

Abstract
Recently, we showed that rivastigmine decreased amyloid-β (Aβ) brain load in aged rats by enhancing its clearance across the blood-brain barrier (BBB) via upregulation of P-glycoprotein (P-gp) and low-density lipoprotein receptor-related protein 1 (LRP1). Here, we extend our previous work to clarify P-gp role in mediating rivastigmine effect on Aβ brain levels and neuroprotection in a mouse model of Alzheimer’s disease (AD) that expresses different levels of P-gp. APPSWE mice were bred with mdr1a/b knockout mice to produce littermates that were divided into three groups; APP+/mdr1+/+, APP+/mdr1+/− and APP+/mdr1−/−. Animals received rivastigmine treatment (0.3 mg/kg/day) or vehicle for 8 weeks using Alzet osmotic mini-pumps. ELISA analysis of brain homogenates for Aβ showed rivastigmine treatment to significantly decrease Aβ brain load in APP+/mdr1+/+ by 25% and in APP+/mdr1+/− mice by 21% compared to their vehicle treated littermates, but not in APP+/mdr1−/− mice. In addition, rivastigmine reduced GFAP immunostaining of astrocytes by 50% and IL-1β brain level by 43% in APP+/mdr1+/+ mice, however its effect was less pronounced in P-gp knockout mice. Moreover, rivastigmine demonstrated a P-gp expression dependent neuroprotective effect that was highest in APP+/mdr1+/+>APP+/mdr1+/−>APP+/mdr1−/− as determined by expression of synaptic markers PSD-95 and SNAP-25 using Western blot analysis. Collectively, our results suggest that P-gp plays important role in mediating rivastigmine non-cholinergic beneficial effects, including Aβ brain load reduction, neuroprotective and anti-inflammatory effects in the AD mouse models.
Keywords: Rivastigmine, P-glycoprotein, Alzheimer’s disease, amyloid-beta, neuroprotection, astrogliosis
1. Introduction
Rivastigmine (Exelon®, Novartis) is a dual acetyl- and butyryl cholinesterase inhibitor used to alleviate symptoms of Alzheimer’s disease (AD). AD is a progressive neurodegenerative disorder mainly characterized by accumulation of amyloid-β (Aβ) peptides in brain parenchyma and neurofibrilary tangles of hyperphosphorylated tau protein [1, 2]. In addition, many neuropathological changes in AD are reported including synaptic failure, astrogliosis at sites of Aβ deposition, disturbance of glutamate excitotoxicity, and blood-brain barrier (BBB) dysfunction [3].
According to the cholinergic hypothesis of AD, loss of cognitive function is largely attributed to depletion of cholinergic neurotransmitters such as acetylcholine (ACh) mainly in the cerebral cortex and caudate nucleus [4, 5]. Rivastigmine is designed to correct loss of presynaptic cholinergic function by inhibiting cholinesterases and thus decreases the degradation of acetylcholine in synaptic cleft and allow for increased cholinergic signaling which improves cognition and memory [6]. However, previous preclinical in vitro and in vivo studies reported that rivastigmine could provide neuroprotective and disease-modifying effects through multiple mechanisms that are not related to its cholinergic effect [7–9]. In one study, rivastigmine has shown to induce presynaptic proteins, preserve neuronal viability and regulate APP processing and Aβ production in vitro [10–12]. Recently, we have demonstrated a novel mechanism for rivastigmine to reduce Aβ brain load by enhancing its clearance through induction of Aβ major transport proteins, low-density lipoprotein receptor related protein-1 (LRP1) and P-glycoprotein (P-gp), at the BBB and liver [13]. These findings suggest that rivastigmine therapeutic effect is exerted through multiple mechanisms that interfere with Aβ pathogenesis and not limited to cholinesterase inhibition. In fact, some studies indicated that cholinesterase inhibition is not enough to alter the course of AD, and clinical efficacy seen with some cholinesterase inhibitors are attributed to other mechanisms that are yet to be identified [14, 15].
The amyloid cascade hypothesis suggests that Aβ is the primary player in the development of AD, and failure of Aβ clearance is a major event that contributes to Aβ brain accumulation [16–19]. Human studies suggest that Aβ accumulation in the brain correlates inversely with the degree of P-gp expression at the BBB [20]. In addition, aging, a high risk factor for AD, is associated with a decline in the expression of P-gp that is correlated with the accumulation of Aβ in the brains of aged non-demented subjects and AD patients [21, 22]. This observation was also reported in rats, where P-gp and LRP1 expressions were decreased with aging and inversely correlated with Aβ brain deposition [23]. Therefore, proteins involved in the transport of Aβ are important in regulating Aβ brain steady state levels and their dysfunction may contribute to Aβ-mediated pathogenesis in conditions such as AD.
Following to our previous findings [13], this study aims to investigate the question “how changes in P-gp expression would affect rivastigmine therapeutic effect, including Aβ lowering effect, neuroprotection and anti-inflammatory effect using APPSW mice that carry APP transgene and P-gp WT (APP/mdr1+/+), P-gp one copy (APP/mdr1+/−), or P-gp knockout (APP/mdr1−/−) mice. Overall findings suggest that P-gp mediates the non-cholinergic effects of rivastigmine, and that understanding various mechanisms of action of rivastigmine is important to optimize its clinical utility for AD patients.
2. Materials and methods
2.1 Materials
Bovine serum albumin (BSA) was purchased from Sigma-Aldrich (St. Louis, MO). Rivastigmine L-tartrate was obtained from Tokyo Chemical Industry Co., LTD (TCI; Portland, OR). Complete mammalian protease inhibitor, donkey serum, Ficoll 400, and sodium dodecylphasphate (SDS) were obtained from (Sigma-Aldrich, MO). Reagents for total protein analysis with the bicinchoninic acid method were obtained from Pierce (Rockford, IL). For Western blot and immunohistochemistry, the mouse monoclonal antibody C-219 against P-gp was obtained from Covance Research Products (Dedham, MA); mouse monoclonal antibody against light chain LRP1 was obtained from Abcam (Cambridge, MA); goat polyclonal antibodies against actin (C-11), HRP-labeled secondary antibodies, rabbit polyclonal anti-GFAP antibody, rabbit polyclonal anti-EAAT (GLT-1) antibody, and CFL594-conjugated donkey anti-rabbit IgG were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Mouse monoclonal anti-PSD-95 antibody and rabbit polyclonal anti-SNAP-25 antibody were obtained from GeneTex, Inc (Irvine, CA). All other chemicals and reagents were of analytical grade and were readily available from commercial sources.
2.2 Animals
All animals used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Louisiana at Monroe, and animal sacrifice procedures were consistent with the IACUC policies and procedures. The transgenic mouse model used is B6;SJL-Tg(APPSWE)2576Kha or APPSWE with a mixed B6;SJL background (Line 1349). This AD model expresses the human APP695 with double mutations (K670N, M671L) that results in amyloid plaque deposition in the brain starting at the age of 9 months [24]. In addition, mdr1a/b knockout mice on FVB background were used. This mouse model carries a disruption in the multi-drug resistance genes Abcb1a (ATP-binding cassette, sub-family B (MDR/TAP), member 1A, a.k.a. Mdr1a) and Abcb1b (ATP-binding cassette, sub-family B (MDR/TAP), member 1B, a.k.a Mdr1b) encoding p-glycoprotein 3 and p-glycoprotein 1, respectively. Both models were purchased from Taconic Biosciences, Inc. (Hudson, NY, USA). APPSWE mice were bred with mdr1a/b knockout mice as reported previously [25], and the offspring of both sexes were distributed equally among the experimental groups. Tail biopsies were sampled for DNA analysis to confirm the presence of the hAPP DNA sequence in offspring, along with either of three different genotypes for mdr1; homozygous (mdr1+/+), heterozygous (mdr1+/−), or double knockout (mdr1−/−), as reported previously [25]. Littermates negative for hAPP transgene were used as controls for PCR analyses, which were performed using primers for APP and mdr1 genes as reported previously [25, 26]. Animals were maintained at 22°C, 35% relative humidity and 12 h dark/light cycle and received water and standard food.
