

Abstract
There is emerging need in new medical products that can mitigate and/or treat the short- and long-term consequences of radiation exposure after a radiological or nuclear terroristic event. The direct effects of ionizing radiation are realized primarily via apoptotic death pathways in rapidly proliferating cells within the initial 1–2 days after the exposure. However later in the course of the radiation disease necrotic cell death may ensue via direct and indirect pathways from increased generation of pro-inflammatory cytokines. Here we evaluated radiomitigative potential of Necrostatin-1 after total body irradiation (TBI) and the contribution of necroptosis to cell death induced by radiation. Circulating TNFα levels were increased starting on d1 after TBI and associated with increased plasmalemma permeability in ileum of irradiated mice. Necrostatin-1 given iv. 48h after 9.5 Gy TBI attenuated radiation-induced receptor interacting protein kinase 3 (RIPK3) serine phosphorylation in ileum and improved survival vs. vehicle. Utilizing apoptosis resistant cytochrome c−/− cells, we showed that radiation can induce necroptosis, which is attenuated by RNAi knock down of RIPK1 and RIPK3 or by treatment with necrostatin-1 or -1s whereas 1-methyl-L-tryptophan, an indoleamine-2,3-dioxygenase inhibitor, did not exhibit radiomitigative effect. This suggests that the beneficial effect of necrostatin-1 is likely through inhibition of RIPK1-mediated necroptotic pathway. Overall, our data indicate that necroptosis, a form of programmed necrosis, may play a significant role in cell death contributing to radiation disease and mortality. This study provides a proof of principle that necrostatin–1 and perhaps other RIPK1 inhibitors are promising therapeutic agents for radiomitigation after TBI.
Keywords: Mitigation, propidium iodide, IL-1α, IL-6, IL-17, 7-Cl-O-Nec-1
1. Introduction
The threat of a nuclear and radiological attack or accident has grown in recent years due to increased activity of global terrorist organizations and a rise in illicit trafficking of radioactive materials. There is emerging need in new medical products that can mitigate and/or treat the short- and long-term consequences of radiation exposure after a radiological/nuclear terrorist event. Ionizing radiation causes radiolysis of water with production of free radicals [1–6]. The immediate burst of radicals upon exposure to irradiation is followed by a dose-dependent and continuous mitochondrial overproduction of reactive oxygen species [7, 8]. The direct effects of ionizing radiation and water radiolysis products are realized primarily via apoptotic cell death pathways in rapidly proliferating hematopoietic cells and gastrointestinal epithelial cells within the initial 1–2 days after the exposure [9] [10, 11]. However later in the course of the radiation disease necrotic cell death may ensue via direct and indirect pathways from increased generation of pro-inflammatory cytokines and translocation of bacteria from intestine to blood stream, which are known consequences of total body irradiation [12]. Thus the therapeutic window of opportunity for inhibitors of necrotic death pathways will likely be 24h or longer after irradiation.
Although previously considered to be an accidental cell death in response to physicochemical insults, recent studies suggest that necrosis is a tightly regulated cell death pathway which includes mitochondrial dysfunction, enhanced reactive oxygen species production, lipid peroxidation, and early plasma membrane rupture [13]. The understanding of a specific cellular program for regulated necrosis was initiated by the discovery that the receptor interacting protein kinase 1 (RIPK1), a serine-threonine kinase, is required for the necrosis of Jurkat human T lymphocyte cells triggered by the death receptor Fas (CD95) [14] and the necrosis of mouse embryonic fibroblasts (MEFs) induced by TNFα [15]. Since then many necroptosis triggers such as interferon γ, LPS-mediated activation of TLR4, genotoxic stress have been described [16]. RIPK1 inhibitors, namely necrostatins, were later developed and proved to be useful in studies of RIPK1-dependent necroptosis [17]. A RIPK family member, RIPK3, was later identified as an essential mediator of necroptosis [18, 19]. Interaction of RIPK3 and RIPK1 via their RIP homotypic interaction motif is required for necroptosis induction [20].
