

Abstract
In this chapter, we introduce interaction networks by describing how they are generated, where they are stored, and how they are shared. We focus on publicly available interaction networks and describe a simple way of utilizing these resources. As a case study, we used Cytoscape, an open source and easy-to-use network visualization and analysis tool to first gather and visualize a small network. We have analyzed this network’s topological features and have looked at functional enrichment of the network nodes by integrating the gene ontology database. The methods described are applicable to larger networks that can be collected from various resources.
Keywords: Interaction networks, Protein–protein interactions, Gene ontology, Cytoscape, Pathways, Network
1. Introduction
A gene interaction network is a set of genes (nodes) connected by edges representing functional relationships among these genes. These edges are named interactions, since the two given genes are thought to have either a physical interaction through their gene products, e.g., proteins, or one of the genes alters or affects the activity of other gene of interest.
The functional products of genes, e.g., proteins, work together to achieve a particular task, and they often physically associate with each other to function or to form a more complex structure. These interactions can be long lasting, such as while forming protein complexes, or brief, when proteins modify each other such as the phosphorylation of a target protein by a protein kinase. Since these interactions are important to carry out most biological processes, knowledge about interacting proteins is crucial for understanding these biological functions, which can be easily done via studying networks of these interactions.
Besides these physical interactions, there are genetic interactions, in which two gene variants have a combined effect that does not manifest itself with only one of them alone. These genetic interactions are also measured at high throughput. There are two general categories of such interactions: synthetic lethal interactions and suppressor interactions. Synthetic lethal interactions are caused when two nonessential genes combine to form an overall lethal effect, and suppressor interactions occur when a lethal variant of one gene is “negated” by that of another gene. These types of interactions are essential in understanding pathways and regulation in model organisms (1–3), as well as providing insight into complex diseases (4).
In recent years, high-throughput methodologies, such as the yeast two-hybrid (Y2H) (5–7), co-immunoprecipitation followed by mass spectrometry (8, 9), or tandem affinity purification (TAP) (10), have been widely used to identify physical protein–protein interactions for a wide range of organisms. In addition, genetic interactions were mapped for humans in high-throughput drug screens (11) and for many organisms (12).
As a result, in the past decade, the number of known protein–protein interactions has increased significantly, and various public databases were created to share these findings. There are also computational approaches that are used to predict protein–protein interactions. These utilize genomic data to establish a structural or evolutionary link among protein pairs (13, 14) or predict novel interactions by analyzing known interactions (15–17).
Almost all of the interactions that are discovered through experiments are collected in public databases. While fairly new, these databases constantly grow in size and present protein–protein interaction data for multiple organisms. Initially, these databases independently collected their datasets. However, now through the International Molecular Exchange Consortium, these databases keep a nonredundant set of protein–protein interaction data from a broad taxonomic range of organisms. Moreover, these databases commit to providing these datasets in standard file formats, such as MITAB or PSI-MI XML 2.5. Currently, the databases listed in Table 1 are actively producing relevant numbers of records curated to these standards and provide these via the Proteomics Standard Initiative Common Query Interface (PSICQUIC) service.
Table 1.
The International Molecular Exchange Consortium Partner Databases: Public databases that provide nonredundant set of protein–protein interactions for a range of organisms are listed (as of January 2011)
| Database | Web site | Number of PPI |
|---|---|---|
| DIP (39) | http://dip.doe-mbi.ucla.edu | 107,619 |
| IntAct (33) | http://www.ebi.ac.uk/intact | 272,410 |
| MINT (40) | http://mint.bio.uniroma2.it/mint | 90,537 |
| MPact (23) | http://mips.gsf.de/genre/proj/mpact | 15,454 |
| MatrixDB (41) | http://matrixdb.ibcp.fr | 845 |
| MPIDB (42) | http://www.jcvi.org/mpidb | 24,295 |
| BioGRID (37) | http://www.thebiogrid.org | 365,574 |
| InnateDB (43) | http://www.innatedb.com | 9,909 |
| BIND (44) | http://www.blueprint.org | 192,961 |
These databases maintain interactions that can be all encompassing, such as IntAct, MINT, and DIP, organism centric, such as BioGrid or MPIDB, or biological domain centric, such as MatrixDB. However, regardless of the database, these interactions are available in standards-compliant, tab-delimited, and XML formats. Currently, these databases carry some redundancy. However, as the data collection pipelines for each database is established and with the implementation of an internal data management system, access to interaction datasets will be more robust.