2.3 Animal experimental conditions
When reached 8 months, animals were divided into three groups; APP+/mdr1+/+ (n =10 mice), APP+/mdr1+/− (n=12 mice) and APP+/mdr1−/− (n=6 mice), and equal number from each group were randomly assigned for rivastigmine treatment (0.3 mg/kg/day) or vehicle (sterile distilled water) for 8 weeks using Alzet osmotic mini-pumps (model 1004; DURECT Corporation, Cupertino, CA). Briefly, mice were anesthetized via isofluorane inhalation and pumps were implanted subcutaneously on the back of mice by making a small cut in the mid-scapulary region, incision was closed with wound clips. Pumps delivered 100 μl of solution at a flow rate of 0.11μl/h for 4 weeks. Pumps were replaced every four weeks by freshly filled pumps to maintain treatment for a period of 8 weeks. At the end of the study, animals were sacrificed, blood samples were collected, and brains were cut into two half for immediate capillary isolation, or snap frozen for subsequent microsectioning and immunohistochemistry experiments. Another set of animals (n =5/group) that underwent similar abovementioned treatments were sacrificed, blood samples were collected, and brains were cut into two half for western blot analysis of protein expression and extraction of Aβ and subsequent analysis with ELISA as shown below.
2.4 Isolation of brain microvessels
Freshly isolated brain hemispheres were immediately homogenized in ice-cold DPBS, phosphate-buffered saline buffer (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, 0.9 mM CaCl2, and 0.5 mM MgCl2 supplemented with 5 mM D-glucose, 1 mM sodium pyruvate, pH 7.4), and microvessels were isolated as described previously [27]. Briefly, Brain homogenate was added to 30% Ficoll in 1:1 volume (final Ficoll concentration was 15%), mixed thoroughly and centrifuged at 5800×g for 15 min at 4°C. Supernatant was discarded and pellet containing microvessels was gently resuspended in ice-cold DPBS containing 1% bovine serum albumin (BSA) and collected by separation over glass beads containing column. Microvessels adhered to glass beads were collected by gentle shaking followed by centrifugation. Pelleted microvessels were lysed in radio-immunoprecipitation assay (RIPA) buffer for subsequent Western blot analysis.
2.5 Western blot analysis
Brain hemispheres were homogenized in RIPA buffer containing 1% protease inhibitor cocktail and centrifuged at 15,000 × g for 10 min at 4°C. Supernatants were stored at −80°C for subsequent Western blot analysis. Twenty five micrograms of protein samples were loaded and resolved using 7.5% SDS-polyacrylamide gel at 140 V for 1 h and transferred electrophoretically onto nitrocellulose membranes at 300 mA for 3 h. After that, membranes were blocked in 2% BSA in PBS solution with rocking for 1 h at room temperature followed by overnight incubation at 4°C with the primary antibodies for P-gp (C-219), LRP1 (light chain), PSD-95, GLT-1, SNAP-25, and β-actin (C-11). Secondary antibodies used were: anti-mouse IgG antibody (1:2000 dilutions) for P-gp and PSD-95, anti-rabbit IgG antibody (1:2000 dilutions) for LRP1, GLT-1 and SNAP-25, and anti-goat IgG antibody (1:1500 dilutions) for β-actin, all labeled with horseradish peroxidase (HRP). Proteins’ blots were developed using a chemiluminescence detection kit (SuperSignal West Femto substrate; Thermo Scientific, Waltham, MA). Bands were visualized using C-DiGit® Blot Scanner (LI-COR Biosciences; Lincoln, NE) and quantified by densitometric analysis.
2.6 Extraction of total Aβ from brain
The extraction of Aβ from the brain tissues was performed as described previously [28]. The SDS fraction of brain samples was used to quantify Aβ oligomers (Aβ-SDS fraction) and the formic acid (FA) extract was collected to quantify insoluble deposited Aβ (FA fraction). Both fractions were neutralized by dilution 1:20 with TB buffer (1 M Tris base, 0.5 M Na2HPO4), followed by further 1:1 dilution with antigen capture buffer provided by the ELISA kit. All fractions were stored at −80°C until the time of analysis. The total amount of Aβ in the brain was calculated by summing the amount of Aβ in all fractions.
2.7 Aβ Enzyme-linked immunosorbent assay (ELISA)
Endogenous Aβ in brain homogenates and plasma samples of mice treated with rivastigmine or vehicle (n=5) were determined using sandwich ELISA. For Aβ detection Sensolyte Anti-human Aβ40 Quantitative ELISA Kit (AnaSpec, Inc., Fremont, CA), and Ultrasensetive anti-human Aβ42 ELISA kit (Novex, Frederick, MD) were used, according to the manufacturers’ instructions. All antibodies used were provided by the kits. For specific detection of Aβ40, a mouse monoclonal antibody-coated plates and a Rabbit HRP-conjugated anti Aβ40 were used as capture and detection antibodies, respectively. For specific detection of Aβ42, a rabbit monoclonal antibody-coated plates and HRP-conjugated anti-rabbit IgG were used as capture and detection antibodies, respectively. The assessment of Aβ levels in in SDS and formic acid fractions by ELISA assay was used as described by us previously [28]. Aβ concentrations were expressed as pg/g brain tissue weight.
2.8 IL-1β Enzyme-linked immunosorbent assay
Brain and plasma levels of IL-1β were determined in mice treated with rivastigmine or vehicle (n = 5) using sandwich ELISA. Quantikine ELISA kit was obtained from R&D Systems (Minneapolis, MN) and used according to the manufacturers’ instructions. Antibodies provided by the kit include, a monoclonal capture antibody specific for mouse IL-1β, and a horseradish peroxidase-linked polyclonal detection antibody specific for mouse IL-1β.
2.9 Immunohistochemistry
Brain sections of 15μm-thick were prepared using Vibratome UltraPro 5000 Cryostat installed with 5040 rotary retracting microtome (Cambridgeshire, UK), sections were then fixed on glass slides, labeled and stored at −80°C. Subsequently, sections were fixed by incubation in acetone for 10 min at −20°C. Sections were washed 3 times in PBS and blocked in TBST containing 10% donkey serum for 1 h at room temperature. For detection of GFAP, a rabbit anti-GFAP polyclonal IgG was used at 1:100 dilution in blocking solution and incubated for 4 h at room temperature. Then, sections were incubated with anti-rabbit IgG-CFL594 secondary antibody for 1 h at room temperature. After washing 4 times with PBS, GFAP immunoreactivity was visualized using Nikon Eclipse Ti-S inverted fluorescence microscope (Norcross, GA) at magnification of 200×. GFAP staining was analyzed using the image J version 1.44 software provided by NIH (http://rsbweb.nih.gov/ij/). After adjusting for threshold, Image J was used to determine the optical density of GFAP in the hippocampus region. Number of sections analyzed were 8 sections/animal for treated and untreated mice (n= 4 mice/group). Data were presented as area fraction of GFAP immunoreactive cells to area of hippocampus region.
2.10 Analysis of acetycholinesterase activity
We determined AChE activity in plasma and brain homogenate from animals treated with rivastigmine or vehicle indirectly by measuring the level of ACh. For this we used choline/acetylcholine assay kit from Abcam. Samples from plasma and brain homogenates were processed according to protocol provided by the kit and detection of choline was performed fluorometrically at excitation/emission of 535/590 nm, respectively. This assay measures either free choline or total choline, which includes free choline and ACh. Thus, ACh level can be determined as follows; ACh = total choline – free choline.