Plasma membrane permeabilization and release of intracellular components are key morphological features of necroptosis [13]. Recent studies suggest that activation of RIPK3 leads to the phosphorylation of mixed lineage kinase domain like (MLKL) protein [21]. Oligomerization and binding of mixed lineage kinase domain like (MLKL) with phosphatidylinositols was shown to allow recruitment of MLKL to the plasma membrane leading to the permeabilization of the latter [22]. Typically necroptosis is induced upon inhibition of caspases but it can occur in the absence of caspase inhibition as seen in TNF-induced systemic inflammatory response syndrome, or during infection [23, 24]. In this study, we evaluated radiomitigative potential of Necrostatin-1 after TBI and the contribution of necroptosis to cell death induced by ionizing radiation. Circulating TNFα levels were increased after TBI and associated with increased plasmalemma permeability in ileum of irradiated mice. Necrostatin-1 given 48h after TBI attenuated irradiation-induced RIPK3 serine phosphorylation in ileum and improved survival vs. vehicle. Furthermore utilizing apoptosis resistant cytochrome c−/− cells, we show that radiation can directly induce necroptosis, which is attenuated by RNAi knock down of RIPK1 and RIPK3 or by treatment with necrostatin-1 or necrostatin-1s.
2. Materials and methods
2.1. Materials
The Annexin V and CytoTox-ONE assays were purchased from Biovision (Mountain View, CA) and Promega (Madison, WI), respectively. Antibodies used in the experiments were follows: RIPK1 from BD Pharmingen (San Diego, CA), RIPK3 from ProSci (Poway, CA), phosphoserine antibody from Abcam (Cambridge, MA). Unless indicated, all other reagents including were from Sigma (St. Louis, MO). Necrostatin-1s (5-((7-Cl-1H-indol-3-yl)methyl)-3- methylimidazolidine-2,4-dione) or 7-Cl-O-Nec-1) was obtained from BioVision (Milpitas, CA)
2.2. Animals
All procedures were pre-approved and performed according to the protocols established by the Institutional Animal Care and Use Committee of the University of Pittsburgh. C57BL/6NTac female mice were anaesthetized with Nembutal (1 mg per 20 gm mouse) and irradiated using a Shepherd Mark 1 Model 68 cesium irradiator at a dose rate of 80 cGy min−1. For analysis of plasma cytokines by multiplex mice were euthanized at either 24 or 48 h after 9.25 Gy TBI (n = 3/time point/group). Mice were euthanized with carbon dioxide inhalation, and blood samples were obtained by cardiac puncture. Multiplex was performed as described previously [25]. For assessment of the radiomitigative effect of necrostatin-1 after TBI, mice (n = 15/group) were irradiated to a total body dose of 9.25 Gy or 9.5 Gy. Mice were divided into separate groups and administered vehicle or Necrostatin-1 (1.65 mg/kg in 10% cremephor el/10% ethanol) via intravascular (i.v.) route at various time points: 1h before, at 24h, at 48h, or at 72h after TBI. The mice were followed for the development of haematopoietic syndrome at which time they were euthanized. For evaluation of RIPK3 phosphorylation, mice were euthanized at 7 d after 9.5 Gy TBI (n = 3/time point). Animals were then transcardially perfused with 5 ml phosphate buffered saline (PBS). Ileum was harvested from each mouse for protein extraction.
2.3. Administration of propidium iodide and detection of propidium iodide and TUNEL positive cells
Propidium iodide (PI; 10 mg/mL; Sigma) was diluted in 0.9% NaCl and 0.2 mg/kg was administered 2 h before killing by intraperitoneal injection in a total volume of not more than 100 μL. TBI mice were killed at 2 d after 9.25 Gy. The Ilea from control and TBI mice were fixed in 2% paraformaldehyde for 1 hour, cryoprotected in 30% sucrose in PBS overnight and then shock frozen in liquid nitrogen cooled isopentane. 7 micron cryostat section were cut and mounted on superfrost slides (Fisher). TUNEL (Terminal deoxynucleotidyl transferase mediated dUPT nick end labeling) staining was conducted as described [26]. In brief, TUNEL staining was conducted with the ApopTag Peroxidase Kit or ApopTag Fluorescein In Situ Apoptosis Detection Kit (Cat# S7101 or S7110, Chemicon International, Temecula, CA). Sections were then counterstained with Dapi and were mounted directly in Gelvatol and coverslipped. Imaging was with a Nikon A1 confocal microscope with a 20x 0.75 NA objective. Three channels were imaged using 405nm (Dapi) 488nm (TUNEL) and 561nm (PI) lasers. Imaging of each channel was sequential to avoid channel bleed through. To derive an overview of labelling a motorized scanning stage was use to collect several images which were stitched using NIS Elements.
2.4. Cells
Mouse embryonic Cyt c−/− cells (ATCC, Manassas, VA) and Cyt c+/+ cells (courtesy of X. Wang, University of Texas, Dallas) were cultured in DMEM supplemented with 15% FBS, 25 mM Hepes, 50 mg/L uridine, 110 mg/L pyruvate, 2mM glutamine, 1× nonessential amino acids, 50 μM β-mercaptoethanol, 100 U/L penicillin, and 100 mg/L streptomycin in a humidified atmosphere of 5% CO2, 95% air at 37°C. In order to induce ferroptosis Cyt c−/− cells were exposed to L-buthionine sulphoximine (BSO) for 48h [27].
