

Findings suggest that North American white-tailed deer commonly harbor cryptic infection with the only known New World mammalian Plasmodium.
Keywords: Malaria parasites, haemosporidians, Plasmodium, Odocoileus
Abstract
Malaria parasites of the genus Plasmodium are diverse in mammal hosts, infecting five mammalian orders in the Old World, but were long considered absent from the diverse deer family (Cervidae) and from New World mammals. There was a description of a Plasmodium parasite infecting a single splenectomized white-tailed deer (WTD; Odocoileus virginianus) in 1967 but none have been reported since, which has proven a challenge to our understanding of malaria parasite biogeography. Using both microscopy and polymerase chain reaction, we screened a large sample of native and captive ungulate species from across the United States for malaria parasites. We found a surprisingly high prevalence (up to 25%) and extremely low parasitemia of Plasmodium parasites in WTD throughout the eastern United States. We did not detect infections in the other ungulate species nor in western WTD. We also isolated the parasites from the mosquito Anopheles punctipennis. Morphologically, the parasites resemble the parasite described in 1967, Plasmodium odocoilei. Our analysis of the cytochrome b gene revealed two divergent Plasmodium clades in WTD representative of species that likely diverged 2.3 to 6 million years ago, concurrent with the arrival of the WTD ancestor into North America across Beringia. Multigene phylogenetic analysis placed these clades within the larger malaria parasite clade. We document Plasmodium parasites to be common in WTD, endemic to the New World, and as the only known malaria parasites from deer (Cervidae). These findings reshape our knowledge of the phylogeography of the malaria parasites and suggest that other mammal taxa may harbor infection by endemic and occult malaria parasites.
INTRODUCTION
Malaria parasites exploit a diverse array of vertebrate hosts (squamate reptiles, birds, and mammals) and dipteran insect vectors (mosquitoes, midges, black flies, among many others) from every terrestrial habitat and every continent except Antarctica (1). The Plasmodium species of avian hosts have been described from most bird orders and families and are geographically cosmopolitan with some species even reaching oceanic islands and displaying a worldwide distribution (2). The Plasmodium species that infect lizards are likewise broadly distributed, also occurring on oceanic islands and large isolated landmasses such as New Zealand (3). Birds are highly mobile and thus can carry parasites over the continents, whereas the distribution of Plasmodium in squamate reptiles may represent repeated lateral transfer between birds and lizards over their evolutionary history (4). In contrast, the Plasmodium species of mammals have a more limited distribution, long thought to be restricted to the Old World where Plasmodium species are known from five mammal orders, namely, Primates (lemurs, monkeys, and apes, including five nominate species in humans), Rodentia (thicket rats, porcupines, and flying squirrels), Chiroptera (bats), Dermoptera (colugos), and Artiodactyla [mouse deer (Tragulidae), antelope, and Asian water buffalo (Bovidae)] (3, 5). Surveys showed Plasmodium parasites to be absent in the diverse deer family Cervidae, which is one of the largest and most widely distributed mammalian families. In addition, no endemic Plasmodium parasite was known from any mammal in the New World. Two species of Plasmodium described from numerous species of South American monkeys are now known to be lateral transfers of two human parasites, Plasmodium vivax and Plasmodium malariae, probably since European contact (6). Thus, Plasmodium parasites have been described from diverse mammal taxa and over a large geographic range including Africa, mainland Asia, and the large islands of Southeast Asia, but not from the New World.
These perplexing patterns, the apparent absence of Plasmodium parasites from mammals in the New World and their absence from the diverse deer family, were challenged by the 1967 report of a new malaria parasite, later described in 1980 as Plasmodium odocoilei, from a single white-tailed deer (WTD) Odocoileus virginianus, from Texas, USA (7). This animal had been splenectomized in an attempt to reveal occult or hidden infections, and even then, prevalence within the blood of the new parasite was extremely low (one parasite per 30,000 red blood cells) (8). Only a single blood smear from the infection was preserved with this smear now deposited at the Natural History Museum in London, UK (9). Despite many studies of blood parasites of WTD, a common big game species in North America, over the past half century, P. odocoilei has not been observed again (8, 10, 11). Many spurious reports of Plasmodium species have entered the parasitological literature, so the WTD parasite may well have been misidentified. However, the illustrations in the species description resemble Plasmodium, including the presence of both multinucleated schizont stages and hemozoin pigment, and one author, P. C. C. Garnham, was one of the field’s eminent scholars and would have been unlikely to misidentify the parasite.