2.11 Statistical analysis
Data were expressed as mean±SEM. The experimental results were statistically analyzed for significant difference using two-tailed unpaired Student’s t-test for two groups and using ANOVA with post hoc test for more than two groups. A P-value of less than 0.05 was considered statistically significant. Data analyses were done using GraphPad Prism, version 5.03.
3. Results
3.1 Effect of rivastigmine treatment on brain and plasma levels of acetylcholine
As expected, APP/mdr1+/+ mice treated with rivastigmine significantly increased Ach brain and plasma levels by 2- to 3-fold compared to control group (P<0.001, Fig. 1). Similarly, rivastigmine was able to enhance brain ACh levels, but to a much lesser extent, in APP/mdr1+/− (by 42%) and APP/mdr1−/− (by 33%) (Fig. 1A), which is consistent with rivastigmine mechanism of action as an acetylcholinesterase inhibitor. Interestingly, however, ACh levels in the brains of vehicle treated APP/mdr1+/− and APP/mdr1−/− mice were significantly less by 52 and 32%, respectively, when compared to APP/mdr1+/+ vehicle treated mice (Fig. 1A). Conversely, ACh plasma levels in vehicle-treated mice showed a trend toward increase in APP/mdr1−/− and APP/mdr1+/− mice compared to APP/mdr1+/+ mice, but was not significant (Fig. 1B).
Figure 1.
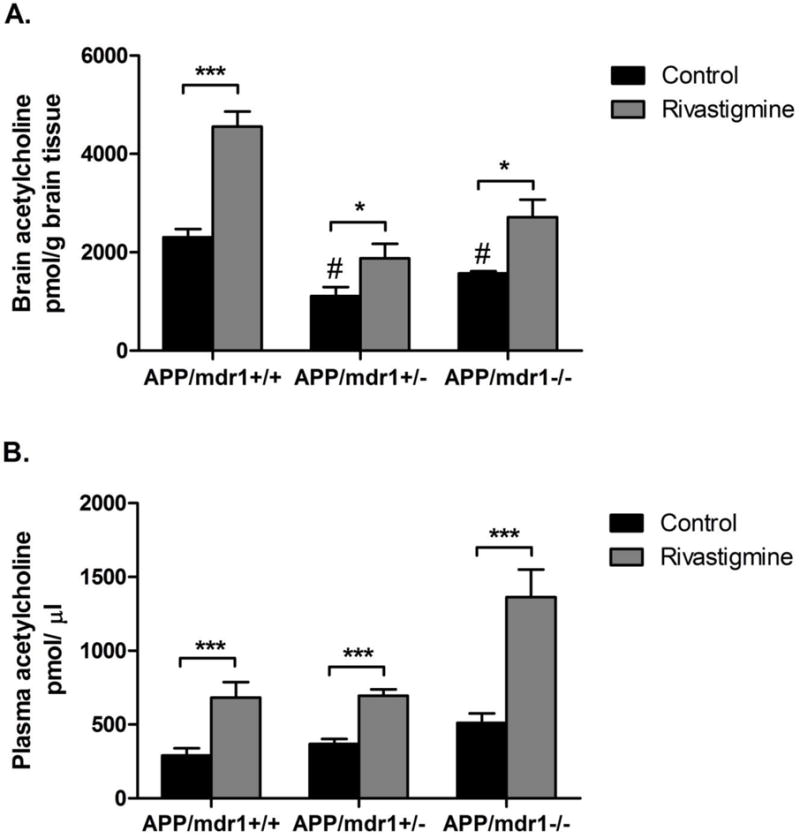
Effect of rivastigmine treatment on brain and plasma levels of acetylcholine (ACh). Levels of ACh after 8 weeks of rivastigmine treatment in APP/mdr1+/+, APP/mdr1+/− and APP/mdr1−/− in brain (A), and plasma (B). Data presented as mean±SEM (n = 3–6, * P<0.05, *** P<0.001 compared to vehicle-treated controls, # P<0.05 compared to vehicle-treated control of APP/mdr1+/+).
3.2 Effect of rivastigmine treatment on Aβ major transport proteins’ expression at the BBB
As shown in Figure 2, 8 weeks treatment with rivastigmine resulted in the up-regulation of both P-gp and LRP1 by 1.5 and 1.34 fold in APP/mdr1+/+ mice and by 1.4 and 1.25 fold in APP/mdr1+/− mice, respectively. No significant effect of rivastigmine was observed on LRP1 expression in APP/mdr1−/− mice. We have noticed that the expression of LRP1 in control mice decreased by 40% in mdr1 double knockout mice compared to mdr1 WT, while LRP1 expression in APP/mdr1+/− was not changed.
Figure 2.
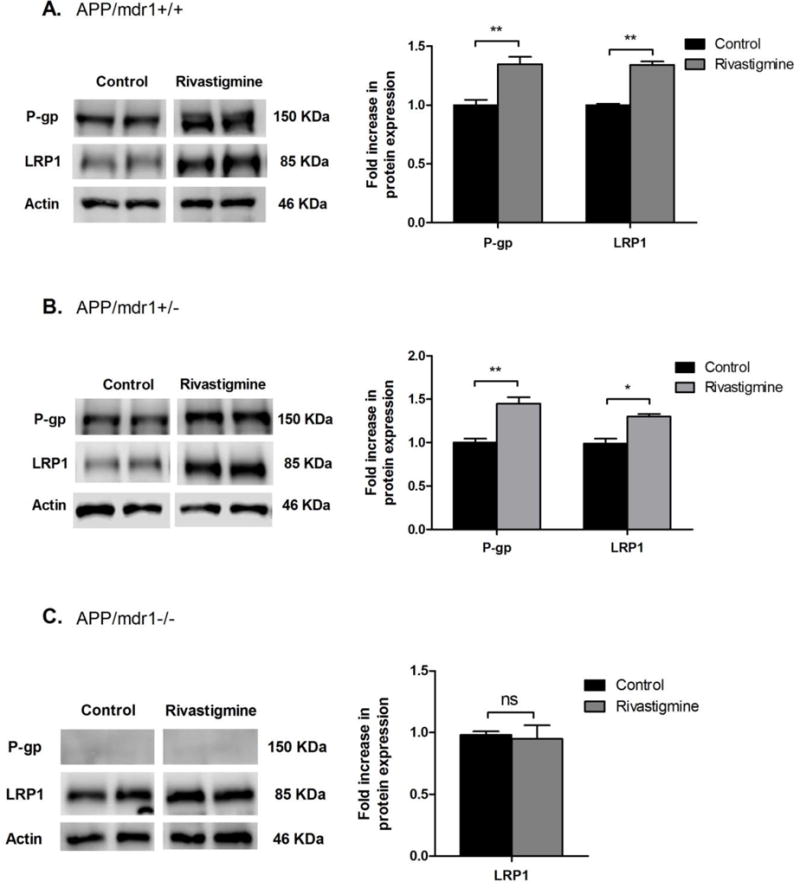
Effect of rivastigmine treatment on expression of Aβ major transport proteins at the BBB. Representative Western blots and densitometry analysis of P-gp and LRP1 from total protein of brain microvessels isolated from APP/mdr1+/+ (A), APP/mdr1+/− (B), and APP/mdr1−/− (C) mice following treatment with rivastigmine or vehicle. Data presented as mean±SEM (n = 3–6, * P<0.05, ** P<0.01, ns = not significant, compared to vehicle-treated controls).