2.5. Exposure of cells to γ-Irradiation
Cells were cultured in fresh medium prior to irradiation and were γ-irradiated with a Shepherd model 143-45A irradiator (J.L. Shepherd & Associates, CA) at indicated dose. Cells were then incubated in the presence or absence of necrostatin-1, necrostatin-1s, ferrostatin-1, zVAD-fmk alone or in combination with necrostatin-1 in complete culture medium at 37°C in 5% CO2 incubator until harvest.
2.6. Measurements of phosphatidylserine externalization, propidium iodide staining and caspase 3/7 activity
Externalization of phosphatidylserine was analyzed by flow cytometry using Annexin V kit (Biovision). In brief, harvested cells were stained with Annexin V-fluorescein isothiocyanate and propidium iodide 5 min in the dark before flow cytometry analysis. Cell debris as represented by distinct low forward and side scatter were gated out for analysis. Ten thousand events were collected on a FACScan flow cytometry (BD Biosciences, Rutherford, NJ) supplied with CellQuest software. Caspase–3/7 activity was measured using a luminescence Caspase–GloTM 3/7 assay kit (Promega) according to the manufacturer’s instructions.
2.7. Lactate dehydrogenase (LDH) assay
LDH release was assessed using CytoTox-ONE kit (Promega) according to manufacturer’s instructions. The enzyme activity from the culture medium and floating cells were used as an index of cell death. The adherent cells were lysed in 0.9% Triton X-100 and assayed for LDH activity. This cell-associated LDH activity was then added to the LDH activity in the culture medium. The amount of LDH present in the medium was calculated as a percentage of the total, which was then used to estimate the percentage of cell death in the corresponding samples.
2.8. Western blotting and immunoprecipitation
Samples were separated on 10% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with antibodies against RIPK1 (BD Pharmingen, 1:2000), RIPK3 (ProSci, 1:500) or actin (loading control, Sigma, 1:5000) followed by horseradish peroxidase-coupled detection. For immunoprecipitation, samples from mouse intestine were homogenized in RIPA buffer supplemented with 1× protease and 1× phosphatase inhibitor cocktails (Sigma and Roche). After pre-clearing with IP matrix (Santa Cruz), lysates were incubated with phosphoserine antibody (Abcam, 1:200) overnight. The resulting immune complexes were washed and resolved on 10% Tris/glycine gels (Invitrogen). Membranes were probed with anti-RIPK3 antibody (ProSci, 1:500) for RIPK3 phosphorylation.
2.9. Statistical Analysis
Data are expressed as mean ± SEM or mean ± S.D. as indicated in figure legends. Changes in variables for different assays were analyzed by a two-tailed Student’s t test for comparisons. Differences were considered significant at p < 0.05. The log-rank test was used for analysis of overall survival that is defined as the time from the date of radiation to the 35 days post radiation between treated vs. the radiation-only control group. Hematopoietic syndrome occurs within the first 30 days post radiation.
3. Results
Previous studies showed that TNFα-induced lethal systemic inflammatory response syndrome in mice is mediated via RIPK1 mediated necroptosis. Furthermore, RIPK1 inhibitor Necrostatin-1 given i.v prior to TNFα administration sensitized C57B6 mice to TNFα-induced hypothermia and shock. To determine optimal timing of Necrostatin-1 administration, we first evaluated plasma levels of TNFα after TBI in mice. TNFα levels were increased starting on day 1 after TBI (Fig. 1a) followed by increases in proinflammatory cytokines IL-1α, IL-6 and IL-17 (Fig. 1b–d), which were shown to mediate TNF-induced systemic inflammatory response syndrome and septic shock in mice [28] [29] [30]. Increases in pro-inflammatory mediators were temporally associated with increased plasmalemma permeability and a small amount of TUNEL staining in ileum of irradiated mice suggesting activation of non-apoptotic cell death pathways after TBI (Fig. 1e).
Figure 1.