During a polymerase chain reaction (PCR)–based survey for the vectors of avian malaria parasites at the Smithsonian’s National Zoological Park (NZP), we recovered an enigmatic malaria parasite sequence. By phylogenetic analysis of the parasite’s cytochrome b gene (cytb), this sequence placed within the overall Plasmodium clade but was divergent from other sequenced Plasmodium taxa. WTD was identified as the blood meal source by analysis of the vertebrate DNA (cytb) isolated from the blood meal of one of the Plasmodium-positive mosquitoes. This finding led us to conduct a major survey of WTD and other wild native and zoo ungulate species from across the United States and to investigate the possible origins of malaria parasites in New World mammals. We report the rediscovery of Plasmodium parasites in WTD and further find that these malaria parasites, which have remained hidden for so long in North America’s most popular game species, the WTD, are common, diverse, and endemic.
RESULTS
Vector and vertebrate host sampling
A total of 1978 individual mosquitoes of 27 species were sampled at two sites, NZP, Washington, DC, and San Diego County, CA (Fig. 1 and table S1). Salivary glands were dissected out of each mosquito and screened by nested PCR for Plasmodium infection using primers that target a fragment of the cytb gene on the parasite’s mitochondrial genome (4). A phylogenetic analysis of the sequences obtained from the salivary glands of Anopheles punctipennis (2 of 35, prevalence of 5%) placed the parasite within the overall Plasmodium clade but not close to other sequenced mammalian Plasmodium taxa. All other parasite sequences retrieved from mosquito salivary glands placed within the avian Plasmodium clade. WTD was identified as the blood meal source from the blood meal of one of the Plasmodium-positive A. punctipennis. This native mosquito species occurs in more mesic habitats throughout North America, taking blood meals from large mammals (12), and historically was a vector of introduced human malaria parasites in North America (13).
Fig. 1. Sampling sites of mosquitoes and wild ungulate hosts for Plasmodium parasites.
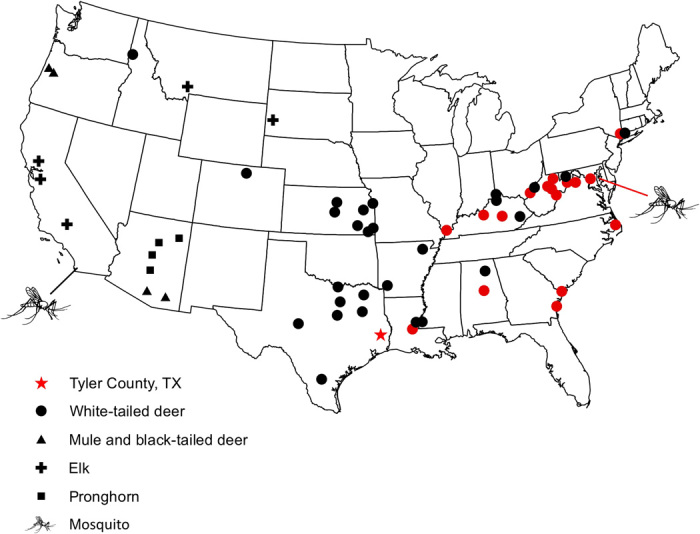
A red star denotes the original sampling location for P. odocoilei from a single, splenectomized WTD in Tyler County, TX. Sites from which positive infections were discovered are indicated in red.
We surveyed WTD across their range as well as two other native cervid species that overlap with the range of the WTD, the mule deer (Odocoileus hemionus, the closest sister taxon to WTD) and elk (Cervus canadensis). We also sampled pronghorn (Antilocapra americana), a distant artiodactylid relative of deer, from parts of their range that overlap with WTD (Fig. 1 and table S2). Because the WTD parasite could have been acquired from an exotic or domestic host, nine captive species of ungulates from NZP campuses in Washington, DC, and Front Royal, VA, were screened for Plasmodium (table S3). All host species were screened for Plasmodium infection by nested PCR of blood or tissue samples. Microscopic examination of blood smears was then carried out for a subset of the samples for confirmation of infection and visualization of parasite morphology.
Parasite screening
The nested PCR-based screening of 308 WTD from 17 states found 41 infected animals from 17 counties throughout 10 states in the eastern and southern United States (Fig. 1 and table S2). Overall prevalence at the Plasmodium-positive sites was 18% but reached approximately 25% at sites in Virginia and West Virginia. None of the other sampled native ungulate species (a total of 151 individuals of three species) or exotic or domestic ungulate species (39 individuals of nine species) were found to harbor Plasmodium infection (tables S2 and S3). A total of four animals produced double-nucleotide peaks on sequence electropherograms indicative of mixed infections (14, 15). Ambiguities were only present at one or two nucleotide positions, and each of these mixed infections could be unequivocally assigned to single parasite infections present in other infected WTD.
