. Science. 1996;272:101–104. doi: 10.1126/science.272.5258.101. [DOI] [PubMed] [Google Scholar]
- 11.Uhnoo I, et al. J Virol. 1990;64:361–368. doi: 10.1128/jvi.64.1.361-368.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho FA, et al. Mucosal Immunol. 2012;5:288–298. doi: 10.1038/mi.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kupz A, et al. Nat Immunol. 2012;13:162–169. doi: 10.1038/ni.2195. [DOI] [PubMed] [Google Scholar]
- 14.Vijay-Kumar M, Carvalho FA, Aitken JD, Fifadara NH, Gewirtz AT. Eur J Immunol. 2010;40:3528–3534. doi: 10.1002/eji.201040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchi L, et al. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 16.Nordlander S, Pott J, Maloy KJ. Mucosal Immunol. 2014;7:775–785. doi: 10.1038/mi.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinnebrew MA, et al. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung S, et al. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pott J, et al. Proc Natl Acad Sci USA. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijay-Kumar M, et al. J Immunol. 2005;174:6322–6331. doi: 10.4049/jimmunol.174.10.6322. [DOI] [PubMed] [Google Scholar]
- 21.Van Maele L, et al. J Immunol. 2010;185:1177–1185. doi: 10.4049/jimmunol.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chassaing B, Ley RE, Gewirtz AT. Gastroenterology. 2014 doi: 10.1053/j.gastro.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parashar UD, Gibson CJ, Bresee JS, Glass RI. Emerg Infect Dis. 2006;12:304–306. doi: 10.3201/eid1202.050006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graff JW, Ettayebi K, Hardy ME. PLOS Pathog. 2009;5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, et al. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.