Plasma TNFα (a), IL-1α (b), IL-6 (c) and (d) IL-17 levels were increased after 9.25 Gy total body irradiation (TBI). Day (d) 0 is prior to irradiation. Mean ± SEM, *p < 0.05 vs. d0. (e) Plasma membrane permeability to propidium iodide (PI) and TUNEL positivity were observed in mouse ileum 2 days after 9.25 Gy TBI. Large area 3D confocal microscopy at high resolution was performed in TBI (A–D, A1) and control (E, E1) mice. PI was given i.p. (0.2 mg/mouse) and mice were sacrificed 2h after injection. Panel A and E are superposed maximum intensity images of nuclei (blue), TUNEL (green) and PI (red). Panel B shows nuclei alone, panel C TUNEL alone (insert: higher magnification of TUNEL positive cells indicated by the arrow, scale bar = 100 μm) and panel D PI alone. Panel A1 and E1 show an enlargement of the inset area in A and E, respectively. TUNEL or PI positivity was not apparent in control.
In order to evaluate whether necroptosis contributes to mortality after TBI we utilized a potent inhibitor of necroptosis, necrostatin-1. Groups of 15 mice received vehicle or one dose of necrostatin-1 i.v. 1 h before or at 24 h, 48 h, or 72 h after 9.25 Gy TBI or 48 h after 9.5 Gy TBI. Necrostatin-1 given 24, 48 or 72 h after TBI was effective in improving survival compared to vehicle (Fig. 2a and b). However pre-treatment with Necrostatin-1 did not improve survival vs. vehicle (Fig. 2b) indicating that Necrostatin-1 mitigates TBI-induced disease process but it does not function as a radioprotector at the dose/time used in these experiments. Previous studies showed that RIPK1 plays an important role in RIPK3phosphorylation, which is required for necroptosis induction [31]. We found that necrostatin-1 attenuated TBI-induced RIPK3 serine phosphorylation in ileum (Fig. 2c), indicating involvement of RIPK1 and RIPK3 activation after TBI.
Figure 2.

Radiation mitigation by necrostatin-1 (Nec-1). (a) C57BL/6 mice injected with 1.65 mg/kg Nec-1 i.v. at 48 h after 9.5 Gy (triangles) had increased survival vs. vehicle (circles) (p = 0. 04 n = 15/group). (b) Nec-1 (1.65 mg/kg) given i.v. 1h before (triangles) 9.25 Gy irradiation did not improve survival vs. vehicle (closed circles). Whereas mice that received Nec-1 (1.65 mg/kg) i.v. at 24h (inverted triangles), at 48h (diamonds) or at 72h (open circles) after irradiation had increased survival vs. vehicle (closed circles) (p = 0.02 for 24h; p = 0. 04 for 48h; p = 0.02 for 72h, n = 15 mice/group). (c) Effect of Nec-1 on RIPK3 phosphorylation induced by irradiation in mouse intestine. Nec-1 attenuated irradiation-induced phosphorylation of RIPK3. Inset: Proteins immunoprecipited with anti-phosphoserine (p-Ser) antibody were probed with RIPK3 antibody. Mean ± SD, *p < 0.01 vs. irradiation (IR).
To further evaluate the role of RIPK1 and RIPK3 in radiation-induced necroptosis, we developed an in vitro model of radiation-induced necroptosis in cytochrome c (cyt c) deficient cells. Activation of caspase 3/7 (~10-fold vs. control, Fig 3a) and externalization of phosphatidylserine (Fig. 3b) were observed in Cyt c+/+ cells exposed to irradiation. On the other hand, Cyt c−/− cells showed attenuated caspase 3/7 activity (~2-fold vs. control), increased PI positivity (~6-fold vs. control) and LDH release (~4-fold vs. control) (Fig. 3a, c, d) upon radiation exposure indicating a switch from apoptotic to necrotic death pathway with permeabilization of plasma membrane. Necrostatin-1 as well as 7-Cl-O-Nec-1 (Necrostatin-1s) decreased irradiation-induced LDH release in a dose dependent manner in Cyt c−/− cells (Fig. 4a). Notably, Necrostatin-1 did not attenuate cell death in MECs (Fig. 5), which are known to undergo primarily apoptotic death upon irradiation [32], although pan-caspase inhibitor zVAD-fmk was effective. Combined ZVAD + necrostatin-1 treatment as effective as ZVAD alone in attenuating irradiation-induced PS externalization (Fig. 5).
Figure 3.

Induction of non-apoptotic cell death by irradiation. (a) Measurement of caspase activity and (b) Flow cytometry analysis of cell death at 48h after 10 Gy irradiation in cytochrome c normal (Cyt c+/+) and deficient (Cyt c−/−) cells. (c) Assessment of PI positivity and (d) LDH release at 48h after 15 Gy irradiation in Cyt c−/− cells. Mean SD, * p < 0.01 vs. Cyt c+/+ IR, #p < 0.05 vs. control.