Examination of blood smears from infected WTD revealed parasites that were diagnosed as Plasmodium by morphology including the diagnostic characters of hemozoin pigment and asexual reproduction in blood cells, and these WTD parasites closely matched the illustrations in the original species description of P. odocoilei (8). Examination of the original type smear [loaned from the Natural History Museum, London, UK (specimen number 742 H of the Garnham collection)] found parasites with similar morphology to those from the newly sampled WTD (Fig. 2), including grossly enlarged infected red blood cells, localized pigment, spiked projections of the gametocytes, and conspicuous vacuoles. When compared to other described mammalian Plasmodium species, the morphology of the WTD parasite was most similar to Plasmodium cephalophi of the grey duiker (Sylvicapra grimmia), an African antelope (3). No blood samples had been stored from the species description, and the original blood smear could not be destructively sampled for molecular analysis.
Fig. 2. Malaria parasites of the WTD as viewed under the light microscope.
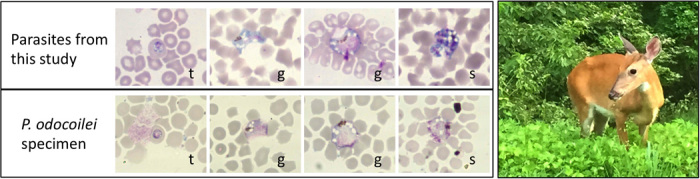
Microphotographs of parasites identified as P. odocoilei of the WTD including blood smears prepared from our study and the original type specimen (Natural History Museum, London, UK) for the species. Shown are trophozoites (t), gametocytes (g), and schizonts (s). Also shown is a picture of a WTD from one of the high-prevalence sites in the study.
Molecular analyses
To determine where the WTD parasites place within a broad phylogeny, four genes from the parasites’ three genomes were amplified and sequenced using primers specific for haemosporidian parasites: cytb and cytochrome oxidase I (coI) from the mitochondrial genome, adenylosuccinate lyase (asl) from the nuclear genome, and caseinolytic protease (clpC) from the plastid genome (4). Haemosporidians, like most parasites from the phylum Apicomplexa, carry a unique organelle, the apicoplast, with its own genome. Bayesian phylogenetic analyses placed the WTD parasites not closely related to Plasmodium of other mammal hosts but rather as sister to Polychromophilus parasites of bats from Eurasia and Africa (Fig. 3). The genus Plasmodium was recovered as polyphyletic in the resulting topology with parasites identified to this genus recovered in three separate clades: P. odocoilei and Polychromophilus, the Plasmodium species of avian and squamate hosts plus Nycteria parasites of Old World bats, and a paraphyletic clade of primate and rodent Plasmodium species plus Hepatocystis parasites of Old World bats (Fig. 3). Thus, the clade of Plasmodium parasites of WTD in North America place most closely related to malaria parasites of Old World bats.
Fig. 3. A multigene phylogeny displaying the relationships between Plasmodium lineages from WTD (clade denoted by black star) and other malaria parasites of vertebrate hosts.

Host taxa are indicated by different symbols and parasite genera by different colors, and size of triangles indicates relative number of parasite taxa contained within a clade. The phylogeny was reconstructed by partitioned Bayesian analyses of mitochondrial (cytb, coI), apicoplast (clpC), and nuclear (asl) genes and rooted with Leucocytozoon taxa. Bayesian posterior probability values are indicated above nodes.
The cytb gene is often used in studies of species-level diversity of malaria parasites, and a phylogenetic analysis of sequence data from this gene revealed a number of parasite haplotypes isolated from WTD (Fig. 4) (16). Two main clades representative of species differed by 3% of 614 base pairs (bp). Individual phylogenetic analysis of each of the other three genes individually (coI, clpC, and asl) also corroborate reproductive isolation of these two main clades and their status as distinct species. Using two independent molecular clock estimates for divergence at the mitochondrial cytb gene for malaria parasites (17, 18), we found that these two clades likely diverged between 2.3 and 6 million years ago (Ma). A recent study evaluating divergence times for mammalian Plasmodium taxa based on analysis of thousands of nuclear genes (19) confirms mitochondrial time estimates for very recent divergences, which would include those estimated for the two WTD Plasmodium species in our study. The ancestor of the WTD is estimated to have traveled across the Bering Land Bridge into North America in the Miocene 4.2 to 5.7 Ma (20). Diversity of cytb haplotypes within one Plasmodium clade (clade 2; Fig. 4) is about 0.5%, estimated with an origin at 0.4 to 1.0 Ma.