Visualization and analysis of these interaction networks is also essential for researchers. In recent years, a variety of software for various platforms has been introduced, such as Cytoscape (18), Osprey (19), Pajek (20), etc. These pieces of software can visualize the networks by employing graph layout algorithms and displaying data attributes as well as visual mappings (e.g., protein images, coloring). Moreover, via filters and plug-ins, they analyze these networks and assist in integration of external data sources such as gene ontology (21, 22).
In this chapter, we will build an interaction network using one of these databases as a resource. We will visualize the network and analyze it using an open source (and free), platform-independent network browser Cytoscape (18). We will first acquire a publicly available interaction dataset. We will then visualize this network, analyze its network properties, and, using available plug-ins, check its functional enrichment. In the near future, we will have access to more centralized systems for maintaining and storing these datasets, so this approach can be easily extended to multiple datasets, extending its use for biomedical research.
2. Methods
IntAct, a network database maintained by EMBL-EBI, hosts almost 300,000 binary interactions shared among more than 50,000 proteins (23). In this chapter, to establish an easier example to follow, a small dataset from IntAct will be acquired and analyzed. The same methodology can be extended to larger datasets as needed.
2.1. Acquiring Datasets
The IntAct database of protein–protein interactions can be accessed freely through the IntAct Web site (http://www.ebi.ac.uk/intact). The whole database can be downloaded in PSI-MI XML or PSI-MI TAB format or can be queried for a list of proteins via a Web interface. Databases mentioned in Table 1 provide similar services. IntAct also provides analysis tools with documentation. In this chapter, a more general approach, applicable to a wider number of datasets, is described.
All interactions in IntAct are derived from literature curation or direct user submissions. IntAct provides these datasets in smaller chunks as well. For a list of highlighted datasets, please visit IntAct’s Dataset of the month archive (24). In this chapter, as an example, we are going to download and analyze the interactions submitted in support of a study that targeted the phosphatidylinositol 3-kinase-mammalian (PI3K-mTOR) pathway (25). As shown in Fig. 1, the dataset can be accessed in multiple formats. The basic plain text version (PSI-MI Tab) will be used for simplicity.
Fig. 1.

An IntAct database of the month archive entry is shown. Like most databases, IntAct provides the data shared in multiple formats. The images in the figure are links to the same dataset in different formats. Internal link to IntAct table page with external links (left); PSI-MI XML (versions 1.0 and 2.5) (middle); and PSI-MI Tab (right).
After downloading the dataset, simply open the dataset in a text editor or spreadsheet editor. The tab-delimited file is organized as a table, where each line describes an interaction (an extended edge list). Each record contains many fields that describe the interaction. For visualization purposes, the gene name field is preferred, since most genes/proteins are annotated with these names. To be able to load this file into Cytoscape, using the Find–Replace function of the editor, remove “uniprotkb:” and “(gene name)” texts from the records. Essentially, an edge list with two columns that have gene names is created. Similar datasets can be generated. Although there are means to import the xml dataset into Cytoscape (see Note 1), the example will focus on the flat file for applicability to other databases or sources.
2.2. Visualizing Interaction Networks
Cytoscape is a platform-independent application (http://www.cytoscape.org) (18). The open source software supports many standard network and annotation file formats, including Simple Interaction Format (SIF), XML-based BioPAX, PSI-MI, SBML, etc. The software can also load delimited text files and MS Excel™ Workbooks. In this chapter, loading text files is described to ensure the applicability of this methodology for various data sources.
Moreover, Cytoscape can also import data files, such as expression profiles, GO annotations generated by other applications or spreadsheet programs, or images for nodes. Using these features, you can load and save arbitrary attributes on nodes, edges, and networks. For instance, you can input a set of custom annotation terms for a protein (26), create a set of confidence values for protein–protein interactions, and filter them later in the software (27).
Cytoscape can establish powerful visual mappings across systems biology, genomics, and proteomics data. It supports advanced analysis and modeling via specific plug-ins that can be installed with one click. Also, visualization and analysis of human-curated pathway datasets such as Reactome or KEGG is possible.