Figure 4.

Induction of necroptotic death by irradiation (IR) in Cyt c−/− cells. (a) Radiomitigative effect necrostatin-1 (Nec-1) and necrostatin-1s (Nec-1s) in Cyt c−/− cells. (b) Effect of 1-Methyl-L-tryptophan (LMT) in irradiated Cyt c−/− cells. (c) RNAi knock down of RIPK1 and RIPK3 in Cyt c−/− cells. (d) RIPK1 and RIPK3 RNAi attenuated irradiation-induced cell death. Cells were γ-irradiated at a dose of 15 Gy, and Nec-1, Nec-1s or LMT was added to cells 30 min after irradiation exposure for 48 h incubation. Mean ± SD. #p < 0.05 vs. IR, *p < 0.01 vs. IR.
Figure 5.
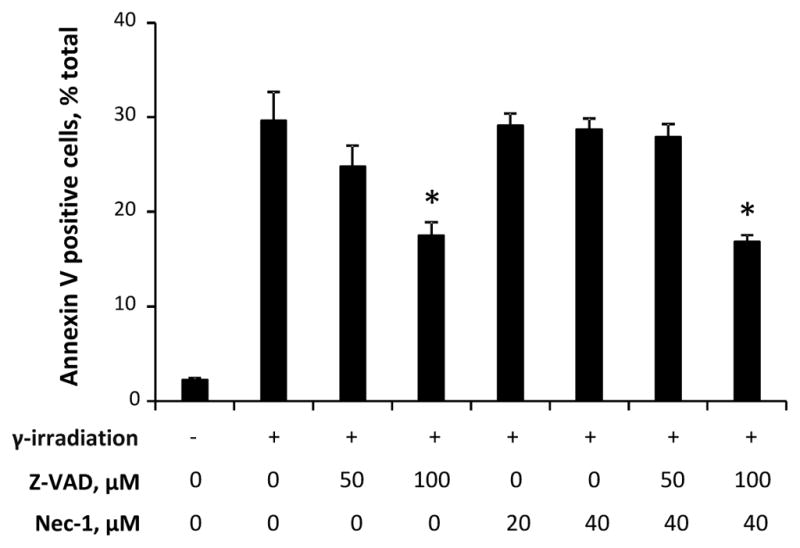
Radiomitigative effect of ZVAD and necrostatin-1 in mouse embryonic cells (MECs). ZVAD but not necrostatin-1 attenuated irradiation induced PS externalization assessed at 48h after 10 Gy irradiation (IR) in MECs. Combined ZVAD + necrostatin-1 treatment was as effective as ZVAD alone. Mean ± SD. #p < 0.05 vs. IR,
Recent data suggest that necrostatin-1 may have pleiotropic effects [33–35] and it can also inhibit indoleamine-2,3-dioxygenase (IDO) [36], which is induced by TNFα [12]. Thus we tested the effect of specific inhibitor of IDO, 1-methyl-L-tryptophan, in cyt c deficient cells. 1-methyl-L-tryptophan did not exhibit any radiomitigative effect (Fig. 4b) suggesting that the radiomitigative effect of necrostatin-1 is likely through inhibition of RIPK1-mediated necroptotic pathway. To further evaluate the role of RIPK1 and RIPK3 in radiation-induced death, we utilized RNAi approach to knock down expression of RIPK1 and RIPK3 (Fig. 4c). Modest but statistically significant decreases in radiation-induced death were observed in Cyt c−/− RIPK1 and RIPK3 knock down cells (Fig. 4d). Combined together these results indicate that necroptosis is activated after irradiation and targeted inhibition of this cell death pathway leads to successful late radiomitigation.
4. Discussion
This study suggests a possible role for necroptosis in the pathogenesis of radiation injury. We show that radiation can lead to direct induction of RIPK1-RIPK3-dependent necroptosis in cytochrome c deficient cells that cannot undergo apoptosis and necrostatin-1 can rescue mice from lethal radiation. Necrostatin-1 has been widely used to evaluate the role of necroptosis since its discovery using an in vitro cell death assay in U937 cells exposed to TNFα and ZVAD to induce necroptosis (Degterev et al, 2005). Recent studies, however suggest that Necrostatin-1 can block apoptosis, ferroptosis and inhibit IDO under certain conditions [27, 36, 37]. Alternative strategies to evaluate the role of RIPK1 and necroptosis include the use of Necrostatin-1s (or 7-Cl-O-NEC), with exclusive selectivity towards RIPK1 in a screen of a >400 kinases, including several other RIP family members [33]. Our data in Cyt c−/− cells showed that both Necrostatin-1s and RNAi knock-down of RIPK1 attenuated radiation-induced cell death.