3.3 Effect of rivastigmine treatment on Aβ brain level
In vehicle treated mice, the effect of P-gp deletion on Aβ oligomers levels was not significantly different between the 3 mice models (P>0.05, Fig. 3A). Treatment with rivastigmine reduced Aβ oligomers only in APP/mdr1+/+ mice by 33%, but not in APP/mdr1+/− or APP/mdr1−/− mice (Fig. 3A). Data from the FA-fraction showed that P-gp deletion resulted in a significant accumulation of Aβ40, but not Aβ42, in the brains of vehicle treated animals, and the effect was higher in P-gp double knockout mice by 33%, and P-gp heterozygous mice by 22% when compared to P-gp WT mice (Fig. 3B & C). For animals treated with rivastigmine, Aβ40 brain load was significantly reduced by 25% and 21% in APP/mdr1+/+ and APP/mdr+/− mice, respectively, compared to their vehicle treated controls (P>0.05, Fig. 3B). For APP/mdr−/− mice, rivastigmine treatment showed a trend toward reduction in Aβ40 level, but it was not statistically significant. For Aβ42 brain level, rivastigmine effect was only seen in APP transgenic, P-gp WT mice (reduced by 30%, P>0.05), while no effect was observed in APP transgenic, P-gp deficient mice (Fig. 3B).
Figure 3.
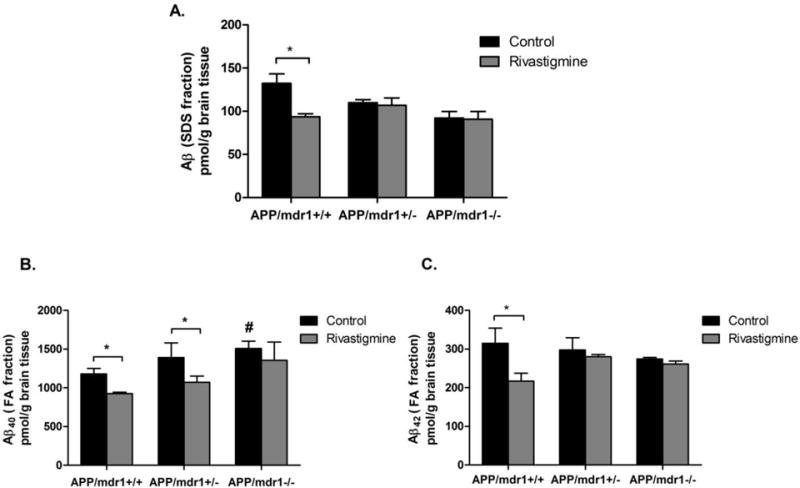
Effect of rivastigmine treatment on brain levels of Aβ. Quantitative analysis of Aβ brain levels using specific ELISA for Aβ-SDS fraction (A), Aβ40-FA fraction (B), and Aβ42-FA fraction (C). Data presented as mean±SEM (n = 3–6, * P<0.05, compared to vehicle-treated controls; #P<0.05 compared to vehicle-treated control of APP/mdr1+/+ mice).
3.4 Effect of rivastigmine treatment on plasma levels of Aβ
Rivastigmine treatment was only able to significantly reduce plasma levels of Aβ40 in APP/mdr1+/+ mice (43%, Fig. 4A; P<0.05); for Aβ42 there was no significant change in response to rivastigmine treatment in APP/mdr1+/+ mice (Fig. 4B). Rivastigmine treatment had no effect on Aβ40 and Aβ42 levels in +/− 42 APP/mdr1 or APP/mdr1−/− (Fig. 4). The effect of P-gp deletion on Aβ plasma levels in control treated mice was differential where Aβ40 levels increased with reduced number of mdr1 copies reaching up to 2-fold in P-gp double knockout mice (Fig. 4A, P<0.05), while Aβ42 levels were not affected by the reduced expression of P-gp (Fig. 4B).
Figure 4.
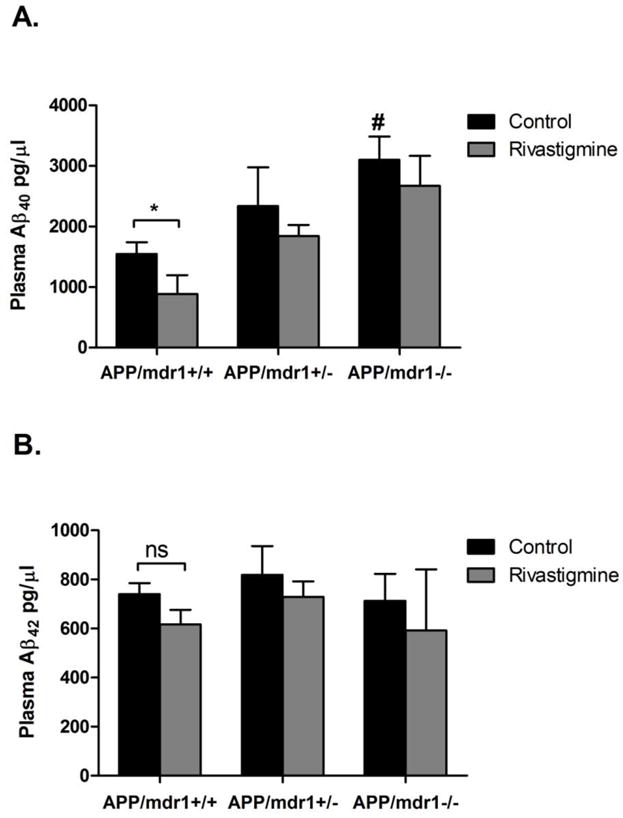
Effect of rivastigmine treatment on plasma levels of Aβ. Quantitative analysis of total Aβ plasma levels using specific ELISA for Aβ40 (A), and for Aβ42 (B). Data presented as mean±SEM (n = 3–6, * P<0.05, ns = not significant, compared to vehicle-treated controls, # P<0.05 compared to vehicle-treated control of APP/mdr1+/+).
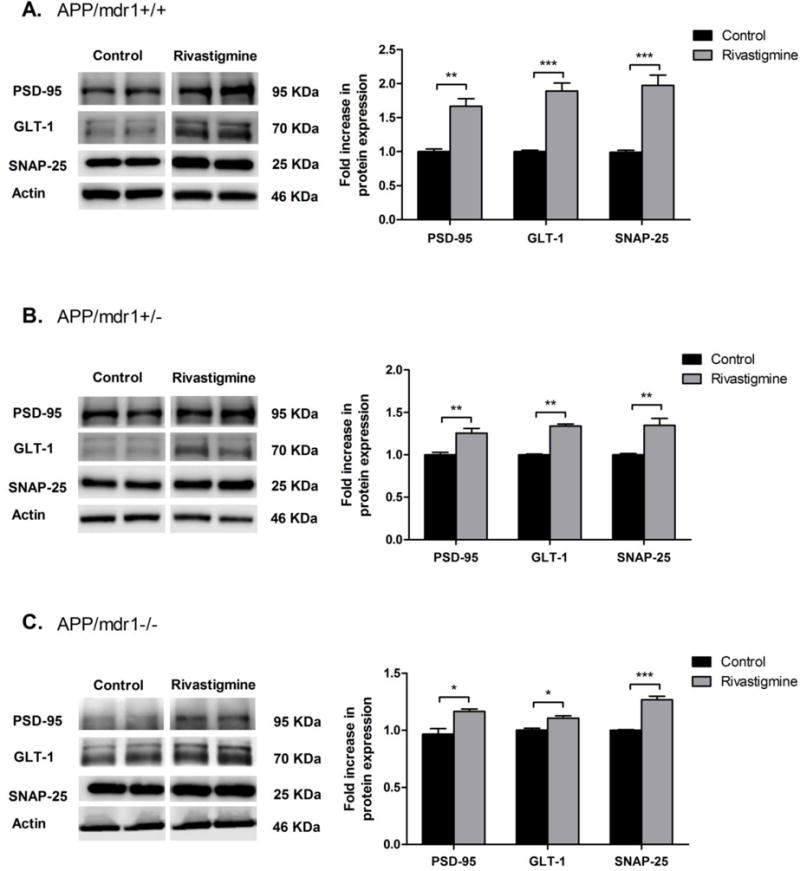
3.5 Effect of rivastigmine treatment on the expression of synaptic markers
Two synaptic markers were evaluated, the pre-synaptic marker synaptosomal-associated protein (SNAP-25), and the post-synaptic marker postsynaptic density protein 95 (PSD-95). As shown in Figure 5, all mice groups treated with rivastigmine showed significant increase in PSD-95 and SNAP-25 protein expression. However, the effect was highest in the APP/mdr1+/+ mouse model with 1.7- and 2-fold increase in PSD-95 and SNAP-25, respectively (Fig. 5A, P<0.01). The effect of rivastigmine on synaptic marker expression was reduced to 1.25- and 1.34-fold in APP/mdr1+/− and 1.2- and 1.3-fold in APP/mdr1−/− for PSD-95 and SNAP-25, respectively (Fig. 5 B and C). This result suggests a possible role for P-gp in mediating rivastigmine effect to preserve synapses through reducing Aβ brain load. In parallel to rivastigmine induction of synaptic markers, rivastigmine significantly enhanced the Na+-dependent glutamate transporter (GLT-1) protein expression by 2-fold in APP/mdr1+/+ mice, and less pronounced, but significant, in APP/mdr1+/− and APP/mdr1−/− by 1.3 and 1.1 fold, respectively.