After launching the software, make sure that the plug-in required for this chapter, Bingo, is loaded. If not installed, Bingo can be installed through the Plug-in Manager under the Plug-ins menu (search for Bingo and click install the latest supported version).
The network prepared in Subheading 2.1 can be loaded by clicking Import > Network From Table (Text/MS Excel)… under the File menu. The dialog box shown in Fig. 2 will ask for columns to be picked. As the file is altered, the gene names will correspond to columns 3 and 4. First, click on the Show Text File Import Options for more options. Since the first line is headers, ignore the first line by incrementing the Start Import Row to 2. Next, select column 3 and column 4 for source interaction and target interaction drop-down lists. This should highlight the preview as shown in the dialog box (Fig. 2). The network should load after the import button is clicked. The user can manipulate the network by selecting and dragging the nodes or alter the layout using the Layout menu. For further details of the available layouts and options, please refer to the software documentation. In this example, the network is shown in yFiles > Organic layout found under the Layout menu.
Fig. 2.
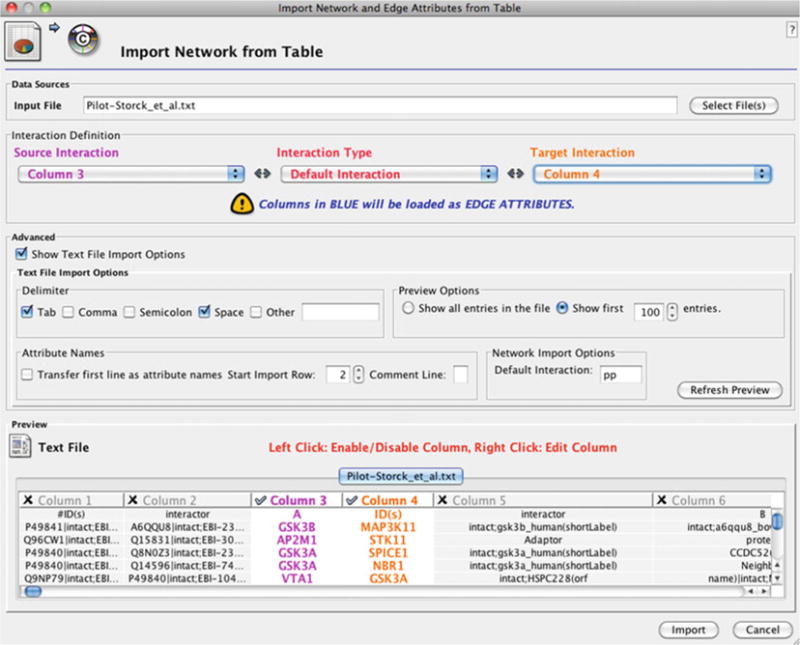
Screen shot of Cytoscape’s import network from table (Text/MS Excel)… dialog box. As input, a flat file is selected, and appropriate columns are marked to import the text file as an edge list into the software.
2.3. Analyzing Interaction Networks
In order to gain more insight about the network, a number of network analyses can be performed. Network Topology gives an overview of network topological features, including diameter, degree distribution, shortest path distribution, and clustering coefficient of the interaction network. A path in a protein–protein interaction network is defined as a list of nodes where each node has an edge to the next node. The shortest path is the shortest path from one node to another in the network. The diameter of the network is defined as the maximum value of distance of the shortest path over all pairs of distinct nodes in a graph. These values are mostly used to see how connected a network is.
The degree distribution measures the proportion of nodes in a graph with a specified number of edges. This distribution is assumed to follow a power law degree distribution, i.e., the degree of the nodes varies as a power of the number of nodes on the network, and has sparked interest on how this distribution has a role in robustness of a system represented as an interaction network (28–30). The clustering coefficient is another measure that describes how well-connected neighbors of the node are, ranging from one to zero. The Network Analysis plug-in displayed under the Plug-ins menu of Cytoscape has various functions that can be utilized for these analyses.
In Fig. 3, we selected the whole network and selected Analyze Network, treating it as an undirected graph. The plug-in displays various simple-to-calculate features of the network, such as the degree distribution (Subheading 2.2), number of connected components, etc. When the Visualize Parameters button is clicked, for instance by selecting the node degree from the drop list named Map node size to, this information can be reflected onto the network for a more visual representation. The next tab on this analysis window is the node degree distribution, where a power law line can be fitted (as seen in Fig. 3) on the degree distribution. Other properties and functions can be accessed by clicking through the tabs on this window, and detailed documentation can be accessed by hitting Help.