There are no reported studies evaluating the role of IDO after TBI. IDO can be induced by TNFα, which is increased in plasma after TBI [12]. In models of systemic inflammatory response, inhibition of IDO pre-insult is deleterious, however, post-insult is beneficial [33, 38]. Our studies suggest that specific IDO inhibitor, 1-methyl-D,L-tryptophan (1-MT) does not exert radiomitigative effect in Cyt c−/− cells. Ferroptosis is a recently discovered form of nonapoptotic oxidative cell death triggered by glutathione peroxidase 4 (Gpx4) deficiency [27, 39]. Necrostatin-1 has been shown to attenuate death of Gpx4 deficient cells suggesting that it might have anti-ferroptotic effects [27]. Ferroptosis is inhibited by the small molecule compounds named ferrostatins [39]. Ferrostatins were reported to potentiate protective effects of necrostatin-1 in renal tubular cell death after ischemia reperfusion [40]. Our experiments using Ferrostatin-1, a potent inhibitor of ferroptosis [39], showed that it was not effective in ameliorating irradiation-induced death of Cyt c−/− cells (Fig. S1a). We reasoned that Cyt c−/− cells might be resistant to ferroptosis, thus we exposed them to BSO that leads to ferroptotic death by Gpx4 inhibition through depletion of glutathione [27]. Cyt c−/− cells were sensitive to BSO-induced death and Ferrostatin-1 was successful in attenuating it (Fig. S1b). Combined these data suggest that the radiomitigative effects of Necrostatin-1 are likely primarily mediated through inhibition of RIPK1-mediated necroptosis after TBI.
Radiation disease is a continuum of biological processes which include i) DNA damage, stress response and apoptosis within minutes to hours; ii) necrotic cell death, inflammation and defects in proliferation and replenishment of damaged or dying cells within days to weeks; and iii) fibrosing inflammation and atrophy of the damaged organs in weeks to months after TBI [41]. Previous studies identified an essential role for RIP kinase in DNA damage-induced NF-κB activation after ionizing radiation [42]. We observed that TNFα levels were higher starting on day 1 after TBI followed by increases in pro-inflammatory cytokines IL-1α and IL-6. Similar findings have been reported by us and others after TBI [43–46]. TNFα induces systemic inflammatory response syndrome and septic shock in mice [28]. Induction of proinflammatory cytokines such as IL-1 [29], IL-6 and IL-17 [30] partially mediates TNF-induced sepsis. TNFα, Fas ligand, and interferon-γ (IFN-γ) can induce regulated necrotic cell death. Loss of membrane integrity and release of intracellular content after necroptosis have the ability to further worsen inflammatory response [13]. The released immunogenic endogenous molecules are termed “damage associated molecular patterns (DAMPs) [13]. They include high-mobility group box-1 (HMGB1) protein, IL-1α, mitochondrial content such as cardiolipin and lipid mediators [13]. These DAMPs then stimulate pattern associated receptors including Toll-like receptors (TLR) thus triggering a vicious cycle of necroptosis → inflammation → necroptosis after TBI. Recently the concept that regulated necrosis may not only follow but also precede or amplify the inflammatory response has been gaining attention [24, 47]. Our data with Cyt c−/− cells suggest that irradiation may cause regulated necrosis independently of an outside signal thus trigger inflammation as has been shown for ischemia/reperfusion injury [48]. Establishing whether regulated necrosis is a trigger or an amplifier of inflammation in vivo after TBI is challenging since the two processes can proceed concurrently with each other making it difficult to infer causality. However experimental studies in mice lacking FAS-associated death domain protein in intestinal epithelial cells show that these mice have increased number of necrotic epithelial cell death and develop a spontaneous inflammatory bowel disease. RIPK3 deficiency prevents epithelial cell death and inflammation in the intestine supporting a proinflammatory function for necroptosis in vivo [49]. Identifying critical steps in this pathway can give hints for the mechanistic and timely strategies for radiomitigation.