Fig. 4. Evolutionary relationships of Plasmodium parasites isolated from WTD and mosquitoes (A. punctipennis) in our study.
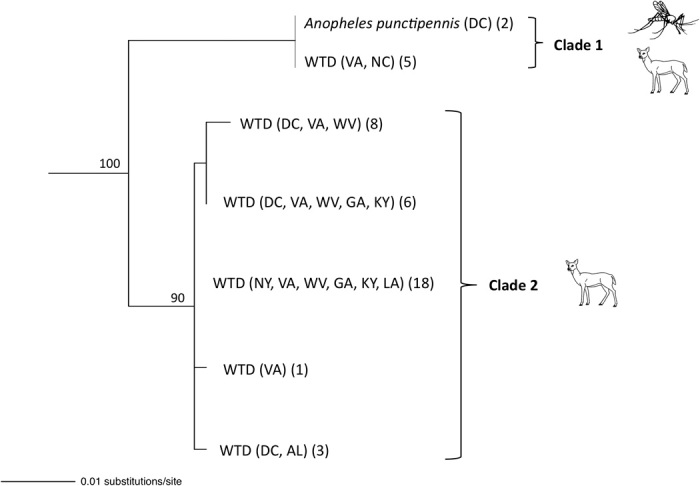
The states from which parasites were sampled and sample sizes are indicated (state abbreviations included in table S2). The phylogeny and bootstrap support values (displayed above nodes) were estimated by maximum likelihood analysis of 614 bp of the cytb gene. Clades 1 and 2 are approximately 3% divergent.
DISCUSSION
We found Plasmodium parasites that are widespread and locally common in WTD, the iconic large game animal of North America. This conclusion is supported by both microscopy of parasite morphology and phylogenetic analysis of gene sequence data. We also isolated Plasmodium DNA from the salivary glands of A. punctipennis, suggesting that this mosquito species is the vector host of the WTD parasites. The parasites clearly concord with the morphology and life history characters that diagnose the genus Plasmodium including vertebrate and dipteran hosts, asexual replication in the blood cells by schizogony, production of male and female gametocytes in the blood cells, and visible hemozoin pigment within the schizonts (meronts) and gametocytes. The parasite matches the morphology of P. odocoilei described five decades ago from a single animal but not observed since. Furthermore, phylogenetic analysis of genes from all three genomes of the parasites placed the WTD parasites within the overall Plasmodium clade. Together, our microscopy and molecular results of both vertebrate and vector hosts corroborate the rediscovery of malaria parasites from the WTD in North America. The malaria parasites, which we regard as the P. odocoilei species group, are confirmed as the only known endemic Plasmodium of a New World mammal and the only Plasmodium known from a deer.
The presence of multiple Plasmodium clades in the WTD argues for a long evolutionary history of Plasmodium parasites and their WTD hosts extending back, perhaps, to the origin of WTD in North America. A few lines of evidence support the passage of these WTD parasites over the Bering Land Bridge within the ancestor of WTD rather than host shifts into WTD from introduced ungulates such as captive animals in zoos. First, at least two likely species of Plasmodium parasites were isolated from WTD (based on their genetic divergence), and these parasites are closely related. Within one of the WTD clades (lineage 2; Fig. 4), we found additional variation (up to 0.5%) that may be indicative of substantial intraspecific variation. The application of multiple independent molecular clock estimates for the malaria parasites (17–19) supports the divergence of these parasite clades to be roughly concordant with the passage of the WTD ancestor through Beringia. In addition, we failed to detect parasite DNA in a sample of nonnative ungulate species in the United States, including domestic species and species housed in collections at zoological parks. Plasmodium parasites have been described from the Asian mouse deer (Tragulidae) and the antelope and Asian water buffalo (Bovidae) in Africa, all of which have been introduced into the New World, but it is unlikely that parasites from these species would jump numerous times into deer and thus into a new host family (Cervidae) (8). Clearly, more research including sampling of Old World ungulate hosts, both native and introduced, and genetic characterization of their Plasmodium parasites are needed to further support or refute hypotheses on the origin of malaria parasites in New World deer. If the parasites did indeed travel over the Bering Land Bridge with a herd of ancestral North American deer, it suggests that suitable temperatures existed in the high arctic for successful transmission of Plasmodium parasites by Anopheline mosquitoes. This potential scenario highlights the importance of the Bering Land Bridge and interglacial periods in the passage of vector-borne pathogens into the New World.