Figure 5.
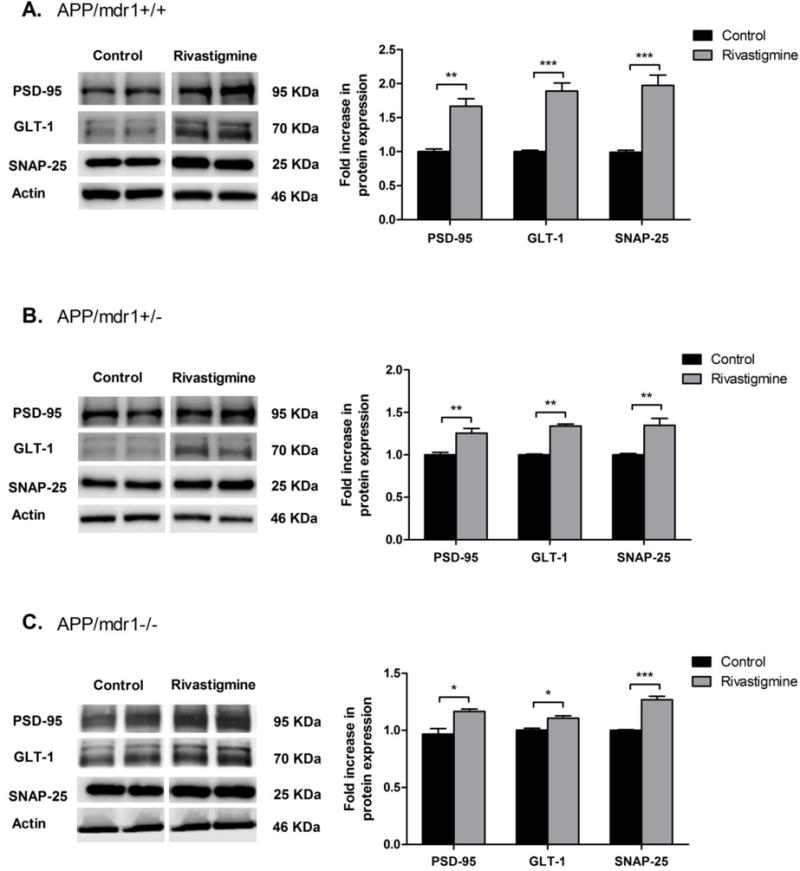
Effect of rivastigmine treatment on protein expression of synaptic markers. Representative Western blots and densitometry analysis of pre-synaptic marker (SNAP-25), postsynaptic marker (PSD-95) and glutamate transporter 1 (GLT1) from brain homogenates of APP/mdr1+/+ (A), APP/mdr1+/− (B) and APP/mdr1−/− (C) mice following treatment with rivastigmine or vehicle. Data presented as mean±SEM (n = 3–6, * P<0.05, ** P<0.01, *** P<0.001 compared to vehicle-treated controls).
3.6 Effect of rivastigmine on hippocampal astrogliosis
Two months treatment with rivastigmine resulted in a significant reduction in astrogliosis in all mice groups, which was determined by measuring the astrocyte reactive marker glial fibrillary acidic protein (GFAP) intensity by immunostaining. Rivastigmine reduced GFAP immunoreactive cells by 45–50% in APP/mdr1+/+ and APP/mdr1+/− mice when compared to control mice (Fig. 6 A–D, G, P<0.001). For APP/mdr1−/−, the effect of rivastigmine was less pronounced but significant with 34% reduction in GFAP intensity compared to controls (Fig 6 E–G, P<0.001). In control treated groups, the effect of P-gp deletion was obvious and was associated with a significant increase in GFAP activation by approximately 30% in both APP/mdr1+/− and APP/mdr1−/− mice when compared to APP/mdr1+/+.
Figure 6.
Effect of rivastigmine on hippocampal astrogliosis. Representative hippocampus sections stained with glial fibrillary acidic protein (GFAP) antibody to detect astrocytes in APP/mdr1+/+ (A, B), APP/mdr1+/− (C, D) and APP/mdr1−/− (E, F). Quantification of GFAP staining in hippocampus of animals treated with rivastigmine or vehicle (G). Data presented as mean±SEM (n = 3–6, *** P<0.001 compared to vehicle-treated controls, # P<0.05 compared to vehicle-treated control of APP/mdr1+/+).
3.7 Effect of rivastigmine on brain and plasma levels of the pro-inflammatory cytokine interleukin-1β
Rivastigmine treatment significantly reduced brain levels of interleukin-1 (IL1-β) by 43% in APP/mdr1+/+ mice and by 33% in APP/mdr1+/− mice, however, this effect was not observed in P-gp double knockout mice (Fig. 7A). Similarly, plasma levels of IL1-β were significantly reduced by rivastigmine by 51 and 31% in APP/mdr1+/+ and APP/mdr1+/− mice, respectively, while APP/mdr1−/− showed no response to the treatment (Fig. 7B). In control treated mice, P-gp deletion caused a significant increase in the brain and plasma levels of the pro-inflammatory mediator IL1-β. IL1-β brain levels were increased by 26 and 51% in APP/mdr1+/− and APP/mdr1−/− mice, respectively, compared to APP/mdr1+/+ (Fig. 7A). The increase in plasma levels of IL1-β was even higher due to P-gp deletion by 75 and 160% in APP/mdr1+/− and APP/mdr1−/− mice, respectively, compared to APP/mdr1+/+ (Fig. 7B).
Figure 7.