Previous studies have shown induction of necroptosis upon gamma irradiation and protection against necroptosis by necrostatin-1 in tumor cells suggesting that activation of necroptosis may improve radiosensitivity. [50]. In the only other published study evaluating the radiomitigative potential of RIPK inhibitors after TBI in normal tissues, C57BL/6 male mice were exposed to a single dose 15 Gy of X-ray that resulted in death by day 4 after irradiation associated with activation of apoptosis, necrosis and autophagy in ileum [51]. Among the three compounds evaluated: autophagy inhibitor 3-methyladenine, caspase inhibitor zVAD-fmk, and necrostatin-1, only necrostatin-1 improved survival by 50% vs. irradiated control when given i.p. at a dose of 33 μg/mouse at 24h after irradiation. This beneficial effect was accompanied by a reduction in irradiation-induced nuclear to cytoplasmic translocation of HMGB1 in ileal epithelium. At higher doses (66 and 132 μg/mouse) however the radiomitigative effect of necrostatin-1 was abolished despite complete inhibition of HMGB1 translocation suggesting that both apoptotic and IDO-mediated effects might have played a role in the benefit observed. It is possible that combined therapies targeting multiple cell death pathways might be more effective than either one alone.
We chose to use 1.65 mg/kg IV dosing for Nec-1 based on previous literature showing benefit after cardiac ischemia-reperfusion when necrostatin-1 is administered at the time of reperfusion via i.v. route [52]. Necrostatin-1 at low doses (0.6 mg/kg i.v.) was shown to sensitize female C57BL/6 mice to TNF-induced systemic inflammatory response that included hypothermia and death if given prior to TNF administration [33]. Similar findings were reported with i.p. administration of necrostatin-1 prior to sepsis induced by cecal ligation and puncture and cerulean-induced pancreatitis [53, 54]. This effect is thought to be due to inhibition of IDO by Necrostatin-1. Higher doses of Necrostatin-1, however, improved survival in TNF-induced shock. We observed that female C57BL/6 mice injected with higher dose Necrostatin-1 (1.65 mg/kg in cremphor el/ethanol) i.v. at 48 h after 9.5 Gy irradiation had increased survival.
In conclusion, the data suggest that necroptosis, a form of programmed necrosis, may play a significant role in cell death contributing to radiation injury and mortality. This study provides a proof of principle that necrostatin-1 and perhaps other RIPK1 inhibitors are promising therapeutic agents for radiomitigation after TBI.
Highlights.
Radiation disease generates pro-inflammatory mediators
Delayed stages of radiation disease include non-apoptotic cell death mechanisms
Necrostatin-1 acts as effective radiomitigator but not as radioprotector
Acknowledgments
Supported, in part, by grants from the NIH (U19AIO68021, NS061817, NS076511)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hall E. Radiation Biology for Radiologist. 4. Philadelphia, PA, USA: J.B. Lippincott, Inc; 1999. [Google Scholar]
- 2.Kang SK, et al. Overexpression of extracellular superoxide dismutase protects mice from radiation-induced lung injury. Int J Radiat Oncol Biol Phys. 2003;57(4):1056–66. doi: 10.1016/s0360-3016(03)01369-5. [DOI] [PubMed] [Google Scholar]
- 3.Neal R, et al. Antioxidant role of N-acetyl cysteine isomers following high dose irradiation. Free Radic Biol Med. 2003;34(6):689–95. doi: 10.1016/s0891-5849(02)01372-2. [DOI] [PubMed] [Google Scholar]
- 4.Anand AJ, et al. Radiation-induced red cell damage: role of reactive oxygen species. Transfusion. 1997;37(2):160–5. doi: 10.1046/j.1537-2995.1997.37297203518.x. [DOI] [PubMed] [Google Scholar]
- 5.Strom E, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2(9):474–9. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- 6.Epperly MW, et al. Modulation of radiation-induced cytokine elevation associated with esophagitis and esophageal stricture by manganese superoxide dismutase-plasmid/liposome (SOD2-PL) gene therapy. Radiat Res. 2001;155(1 Pt 1):2–14. doi: 10.1667/0033-7587(2001)155[0002:morice]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 7.Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med. 2013;65:607–19. doi: 10.1016/j.freeradbiomed.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Coelho D, et al. Caspase-3-like activity determines the type of cell death following ionizing radiation in MOLT-4 human leukaemia cells. Br J Cancer. 2000;83(5):642–9. doi: 10.1054/bjoc.2000.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams JP, McBride WH. After the bomb drops: a new look at radiation-induced multiple organ dysfunction syndrome (MODS) Int J Radiat Biol. 2011;87(8):851–68. doi: 10.3109/09553002.2011.560996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potten CS, Booth C. The role of radiation-induced and spontaneous apoptosis in the homeostasis of the gastrointestinal epithelium: a brief review. Comp Biochem Physiol B Biochem Mol Biol. 1997;118(3):473–8. doi: 10.1016/s0305-0491(97)00219-8. [DOI] [PubMed] [Google Scholar]