WTD may have carried multiple parasite lineages from their ancestor population during the migration across Beringia (20), or Pleistocene glaciations forced WTD into ice-free refugia, as in the mule deer O. hemionus (21, 22), leading to the diversification of both the WTD and parasites. WTD were nearly extirpated throughout much of the eastern and southern portions of their range due to overhunting, exploitation, and other causes, with reintroductions into these areas in the 1920s to the 1970s (23). Thus, it is likely that any biogeographic signals of the parasites would have been lost. In summary, the absence of parasites in the exotic host species we sampled and the finding of multiple closely related Plasmodium lineages within the WTD support the endemic nature of these WTD malaria parasites in North America.
Our results beg the question of why diverse malaria parasites that are specialized to WTD remained cryptic in a large game mammal that has long been the center of study for wildlife biologists and managers. Most likely, the answer is the occult nature of all of the infections observed for P. odocoilei, with extremely low parasite density (an estimated ~1/65,000 red blood cells infected). This is not unusual for the Plasmodium infecting mammals including the malaria parasites of humans. Examples include P. cephalophi and Plasmodium brucei in the common duiker (S. grimmia), an African antelope, Plasmodium traguli from the chevrotain (Tragulus javanicus) of Southeast Asia, Plasmodium inui in Asian macaque monkeys, and Plasmodium girardi in lemurs (3). Some of these species were discovered only from hosts that had been splenectomized as an experimental method to reveal low-level blood infections. Very-low-level infections, although not as extreme, are also common in many avian malaria parasites (2, 24). This life history trait, of producing few parasites in the blood of the vertebrate host, is clearly sufficient for the successful transfer to the insect host and shows that the gametes produced by the parasite are able to find what are likely rare mates in the vector blood meal. When feeding, Anopheles take approximately 3 μl of blood, and WTD blood contains approximately 8 × 106 erythrocytes/μl; a blood meal would therefore contain hundreds of parasite cells, even for such occult infections (25, 26). Together, these findings suggest that for some species of Plasmodium, low-level infections are successful in transmission.
Our findings highlight the importance of molecular methods in parasite discovery. Through the application of sensitive PCR-based screening, we detected low parasitemia infections in WTD previously undetected under the light microscope for a half century. Phylogenetic methods also allowed for the identification of multiple cryptic Plasmodium species in WTD. The discovery of Plasmodium parasites that are common and widespread in such a well-studied game species as the WTD, yet not recognized for so long, argues that other malaria parasites of mammals are yet to be discovered, perhaps in surprising host species within plain sight. The coupling of light microscopy and molecular methods and increased surveillance for mammalian Plasmodium parasites promises to further expand our knowledge of the systematic diversity and phylogeography of the malaria parasites.
MATERIALS AND METHODS
Experimental design
The main objectives of our study were to examine the distribution of the Plasmodium parasites of New World mammals including their vector and vertebrate hosts, geographic range, and possible origin in the New World. We sampled hundreds of vertebrate hosts from across the continental United States, including both wild and native ungulate species as well as nonnative and domestic ungulate species. Samples were screened by PCR, which allowed for detection of low-level infections, and additionally by microscopy for the WTD samples, which allowed for visualization of the parasites and confirmation of development in the vertebrate host. Thousands of mosquitoes were screened by PCR for malaria parasites as well. Analysis of sequence data from the parasites then allowed us to investigate the diversity of parasites, their phylogenetic relationships with other malaria parasites, and to estimate divergence dates.
Sampling of vertebrate hosts
WTD samples were obtained during necropsy of dead animals, health assessments, or other research purposes by the Smithsonian Conservation Biology Institute, the Southeastern Cooperative Wildlife Disease Study, and the American Museum of Natural History. We screened a total of 308 WTD samples from 25 counties in 17 states across the range of the species in the United States (table S2 and Fig. 1). Elk were sampled from five counties in three states, mule deer from four counties in two states, and pronghorn from four counties in one state (table S2 and Fig. 1). Mule deer, elk, and pronghorn samples were obtained from areas that overlap with the geographic range of the WTD. A total of 39 collection animals of nine species were screened from the NZP campuses in Washington, DC, and Front Royal, VA, including two antelope species and two deer species (table S3). Samples screened in this study include spleen and liver tissue and blood collected during the warm months in which biting mosquitoes are active (May through October). DNA extractions were obtained from heart and ear tissues for the Connecticut and New York samples. All sampling methods were approved by the Institutional Animal Care and Use Committee.
As part of other research studies, WTD were live caught at the Front Royal Campus of the NZP, which enabled zoo veterinary staff the opportunity to prepare blood smears from the animals in addition to blood collection for PCR screening. Blood was collected from the jugular vein into heparinized syringes, and thin blood smears were immediately prepared in the field. Blood smears were then fixed in methanol, stained with Giemsa, and examined for blood parasites under ×100 magnification.