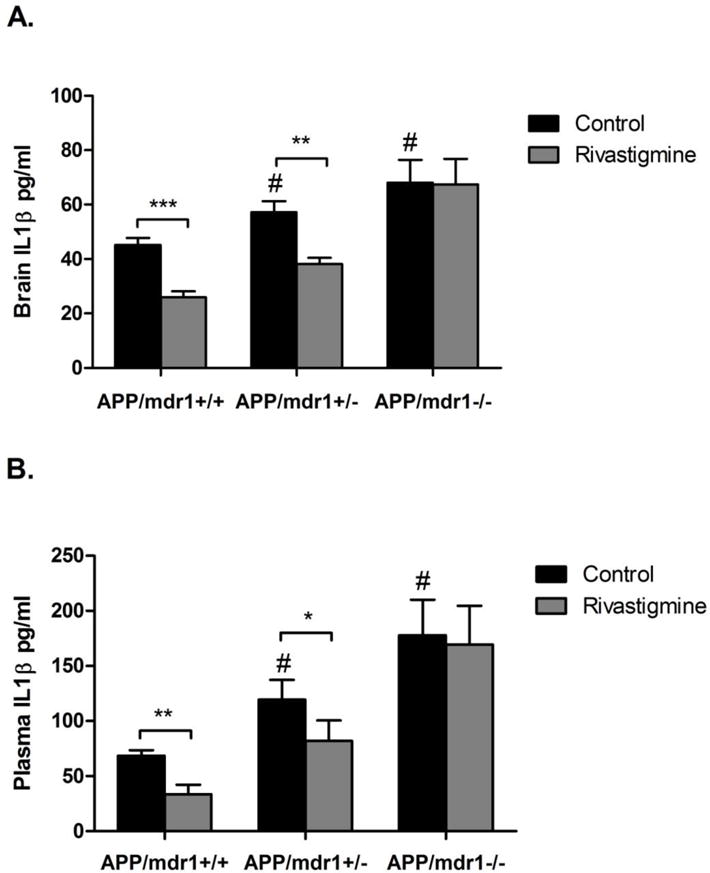
Effect of rivastigmine on brain and plasma levels of the pro-inflammatory cytokine interleukin 1-β (IL1-β). Levels of IL1-β after 8 weeks of rivastigmine treatment in APP/mdr1+/+, APP/mdr1+/− and APP/mdr1−/− brain (A), and plasma (B). Data presented as mean±SEM (n = 3–6, * P<0.05, ** P<0.01, *** P<0.001 compared to vehicle-treated controls, # P<0.05, compared to vehicle-treated control of APP/mdr1+/+).
4. Discussion
Currently, AD has no treatment and available medications provide some cognitive improvements and symptomatic relief, but they do not stop the progression of AD. Rivastigmine is one of the FDA-approved drugs for AD and dementia [28, 29]. Rivastigmine is a dual cholinesterase inhibitor which acts by inhibiting degradation of ACh at synapses and improving cholinergic transmission that is known to be impaired in AD. Recently, we have shown that FDA-approved drugs for AD modulate Aβ transport by up-regulating the expression of P-gp and LRP1 at the BBB and hepatocytes [13]. Among those drugs, we found that rivastigmine was most efficient to induce the expression of P-gp and LRP1 in the brains of aged rats and were associated with reduced levels of endogenous Aβ40 and Aβ42 [13].
With aging and in AD, Aβ accumulation has been reported and was associated with decline in Aβ clearance [30]. This observation suggests that enhancing the clearance of Aβ may protect the brain from Aβ-mediated neuropathology [31, 32]. We have shown previously that upregulation of P-gp and LRP1 induces Aβ clearance across the BBB in vitro and in vivo [33]. P-gp is a primary transporter involved in brain clearance of Aβ across the BBB [13, 34]. Vogelgesang and colleagues reported that accumulation of Aβ in the brain correlates inversely with the degree of P-gp expression at the BBB [22]. These findings suggest that high levels of P-gp at the BBB may be protective from Aβ deposition by eliminating Aβ out of the brain and preventing re-entry of Aβ from peripheral circulation. Previous findings from human studies suggested that peripheral Aβ can cross a defective BBB and accumulates in the brain [35]. Thus, a healthy and functional BBB has an important role in preserving Aβ brain homeostasis via expression and activity of proteins involved in the transport of Aβ.
The aim of the current study was to investigate the role of P-gp in mediating effect of rivastigmine treatment on Aβ brain level and related pathology in AD mouse model express different levels of P-gp. This model was previously used and characterized by Cirrito and colleagues [25] who reported that P-gp deletion in APP mouse model did not alter the expression of receptor for advanced glycation endproducts (RAGE), an important receptor for influx of Aβ from blood to brain, but increased multidrug resistance associated protein1 (MRP1) expression as a compensatory mechanism for P-gp deletion in APP/mdr1−/− mice [25]. Role of MRP1 in Aβ clearance was previously reported in Abcc1, gene encode for Mrp1, deficient AD mouse model where its complete knockout increased Aβ brain deposit suggesting, like P-gp, its role in controlling Aβ brain levels [36]. Yet, findings from our study showed that Mrp1 couldn’t rescue the absence of P-gp as evident by Aβ increased load and related pathology with or without rivastigmine treatment. At the BBB, compared to other ABC transporters the expression of P-gp is reported to be significantly higher than MRP1 and ABCG2 [37, 38], demonstrating P-gp important function at the BBB. With regard to ABCG2, it is reported that in mdr1−/− mice the expression of Abcg2 was not altered by the deletion of P-gp [38], thus its expression in our model is not expected to change. In addition, further characterization showed P-gp deletion has no effect on the expression of ABCA1, ApoE and LXR, important proteins involved in Aβ lipidation and degradation pathway (data not shown).
Our findings support P-gp as important component at the BBB to regulate Aβ brain levels and its related pathology, and in modulating the neuroprotective effect of rivastigmine in a P-gp dependent manner. As expected, rivastigmine increased ACh brain level similarly (by1.7- to 2-fold) in all treated APP transgenic mice expressing different levels of P-gp. Also, in plasma, rivastigmine was able to increase ACh level in all mice and this increase, while was higher than that observed in brain, was comparable between the groups, approximately 2- to 2.5-fold increase. These results indicate that P-gp deletion did not interfere with rivastigmine cholinergic stimulation. However, in vehicle treated mice, P-gp deletion significantly reduced brain ACh compared to P-gp WT mice. This decrease in brain level of ACh could be indirectly attributed to P-gp deletion through increasing Aβ brain accumulation (Fig. 3). The degree of P-gp expression in the brain correlated inversely with Aβ brain levels, which was reflected on Ach levels where less P-gp was associated with more Aβ, due to reduced efflux across the BBB, and less Ach levels. This finding is consistent with previous reports indicated that Aβ inhibits ACh synthesis and release in rat hippocampal slices and in vivo in young and aged rats thus causing cholinergic hypo-function [39–41]. This result supports role for P-gp in the development of AD and cognitive impairment by depleting Ach levels that is important for learning and memory [4, 5]. Interestingly, in plasma, among the 3 models rivastigmine effect on ACh level was highest in P-gp deleted mice, an observation that may link rivastigmine to its peripheral cholinergic adverse effects in AD patients. As the expression of P-gp declines at the BBB and Aβ brain burden increases, this could induce rivastigmine peripheral cholinergic stimulation and exacerbate its peripheral adverse events [42].
Consistent with our previous study [13], rivastigmine induced the expression of P-gp and LRP1 in isolated brain capillaries of APP/mdr1+/+ and APP/mdr1+/− mice, and this increase was equivalent for both models. However, in P-gp double knockout mice, rivastigmine could not induce LRP1 expression. In control group, complete deletion of P-gp in APP transgenic mice reduced LRP1 expression by 40% when compared to APP/mdr1+/+ group, which is consistent with previously reported 51% reduction in the same mouse model [25]. In addition, the effect of P-gp levels on Aβ peptides was deferential where P-gp deletion caused a significant increase in parenchymal accumulation of Aβ40, but not Aβ42, when compared to APP/P-gp WT mice. This inverse correlation between Aβ40 deposition and amount of P-gp expression was expected and suggest the important role of P-gp in the removal of Aβ across the BBB to maintain Aβ brain homeostasis. The isoform specific accumulation of Aβ40 could be attributed to higher brain levels of Aβ40 than Aβ42, and the clearance rate of Aβ across the BBB is higher for Aβ40 than Aβ42 [43, 44]. Therefore, interruption of Aβ clearance across the BBB by P-gp deletion could be reflected more likely on Aβ40 than Aβ42. Nonetheless, higher Aβ42 depositions might be observed at a later age where aggressive Aβ deposition occurs. Similar pattern was observed in the plasma where P-gp deletion increased Aβ40 levels, but not Aβ42. This observation is consistent with P-gp function in the biliary excretion of Aβ40 as we reported previously [45]. P-gp deletion in the liver could thus lead to Aβ increased levels in the plasma due to reduced Aβ hepatic clearance, which in turn could further contribute to Aβ accumulation in the brain as a result of decreased extrusion at the BBB. This plasma to brain influx of Aβ possibly hindered the cholinergic and non-cholinergic effects of rivastigmine in APP/mdr1−/− mice. On the other hand, treatment with rivastigmine decreased both Aβ40 and Aβ42, and SDS stable oligomers in P-gp WT mice. It is convincing that rivastigmine inductive effect on P-gp and LRP1 stimulates the removal of Aβ from the brain and reduces its accumulation, and the deletion of P-gp impairs this mechanism, suggesting that rivastigmine lowering effect on Aβ40 is proportional to the degree of P-gp expression.