- 11.Tyurina YY, et al. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44(3):299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Fukumoto R, et al. Ciprofloxacin modulates cytokine/chemokine profile in serum, improves bone marrow repopulation, and limits apoptosis and autophagy in ileum after whole body ionizing irradiation combined with skin-wound trauma. PLoS One. 2013;8(3):e58389. doi: 10.1371/journal.pone.0058389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 15.Lin Y, et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279(11):10822–8. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 16.Vanden Berghe T, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 17.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 18.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 19.Zhang DW, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 20.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–97. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 22.Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 23.Duprez L, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 25.Shein SL, et al. Hemorrhagic shock shifts the serum cytokine profile from pro- to anti-inflammatory after experimental traumatic brain injury in mice. J Neurotrauma. 2014;31(16):1386–95. doi: 10.1089/neu.2013.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci Rep. 2015;5:8566. doi: 10.1038/srep08566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tracey KJ, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234(4775):470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 29.Everaerdt B, Brouckaert P, Fiers W. Recombinant IL-1 receptor antagonist protects against TNF-induced lethality in mice. J Immunol. 1994;152(10):5041–9. [PubMed] [Google Scholar]
- 30.Takahashi N, et al. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205(8):1755–61. doi: 10.1084/jem.20080588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu XN, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21(11):1709–20. doi: 10.1038/cdd.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang J, et al. Designing inhibitors of cytochrome c/cardiolipin peroxidase complexes: mitochondria-targeted imidazole-substituted fatty acids. Free Radic Biol Med. 2014;71:221–30. doi: 10.1016/j.freeradbiomed.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi N, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20(2):366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandenabeele P, et al. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013;20(2):185–7. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller AJ, et al. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–9. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 38.Jung ID, et al. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J Immunol. 2009;182(5):3146–54. doi: 10.4049/jimmunol.0803104. [DOI] [PubMed] [Google Scholar]
- 39.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linkermann A, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836–41. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okunieff P, et al. Molecular markers of radiation-related normal tissue toxicity. Cancer Metastasis Rev. 2008;27(3):363–74. doi: 10.1007/s10555-008-9138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hur GM, et al. The death domain kinase RIP has an essential role in DNA damage-induced NF-kappa B activation. Genes Dev. 2003;17(7):873–82. doi: 10.1101/gad.1062403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Epperly MW, et al. Manganese [correction of Magnesium] superoxide dismutase (MnSOD) plasmid/liposome pulmonary radioprotective gene therapy: modulation of irradiation-induced mRNA for IL-I, TNF-alpha, and TGF-beta correlates with delay of organizing alveolitis/fibrosis. Biol Blood Marrow Transplant. 1999;5(4):204–14. doi: 10.1053/bbmt.1999.v5.pm10465100. [DOI] [PubMed] [Google Scholar]
- 44.Girinsky TA, et al. Peripheral blood corticotropin-releasing factor, adrenocorticotropic hormone and cytokine (interleukin beta, interleukin 6, tumor necrosis factor alpha) levels after high- and low-dose total-body irradiation in humans. Radiat Res. 1994;139(3):360–3. [PubMed] [Google Scholar]
- 45.Linard C, et al. Acute induction of inflammatory cytokine expression after gamma-irradiation in the rat: effect of an NF-kappaB inhibitor. Int J Radiat Oncol Biol Phys. 2004;58(2):427–34. doi: 10.1016/j.ijrobp.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 46.Zhang M, et al. Response patterns of cytokines/chemokines in two murine strains after irradiation. Cytokine. 2012;58(2):169–77. doi: 10.1016/j.cyto.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 47.Linkermann A, et al. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759–67. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 48.Linkermann A, et al. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13(11):2797–804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- 49.Welz PS, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–4. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 50.Nehs MA, et al. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery. 2011;150(6):1032–9. doi: 10.1016/j.surg.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 51.Chen Q, et al. Apoptosis, necrosis, and autophagy in mouse intestinal damage after 15- Gy whole body irradiation. Cell Biochem Funct. 2014;32(8):647–56. doi: 10.1002/cbf.3068. [DOI] [PubMed] [Google Scholar]
- 52.Lim SY, et al. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21(6):467–9. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linkermann A, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol Med. 2012;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNeal SI, et al. The dual functions of receptor interacting protein 1 in fas-induced hepatocyte death during sepsis. Shock. 2011;35(5):499–505. doi: 10.1097/SHK.0b013e31820b2db1. [DOI] [PMC free article] [PubMed] [Google Scholar]