Sampling of invertebrate hosts
Mosquitoes were sampled at the NZP in Washington, DC, from mid April through late October and from sites throughout San Diego County in California in April and May using Centers for Disease Control (CDC) miniature light traps baited with CO2 and gravid traps baited with rabbit pellet–infused water. Male mosquitoes were discarded, while blood-fed, unfed, and gravid females were separated from each other. Approximately 20% of gravid females from each mosquito species and all blood-fed individuals were then screened for malaria parasite infection by nested PCR (table S1). Mosquitoes were determined to be gravid by dissection of ovaries under the dissection scope and visual inspection of ovaries on glass microscope slides under the light microscope at 10×. All blood-fed and selected parous mosquitoes were identified to species based on morphology with further differentiation of Culex species carried out by PCR for infected individuals (27). The salivary glands of each mosquito were then carefully extracted under a dissection scope using sterile instruments, and DNA was extracted using the Qiagen BioSprint 96 System following the manufacturer’s guidelines. The abdomen from each blood-fed individual was removed using sterile instruments and transferred to a microcentrifuge tube with buffer ATL where it was agitated with a sterile plastic pestle before DNA extraction using the Qiagen BioSprint 96 System following the manufacturer’s instructions.
Parasite screening and phylogenetic analysis
DNA was extracted from blood or tissues using a Qiagen DNeasy Blood and Tissue Kit or the Qiagen BioSprint 96 System following the manufacturer’s guidelines. A 614-bp fragment of the cytb gene in malaria parasites was amplified by nested PCR using the conserved primers DW2/DW4 followed by DW1/DW3 (28). Negative controls were included for each round of PCRs, and no contamination was detected. PCR products were visualized by gel electrophoresis, purified using ExoSAP-IT (Affymetrix), and cycle-sequenced using BigDye Terminator v3.1, and the sequencing products were cleaned with Sephadex G-50 columns and then sequenced on an ABI 3130xl Sequencer. Sequences were edited and aligned using Sequencher version 5.0 (Gene Codes). Using Geneious, maximum likelihood methods were used to reconstruct a phylogeny and estimate nodal support values for the cytb data set.
For the four-gene phylogeny reconstruction, DNA from four genes across three genomes was amplified and sequenced, including cytb and coI from the mitochondrial genome, clpC from the plastid genome, and asl from the nuclear genome (4). All sequences were edited and aligned by eye using Sequencher v5.0. BEAST v1.8.0 (29) was used to generate a Bayesian tree and posterior probability values. Mitochondrial (cytb and coxI) data were partitioned into first plus second and third codon positions, where clpC and asl data were not, as determined by PartitionFinder (30). A GTR + I + G model was selected with equal nucleotide frequencies and estimated substitution rate matrix. The starting tree topology was obtained via the graphical user interface implementation of RAxML (31), and a Yule speciation process was applied. An uncorrelated relaxed clock (32) was used, and a Markov chain Monte Carlo with chain length of 50,000,000 was run. The first 25% of trees were discarded as burn-in, and the tree was summarized with TreeAnnotator (33).
Extraction products from the blooded mosquito abdomens were used to amplify a short fragment of the vertebrate cytb gene using primers SteeriF1/SteeriR1 (34). PCR products were visualized on agarose gels, cleaned using ExoSAP-IT, and sequenced as described above. Using the BLAST function in the GenBank and BOLD databases, blood meal origin was determined.
Supplementary Material
Funding
This work was supported by the Morris Animal Foundation grant D10ZO-043 (E.S.M. and R.C.F.), the National Science Foundation (NSF) DEB-1241041 (E.S.M. and R.C.F), and an NSF Postdoctoral Fellowship (E.S.M.). The collection of some samples was supported by the 18 member states of the Southeastern Cooperative Wildlife Disease Study and through a cooperative agreement with the U.S. Department of Agriculture, Animal and Plant Health Inspection Service, Veterinary Services (15-9100-1407). Author contributions: E.S.M., T.W., S.L.P., E.K.L., M.J.Y., J.J.S., and R.C.F. designed the study. E.S.M., H.B., K.F., T.W., W.J.M., T.D.F., L.W., P.H.J., E.K.L., and M.J.Y. collected samples. E.S.M., N.M., H.B., S.L.P., E.K.L., and M.J.Y. performed laboratory work. E.S.M. and S.L.P. analyzed the data. E.S.M., S.L.P., E.K.L., M.J.Y., J.J.S., and R.C.F. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors or are deposited into the GenBank database (accession numbers presented in table S4).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501486/DC1
Table S1. Mosquito species screened by nested PCR for Plasmodium infection for the study.