The effect of P-gp in controlling the neuroprotective effect of rivastigmine was also observed. Rivastigmine increased the expression of synaptic proteins including SNAP-25, a pre-synaptic marker, and PSD-95, a post-synaptic marker, in a P-gp dependent manner. This increase in synaptic proteins expression could be attributed indirectly to rivastigmine effect on reducing Aβ brain load, thereby, decreasing Aβ-mediated synaptotoxicity. High brain levels of Aβ could interfere with synaptic plasticity and induce endocytosis of synaptic transporters and receptors and cause synaptic loss [3]. Supportive to rivastigmine induction of synaptic markers and protection of synapses, rivastigmine significantly enhanced GLT-1 protein expression in brain. GLT-1 is a Na+-dependent glutamate transporter (EAATs) expressed on the plasma membrane of neurons and glial cells and has important role in regulating neurotransmission and intersynaptic cross-talk [46], and dysfunction in this transporter is reported in neurological diseases including AD [47]. This neuroprotective effect of rivastigmine, while attenuated, was significant in P-gp deleted mice, which suggest the importance of functional BBB to mediate rivastigmine beneficial effects. Nonetheless, rivastigmine partial neuroprotective effect in the P-gp deficient mice could be attributed to its classical effect in maintaining ACh level at synapses and improving synaptic cholinergic function [6].
Astrocytes are major cell type involved in degradation of Aβ and maintenance of synaptic glutamate concentrations [48]. It is suggested that accumulation of Aβ inside the brain causes a cascade of inflammatory events that lead to astrogliosis that localize to Aβ deposits [49]. Astrocytes activation enhances degradation of Aβ; however, their chronic activation has shown to initiate the release of damaging cytokines including, tumor necrosis factor α, IL-1 and IL-6. The release of such cytokines could stimulate neuronal dysfunction and death, and thus contributing to cognitive dysfunction and behavioral abnormalities seen in AD [50, 51]. These Aβ-mediated inflammatory events concurrently could contribute to cerebrovascular alterations and lead to BBB dysfunction observed in AD. In our AD mouse model, gliosis is evident at early age (5 months of age) and deletion of P-gp caused a significant increase in GFAP immunoreactivity by approximately 30% compared to APP transgenic, P-gp WT mice. As part of rivastigmine neuroprotective effect, we demonstrated that chronic treatment with rivastigmine significantly reduced GFAP immunoreactivity in P-gp WT and P-gp heterozygous mice (by ~50%) compared to P-gp deleted mice (by 36%). Concomitantly, deletion of P-gp resulted in a significant increase in the brain and plasma levels of the pro-inflammatory mediator IL1-β, and rivastigmine treatment decreased IL1-β brain and plasma levels, which was consistent with its anti-astrogliosis effect and support a role for P-gp in this effect. The significant increase in IL1-β brain and plasma level in P-gp deleted mice could be attributed to the higher Aβ brain and plasma levels which stimulate inflammation and secretion of inflammatory markers. This increased level of IL1-β was significantly reduced by rivastigmine in P-gp expression dependent manner, however, this effect was not observed in the P-gp knockout mice. While an explanation for this observation is not clear, it could be related to the increased Aβ levels in this mouse model that was not altered by rivastigmine treatment (Fig. 3).
Taken together, despite the fact that rivastigmine was able to increase brain and plasma levels of ACh, and showed some anti-inflammatory effects in P-gp deleted mice, it could not decrease Aβ brain levels. The reduced neuroprotective and anti-inflammatory effects of rivastigmine in P-gp deleted mice highlight the significant contribution of P-gp to the neuroprotective mechanism of rivastigmine. Aβ plays a central role in the degenerative process of neurons that lead to neuronal dysfunction in AD [52]. Also, Aβ decreases the release of ACh from presynaptic terminals and impairs nicotinic ACh receptor (nAChr) signaling [3]. This suggests that the cholinergic and amyloid hypotheses are not independent and proposes a possible cross-talk between the two pathways. Accordingly, it is possible that rivastigmine could boost its cholinergic effect and increase ACh level at synapses via two mechanisms; cholinesterase inhibition and decreasing Aβ brain load.
5. Conclusion
Collectively, our findings suggest that rivastigmine lowering effect on Aβ and its neuroprotective effect depend largely on P-gp function at the BBB. In AD, inflammation and oxidative stress alter the expression of BBB transporters which in turn promotes accumulation of Aβ inside the brain [53, 54]. P-gp is downregulated in AD and with aging and is associated with higher accumulation of brain Aβ [13, 20–22]. Malfunction in major transporters of Aβ, like P-gp, at the BBB could thus influence the therapeutic efficiency of drugs that depend largely on those molecules to clear Aβ out of the brain. This could be true for rivastigmine, where the deletion of P-gp associated with reduction in LRP1 expression deteriorated the ability of rivastigmine to lower Aβ and negatively affected its anti-inflammatory and neuroprotective effects. Besides, our findings showed rivastigmine to up-regulate P-gp at the BBB, thus further stimulating Aβ clearance. Simultaneously, however, it is expected that such increase in P-gp expression to limit brain access of concomitantly administered CNS drugs that are P-gp substrates and may affect their therapeutic outcome.
Highlights.
P-gp could play role in the development of AD by depleting Ach levels
Rivastigmine lowers Aβ brain load, at least in part, by upregulating P-gp and LRP1 at the BBB.
P-gp deletion attenuates rivastigmine effect on Aβ and its related pathology.
Rivastigmine’s neuroprotective effect is P-gp expression dependent (APP/mdr1+/+> APP/mdr1+/−>APP/mdr1−/−)
Acknowledgments
This project was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103424.
Abbreviations
- ABCA1
ATP-binding cassette sub-family A, member 1
- APP
amyloid precursor protein
- ACh
acetylcholine
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- BBB
blood-brain barrier
- BSA
bovine serum albumin
- ChEIs
Cholinesterase inhibitors
- ELISA
enzyme-linked immunosorbent assay
- FA
formic acid
- GFAP
glial fibrillary acidic protein
- GLT-1
Na+-dependent glutamate transporter
- IL1
interleukin-1
- LRP1
low density lipoprotein receptor related protein 1
- LTP
long term potentiation
- P-gp
P-glycoprotein
- PSD-95
postsynaptic density protein 95
- SDS
sodium dodecylsulfate
- SNAP-25
synaptosomal-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interest.