Fig. 3.
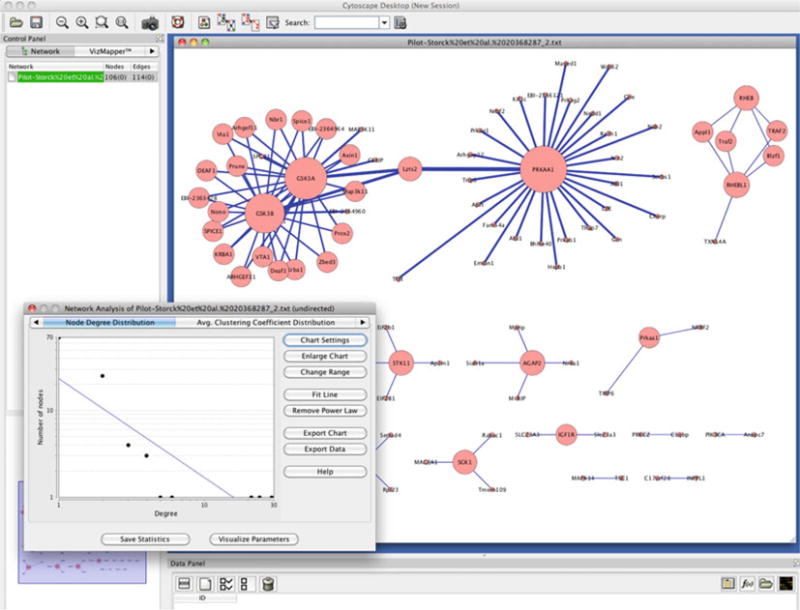
Cytoscape (18) and network analysis plug-in is shown. The plot on the left bottom shows that the degree distribution of the analyzed network likely follows a power law. The nodes in the networks are sized by their node degree.
2.4. Functional Enrichment Analysis
We further analyze the network via querying the network genes for overrepresentation of gene ontology annotations. The network we established is the result of a study that targeted the PI3K-mTOR pathway. This pathway plays pivotal roles in cell survival, growth, and proliferation downstream of growth factors (25). Moreover, its perturbations have been associated with cancer progression, type 2 diabetes, and neurological disorders. In Fig. 4, we show how one of the plug-ins, Bingo (22), can be used to investigate this network. We first selected the biggest component of the interaction network we have established (Fig. 3). The 56 selected nodes are then passed onto the Bingo plug-in by launching the plug-in from the Plug-ins menu. Before running the plug-in selecting, adjust the input form, as described below:
Give a name to your new analysis in the Cluster name text box.
Leave the Get Cluster from Network box checked.
Select your default species name by selecting the organism of the network from the Select organism/annotation drop-down list. Select Homo sapiens for this example. If, in the future, you require a different species, you will need a Gene Association file for that species. You can download one from The Gene Ontology Project (21) and load it as a custom file.
Select the statistical test and corrections that are required. In this example, we used Hypergeometric test. Binomial testing is preferred when the amount of data is very large.
Select the statistical correction that will be used. You can choose Benjamini and Hochberg False Discovery Rate (FDR). This is the correction to use in most cases, as the Bonferroni correction is too conservative. Bonferroni correction can be used when you want to demonstrate that your results are significant without doubt.
Choose a significance level. You can leave the default 0.05. This threshold controls which nodes are detailed.
Under select the categories to be visualized, choose the overrepresented categories after correction so that they are visualized.
Select the ontology file that will be used for the analysis. The three main categories, Biological Process, Cellular Component, and Molecular Function, and a combined list are available. In this example, we will use the GO_Biological_Process file.
Note that if you want to save these settings, you can hit the Save settings as default button at this stage.
Start BingoK:\P26335\JSEN\16\00\1033251\final-proof1\jsen-nikolic-2529685.
A smaller window named Parsing Annotation… should appear, showing progress, listing the number of entities (proteins) and classifications (assignments of protein to term).
Fig. 4.