Table S2. County and state locations for the wild ungulate species sampled in our study and proportion found to be infected with malaria parasites at each site by nested PCR.
Table S3. Collection animals screened at the NZP’s campuses in Washington, DC, and Front Royal, VA, for malaria parasite infection by nested PCR.
Table S4. GenBank accession numbers for Plasmodium sequences generated from WTD and A. punctipennis and used in the phylogenetic analyses in this study.
REFERENCES AND NOTES
- 1.N. D. Levine, The Protozoan Phylum Apicomplexa (CRC Press, Boca Raton, FL, 1988). [Google Scholar]
- 2.G. Valkiunas, Avian Malaria Parasites and other Haemosporidia (CRC Press, Boca Raton, FL, 2005). [Google Scholar]
- 3.P. C. C. Garnham, Malaria parasites and other Haemosporidia (Blackwell Scientific Publication, Oxford, 1966). [Google Scholar]
- 4.Martinsen E. S., Perkins S. L., Schall J. J., A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): Evolution of life-history traits and host switches. Mol. Phylogenet. Evol. 47, 261–273 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Schaer J., Perkins S. L., Decher J., Leendertz F. H., Fahr J., Weber N., Matuschewskia K., High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc. Natl. Acad. Sci. U.S.A. 110, 17415–17419 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu W., Li Y., Shaw K. S., Learn G. H., Plenderleith L. J., Malenke J. A., Sundararaman S. A., Ramirez M. A., Crystal P. A., Smith A. G., Bibollet-Ruche F., Ayouba A., Locatelli S., Esteban A., Mouacha F., Guichet E., Butel C., Ahuka-Mundeke S., Inogwabini B.-I., Ndjango J.-B. N., Speede S., Sanz C. M., Morgan D. B., Gonder M. K., Kranzusch P. J., Walsh P. D., Georgiev A. V., Muller M. N., Piel A. K., Stewart F. A., Wilson M. L., Pusey A. E., Cui L., Wang Z., Färnert A., Sutherland C. J., Nolder D., Hart J. A., Hart T. B., Bertolani P., Gillis A., LeBreton M., Tafon B., Kiyang J., Djoko C. F., Schneider B. S., Wolfe N. D., Mpoudi-Ngole E., Delaporte E., Carter R., Culleton R. L., Shaw G. M., Rayner J. C., Peeters M., Hahn B. H., Sharp P. M., African origin of the malaria parasite Plasmodium vivax. Nat. Commun. 5, 3346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuttler K. L., Robinson R. M., Rogers W. P., Exacerbation of latent erythrocytic infections in deer following splenectomy. Can. J. Comp. Med. Vet. Sci. 31, 317–319 (1967). [PMC free article] [PubMed] [Google Scholar]
- 8.Garnham P. C. C., Kuttler K. L., A malaria parasite of the white-tailed deer (Odocoileus virginianus) and its relation with known species of Plasmodium in other ungulates. Proc. R. Soc. Lond. B. Biol. Sci. 206, 395–402 (1980). [DOI] [PubMed] [Google Scholar]
- 9.P. C. C. Garnham, A. J. Duggan, Catalogue of the Garnham collection of malaria parasites and other haemosporidia (Cambridge Univ. Press, Cambridge, 1986). [Google Scholar]
- 10.Davidson W. R., Crum J. M., Blue J. L., Sharp D. W., Phillips J. H., Parasites, diseases, and health status of sympatric populations of fallow deer and white-tailed deer in Kentucky. J. Wildl. Dis. 21, 153–159 (1985). [DOI] [PubMed] [Google Scholar]
- 11.Davidson W. R., Crow C. B., Crum J. M., Gerrish R. R., Observations on Theileria cervi and Trypanosoma cervi in white-tailed deer (Odocoileus virginianus) from the Southeastern United States. Proc. Helminthol. Soc. Wash. 50, 165–169 (1983). [Google Scholar]
- 12.L. O. Howard, H. G. Dyar, F. Khab, The Mosquitoes of North and Central America and the West Indies (Carnegie Institution of Washington, Washington, DC, 1912). [Google Scholar]
- 13.King W. V., Experiments on the development of malaria parasites in three American species of Anopheles. J. Exp. Med. 23, 703–716 (1916). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valkiūnas G., Bensch S., Iezhova T. A., Križanauskienė A., Hellgren O., Bolshakov C. V., Nested cytochrome b polymerase chain reaction diagnostics underestimate mixed infections of avian blood haemosporidian parasites: Microscopy is still essential. J. Parasitol. 