References
- 1.Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Annals of the New York Academy of Sciences. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England journal of medicine. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 4.Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb Cortex. 1996;6:165–177. doi: 10.1093/cercor/6.2.165. [DOI] [PubMed] [Google Scholar]
- 5.Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends in neurosciences. 1999;22:273–280. doi: 10.1016/s0166-2236(98)01361-7. [DOI] [PubMed] [Google Scholar]
- 6.Babic T. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. Journal of neurology, neurosurgery, and psychiatry. 1999;67:558. doi: 10.1136/jnnp.67.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen H, Kihara T, Hongo H, Wu X, Kem WR, Shimohama S, Akaike A, Niidome T, Sugimoto H. Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of alpha7 nicotinic receptors and internalization of NMDA receptors. British journal of pharmacology. 2010;161:127–139. doi: 10.1111/j.1476-5381.2010.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HG, Moon M, Choi JG, Park G, Kim AJ, Hur J, Lee KT, Oh MS. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology. 2014;40:23–32. doi: 10.1016/j.neuro.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Ballard CG, Chalmers KA, Todd C, McKeith IG, O’Brien JT, Wilcock G, Love S, Perry EK. Cholinesterase inhibitors reduce cortical Abeta in dementia with Lewy bodies. Neurology. 2007;68:1726–1729. doi: 10.1212/01.wnl.0000261920.03297.64. [DOI] [PubMed] [Google Scholar]
- 10.Bailey JA, Lahiri DK. A novel effect of rivastigmine on pre-synaptic proteins and neuronal viability in a neurodegeneration model of fetal rat primary cortical cultures and its implication in Alzheimer’s disease. Journal of neurochemistry. 2010;112:843–853. doi: 10.1111/j.1471-4159.2009.06490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey JA, Ray B, Greig NH, Lahiri DK. Rivastigmine lowers Abeta and increases sAPPalpha levels, which parallel elevated synaptic markers and metabolic activity in degenerating primary rat neurons. PloS one. 2011;6:e21954. doi: 10.1371/journal.pone.0021954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang HQ, Sun ZK, Yang WM, Han HM, Ma JJ, Li W. Effects of rivastigmine on secreted amyloid precursor protein and beta-amyloid secretion in neuroblastoma SK-N-SH cells. Neurochem J. 2013;7:215–220. [Google Scholar]
- 13.Mohamed LA, Qosa H, Kaddoumi A. Age-Related Decline in Brain and Hepatic Clearance of Amyloid-Beta is Rectified by the Cholinesterase Inhibitors Donepezil and Rivastigmine in Rats. ACS chemical neuroscience. 2015 doi: 10.1021/acschemneuro.5b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Donepezil Study Group. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 15.Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Current drug targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- 16.Sommer B. Alzheimer’s disease and the amyloid cascade hypothesis: ten years on. Current opinion in pharmacology. 2002;2:87–92. doi: 10.1016/s1471-4892(01)00126-6. [DOI] [PubMed] [Google Scholar]
- 17.Sagare AP, Bell RD, Zlokovic BV. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease, Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 20.Lee G, Bendayan R. Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharmaceutical research. 2004;21:1313–1330. doi: 10.1023/b:pham.0000036905.82914.8e. [DOI] [PubMed] [Google Scholar]
- 21.Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Current Alzheimer research. 2004;1:121–125. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:535–541. doi: 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, Donahue JE, Johanson CE. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. Journal of neuropathology and experimental neurology. 2010;69:1034–1043. doi: 10.1097/NEN.0b013e3181f46e25. [DOI] [PubMed] [Google Scholar]
- 24.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 25.Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. The Journal of clinical investigation. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 27.Qosa H, Abuznait AH, Hill RA, Kaddoumi A. Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2012;31:151–165. doi: 10.3233/JAD-2012-120319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clinical pharmacokinetics. 2002;41:719–739. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- 29.Birks J, Grimley Evans J, Iakovidou V, Tsolaki M. Rivastigmine for Alzheimer’s disease. The Cochrane database of systematic reviews. 2000:CD001191. doi: 10.1002/14651858.CD001191. [DOI] [PubMed] [Google Scholar]
- 30.Chiu C, Miller MC, Monahan R, Osgood DP, Stopa EG, Silverberg GD. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: preliminary observations. Neurobiology of aging. 2015 doi: 10.1016/j.neurobiolaging.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 31.Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends in neurosciences. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Abuznait AH, Qosa H, Busnena BA, El Sayed KA, Kaddoumi A. Olive-oil-derived oleocanthal enhances beta-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: in vitro and in vivo studies. ACS chemical neuroscience. 2013;4:973–982. doi: 10.1021/cn400024q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. beta-Amyloid efflux mediated by p-glycoprotein. Journal of neurochemistry. 2001;76:1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- 35.Clifford PM, Zarrabi S, Siu G, Kinsler KJ, Kosciuk MC, Venkataraman V, D’Andrea MR, Dinsmore S, Nagele RG. Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain research. 2007;1142:223–236. doi: 10.1016/j.brainres.2007.01.070. [DOI] [PubMed] [Google Scholar]
- 36.Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Bruning T, Plath AS, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. The Journal of clinical investigation. 2011;121:3924–3931. doi: 10.1172/JCI57867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yousif S, Marie-Claire C, Roux F, Scherrmann JM, Decleves X. Expression of drug transporters at the blood-brain barrier using an optimized isolated rat brain microvessel strategy. Brain research. 2007;1134:1–11. doi: 10.1016/j.brainres.2006.11.089. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug metabolism and disposition: the biological fate of chemicals. 2012;40:1164–1169. doi: 10.1124/dmd.112.044719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaucher E, Aumont N, Pearson D, Rowe W, Poirier J, Kar S. Amyloid beta peptide levels and its effects on hippocampal acetylcholine release in aged, cognitively-impaired and -unimpaired rats. Journal of chemical neuroanatomy. 2001;21:323–329. doi: 10.1016/s0891-0618(01)00120-x. [DOI] [PubMed] [Google Scholar]
- 40.Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R. Amyloid beta-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. Journal of neurochemistry. 1998;70:2179–2187. doi: 10.1046/j.1471-4159.1998.70052179.x. [DOI] [PubMed] [Google Scholar]
- 41.Lane RM, Potkin SG, Enz A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J Neuropsychopharmacol. 2006;9:101–124. doi: 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- 42.Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. International journal of clinical practice Supplement. 2002:45–63. [PubMed] [Google Scholar]
- 43.Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid beta-peptide from brain: transport or metabolism? Nature medicine. 2000;6:718–719. doi: 10.1038/77397. [DOI] [PubMed] [Google Scholar]
- 44.Ito S, Ohtsuki S, Terasaki T. Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1–40) across the rat blood-brain barrier. Neuroscience research. 2006;56:246–252. doi: 10.1016/j.neures.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Mohamed LA, Kaddoumi A. In Vitro Investigation of Amyloid-beta Hepatobiliary Disposition in Sandwich-Cultured Primary Rat Hepatocytes. Drug metabolism and disposition: the biological fate of chemicals. 2013;41:1787–1796. doi: 10.1124/dmd.113.052514. [DOI] [PubMed] [Google Scholar]
- 46.Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain research Brain research reviews. 2004;45:250–265. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Danbolt NC. Glutamate uptake. Progress in neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 48.Theodossiadis GP, Grigoropoulos VG, Liarakos VS, Rouvas A, Emfietzoglou I, Theodossiadis PG. Restoration of the photoreceptor layer and improvement of visual acuity in successfully treated optic disc pit maculopathy: a long follow-up study by optical coherence tomography. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2012;250:971–979. doi: 10.1007/s00417-011-1918-z. [DOI] [PubMed] [Google Scholar]
- 49.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 50.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 51.Cummings JL, Vinters HV, Cole GM, Khachaturian ZS. Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology. 1998;51:S2–17. doi: 10.1212/wnl.51.1_suppl_1.s2. discussion S65–17. [DOI] [PubMed] [Google Scholar]
- 52.Mucke L, Selkoe DJ. Neurotoxicity of Amyloid beta-Protein: Synaptic and Network Dysfunction. Cold Spring Harbor perspectives in medicine. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, Banks WA, Butterfield DA. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Abeta accumulation in AD brain. Free radical biology & medicine. 2010;49:1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nature medicine. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]