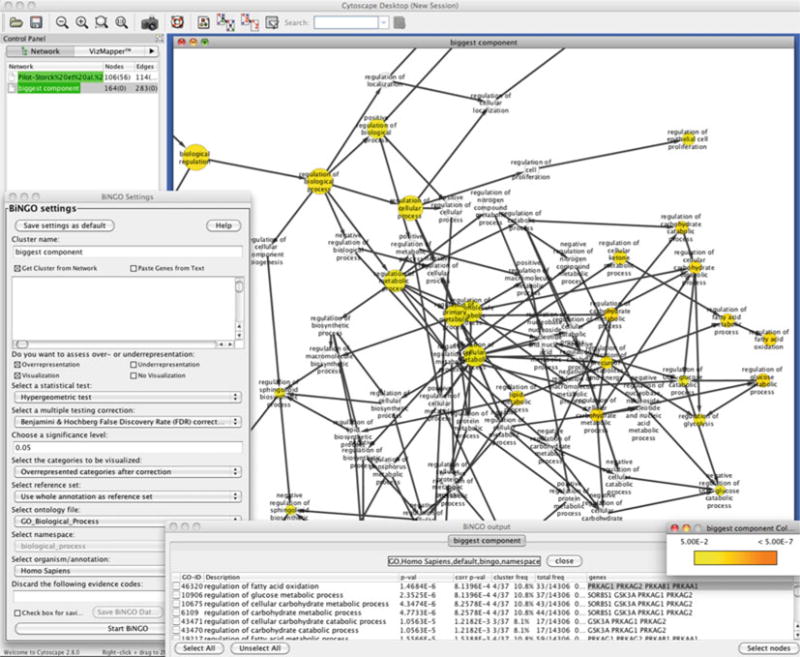
Enrichment analysis of the biggest component of the network analyzed (Fig. 3) is shown. A section of the gene ontology term graph is shown in the background. The Bingo plug-in settings and the output are shown in the foreground.
The plug-in then generates an acyclic graph of gene ontology (GO) terms and is labeled accordingly. The network shows the terms that are actually mostly associated (overrepresented) among the network nodes (note the gradient from white to yellow). Since the GO graph is quite large, branches of the GO with no significant terms would not appear in this network. As shown in Fig. 4, terms under biological regulation, such as fatty acid metabolic processes or regulation of cell proliferation, have been highlighted, which is relevant to previous associations of the PI3K-mTOR pathway (see Note 2 for other sources to use).
Cytoscape has a node/edge/network attribute browser named Data Panel that is displayed below the networks. Select a node from the GO network and browse the Node Attribute field. Some of the fields populated under the node attributes include
description_test: the name of the GO biological process
adjustedPValue_test: the P-value for the node, adjusted for multiple hypothesis testing (for comparison, the un-adjusted P-value is also there, with the name pValue_test)
n_test: the number of network nodes in your selection set
x_test: the number of nodes in your selection set mapping to the term
Moreover, the Bingo plug-in will produce an output window listing the P-values of all nodes with significant enrichment (Fig. 4, bottom). You can select these terms, and the Select nodes button will highlight the nodes in the network that are associated with those terms. This window also links the GO terms to Amigo (31), the gene ontology browser.
Footnotes
As data integration and analysis can become challenging, datasets may not always be grabbed and used as easily. For those who are not as computer savvy, the data collection and visualization described can be facilitated through additional plug-ins.
For instance, Cytoscape can also work as a web service client. This means Cytoscape can directly connect to external public databases and import network and annotation data. Currently, Pathway Commons (32), IntAct (33), BioMart (34), NCBI Entrez Gene (35), and The Protein Identifier Mapping Service (PICR) (36) are supported through Cytoscape. As more standards are established and accepted throughout the research community, the number of databases accessible likewise should increase. Moreover, databases such as the BioGRID (37) developed their own plug-in for Cytoscape, BiogridPlugin2, to import interaction data sets into Cytoscape (note that BioGRID is a Prospective IMEx consortium member).
As mentioned earlier, if the additional plug-ins to analyze the networks do not work, there are always additional web services that can accomplish similar tasks. Although utilizing these will make life harder, similar results should be generated as most of these resources use similar techniques.
There are other Cytoscape plug-ins, such as Pingo (45) that can identify significantly associated user-defined target Gene Ontology terms. Also, there are services independent of Cytoscape (e.g., FuncAssociate (38)) that are capable of providing functions similar to Bingo. Researchers can consider using these tools for verification and extended analysis of their networks.