92, 418–422 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Valkiūnas G., Palinauskas V., Ilgūnas M., Bukauskaitė D., Dimitrov D., Bernotienė R., Zehtindjiev P., Ilieva M., Iezhova T. A., Molecular characterization of five widespread avian haemosporidian parasites (Haemosporida), with perspectives on the PCR-based detection of haemosporidians in wildlife. Parasitol. Res. 113, 2251–2263 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Bensch S., Pérez-Tris J., Waldenström J., Hellgren O., Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: Multiple cases of cryptic speciation? Evolution 58, 1617–1621 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Ricklefs R. E., Outlaw D. C., A molecular clock for malaria parasites. Science 329, 226–229 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Pacheco M. A., Battistuzzi F. U., Junge R. E., Cornejo O. E., Williams C. V., Landau I., Rabetafika L., Snounou G., Jones-Engel L., Escalante A. A., Timing the origin of human malarias: The lemur puzzle. BMC Evol. Biol. 11, 299–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva J. C., Egan A., Arze C., Spouge J. L., Harris D. G., A new method for estimating species age supports the coexistence of malaria parasites and their mammalian hosts. Mol. Biol. Evol. 32, 1354–1364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilbert C., Ropiquet A., Hassanin A., Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): Systematics, morphology, and biogeography. Mol. Phyl. Evol. 40, 101–117 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Latch E. K., Heffelfinger J. R., Fike J. A., Rhodes O. E. Jr, Species-wide phylogeography of North American mule deer (Odocoileus hemionus): Cryptic glacial refugia and postglacial recolonization. Mol. Ecol. 18, 1730–1745 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Latch E. K., Reding D. M., Heffelfinger J. R., Alcalá-Galván C. H., Rhodes O. E. Jr, Range-wide analysis of genetic structure in a widespread, highly mobile species (Odocoileus hemionus) reveals the importance of historical biogeography. Mol. Ecol. 23, 3171–3190 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Ellsworth D. L., Honeycutt R. L., Silvy N. J., Bickham J. W., Klimstra W. D., Historical biogeography and contemporary patterns of mitochondrial DNA variation in white-tailed deer from the Southeastern United States. Evolution 48, 122–136 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Martinsen E. S., Waite J. L., Schall J. J., Morphologically defined subgenera of Plasmodium from avian hosts: Test of monophyly by phylogenetic analysis of two mitochondrial genes. Parasitology 134, 483–490 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Jeffery G. M., Blood meal volume in Anopheles quadrimaculatua, A. albimanus and A. aegypti. Exp. Parasitol. 5, 371–375 (1956). [DOI] [PubMed] [Google Scholar]
- 26.Powell M. C., DelGiudice G. D., Birth, morphologic, and blood characteristics of free-ranging white-tailed deer neonates. J. Wildl. Dis. 41, 171–183 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Crabtree M. B., Savage H. M., Miller B. R., Development of a species-diagnostic polymerase chain reaction assay for the identification of Culex vectors of St. Louis Encephalitis virus based on interspecies sequence variation in ribosomal DNA spacers. Am. J. Trop. Med. Hyg. 53, 105–109 (1995). [PubMed] [Google Scholar]
- 28.Martinsen E. S., Paperna I., Schall J. J., Morphological versus molecular identification of avian Haemosporidia: An exploration of three species concepts. Parasitology 133, 279–288 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Drummond A. J., Rambaut A., BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214–222 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanfear R., Calcott B., Ho S. Y. W., Guindon S., PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Silvestro D., Michalak I., raxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012). [Google Scholar]
- 32.Drummond A. J., Ho S. Y. W., Phillips M. J., Rambaut A., Relaxed phylogenetics and dating with confidence. PLOS Biol. 4, e88 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.A. Rambaut, A. J. Drummond, TreeAnnotator (2007); available at http://beast.bio.ed.ac.uk/TreeAnnotator.
- 34.den Tex R.-J., Thorington R., Maldonado J. E., Leonard J. A., Speciation dynamics in the SE Asian tropics: Putting a time perspective on the phylogeny and biogeography of Sundaland tree squirrels, Sundasciurus. Mol. Phylogenet. Evol. 55, 711–720 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501486/DC1
Table S1. Mosquito species screened by nested PCR for Plasmodium infection for the study.
Table S2. County and state locations for the wild ungulate species sampled in our study and proportion found to be infected with malaria parasites at each site by nested PCR.
Table S3. Collection animals screened at the NZP’s campuses in Washington, DC, and Front Royal, VA, for malaria parasite infection by nested PCR.
Table S4. GenBank accession numbers for Plasmodium sequences generated from WTD and A. punctipennis and used in the phylogenetic analyses in this study.