References
- 1.Avery L, Wasserman S. Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 1992;8:312–316. doi: 10.1016/0168-9525(92)90263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guarente L. Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet. 1993;9:362–366. doi: 10.1016/0168-9525(93)90042-g. [DOI] [PubMed] [Google Scholar]
- 3.Sham P. Shifting paradigms in gene-mapping methodology for complex traits. Pharmacogenomics. 2001;2:195–202. doi: 10.1517/14622416.2.3.195. [DOI] [PubMed] [Google Scholar]
- 4.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 5.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 6.Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 7.Ito T, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman JL, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291:1001–1004. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- 9.Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 10.Rigaut G, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 11.Huang LS, Sternberg PW. Genetic dissection of developmental pathways. Worm-Book. 2006:1–19. doi: 10.1895/wormbook.1.88.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;30:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 13.Goh CS, Cohen FE. Co-evolutionary analysis reveals insights into protein-protein interactions. J Mol Biol. 2002;324:177–192. doi: 10.1016/s0022-2836(02)01038-0. [DOI] [PubMed] [Google Scholar]
- 14.Overbeek R, et al. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bebek G, Yang J. PathFinder: mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinformatics. 2007;8:335. doi: 10.1186/1471-2105-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng SK, Zhang Z, Tan SH. Integrative approach for computationally inferring protein domain interactions. Bioinformatics. 2003;19:923–929. doi: 10.1093/bioinformatics/btg118. [DOI] [PubMed] [Google Scholar]
- 17.Aloy P, et al. Structure-based assembly of protein complexes in yeast. Science. 2004;303:2026–2029. doi: 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- 18.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol. 2003;4:R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batagelj V, Mrvar A. Pajek – Program for Large Network Analysis. Connections. 1998;21:47–57. [Google Scholar]
- 21.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 23.Guldener U, et al. MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res. 2006;34:D436–441. doi: 10.1093/nar/gkj003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.IntAct. Datasets of the month Archive. 2011 http://www.ebi.ac.uk/intact/pages/dotm/dotm_archive.xhtml.
- 25.Pilot-Storck F, et al. Interactome mapping of the phosphatidylinositol 3-kinase-mammalian target of rapamycin pathway identifies deformed epidermal autoregulatory factor-1 as a new glycogen synthase kinase-3 interactor. Mol Cell Proteomics. 2010;9:1578–1593. doi: 10.1074/mcp.M900568-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smoot ME, et al. Cytoscape 2.8: New Features for Data Integration and Network Visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linderman G, Chance M, Bebek G. Magnet: Micro Array Gene Expression Network Evaluation Toolkit. 2011 doi: 10.1093/nar/gks526. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 29.Jeong H, et al. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 30.Bebek G, et al. The degree distribution of the generalized duplication model. Theoretical Computer Science. 2006;369:239–249. [Google Scholar]
- 31.Carbon S, et al. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerami EG, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aranda B, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haider S, et al. BioMart Central Portal–unified access to biological data. Nucleic Acids Res. 2009;37:W23–27. doi: 10.1093/nar/gkp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maglott D, et al. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cote RG, et al. The Protein Identifier Cross-Referencing (PICR) service: reconciling protein identifiers across multiple source databases. BMC Bioinformatics. 2007;8:401. doi: 10.1186/1471-2105-8-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stark C, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berriz GF, et al. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–2504. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- 39.Salwinski L, et al. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ceol A, et al. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chautard E, et al. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2011;39:D235–240. doi: 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goll J, et al. MPIDB: the microbial protein interaction database. Bioinformatics. 2008;24:1743–1744. doi: 10.1093/bioinformatics/btn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynn DJ, et al. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4:218. doi: 10.1038/msb.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Isserlin R, El-Badrawi RA, Bader GD. The Biomolecular Interaction Network Database in PSI-MI 2.5. Database (Oxford) 2011. 2011:baq037. doi: 10.1093/database/baq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smoot M, Ono K, Ideker T, Maere S. PiNGO: a Cytoscape plugin to find candidate genes in biological networks. Bioinformatics. 2011;27:1030–1031. doi: 10.1093/bioinformatics/btr045. [DOI] [PMC free article] [PubMed] [Google Scholar]