

Abstract
Zidovudine (AZT), the first drug approved by the US Food and Drug Administration for the treatment of human immunodeficiency virus (HIV) infection, is metabolized in the host cells to 5′-AZT triphosphate (AZT-TP) which inhibits HIV reverse transcriptase. As the pharmacokinetics of AZT and its phosphorylated metabolites in human peripheral blood mononuclear cells (hPBMCs) is limited, the aim of this study was to determine the pharmacokinetic parameters of AZT and its phosphorylated metabolites in hPBMCs from 12 healthy Chinese male subjects after a single oral dose of 600 mg of AZT. Blood samples were collected prior to drug administration, then at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8 and 10 h after drug administration. Mononuclear cells collected by Ficoll-Hypaque density gradient centrifugation were used for determination of AZT and metabolites [AZT monophosphate (AZT-MP), AZT diphosphate (AZT-DP) and AZT-TP] and the plasma was used to evaluate the pharmacokinetics of AZT. Plasma concentration of AZT peaked within 0.583 h and intracellular concentrations of AZT, AZT-MP, AZT-DP and AZT-TP peaked within 1.083, 1.500, 1.417 and 1.583 h, respectively. AZT in plasma was eliminated rapidly with t1/2 of 2.022 h, and AZT-MP, AZT-DP and AZT-TP were eliminated with t1/2 of 13.428, 8.285 and 4.240 h, respectively. The plasma concentration of the phosphorylated metabolites was not quantifiable.
KEY WORDS: LC–MS/MS, Zidovudine, Metabolites, Phosphates, hPBMCs, hPBMCs, Pharmacokinetics, Intracellular kinetic
Graphical abstract
A pharmacokinetic experiment has been performed to evaluate the pharmacokinetics of zidovudine in plasma and pharmacokinetics of zidovudine and its phosphate metabolites in human peripheral blood mononuclear cells.
1. Introduction
Nucleoside reverse transcriptase inhibitors (NRTIs) were the first agents shown to be safe and effective for the treatment of patients infected with HIV, and they remain cornerstones of most treatment regimens today. These drugs are selectively phosphorylated to their active triphosphate moieties (NRTI-TP) in human peripheral blood mononuclear cells (hPBMCs)1. Zidovudine (3'-azido-3'-deoxythymidine, AZT) was the first drug for the treatment of human immunodeficiency virus (HIV) infection approved by the US Food and Drug Administration (FDA). The parent compound AZT must be metabolized in the host cells to 5'-AZT triphosphate (AZT-TP), which acts as a competitive inhibitor of HIV reverse transcriptase or is incorporated into the viral genome, terminating DNA chain elongation2, 3. It remains an important NRTI component of many first-line highly active antiretroviral therapies (HAART) used today4.
AZT is converted inside the cell into AZT monophosphate (AZT-MP) by thymidine kinase. Thymidylate kinase in turn converts AZT-MP into AZT diphosphate (AZT-DP), which is further phosphorylated to AZT-TP, presumably by pyrimidine nucleoside diphosphate kinase. AZT monophosphate is the predominant compound in human cells exposed to AZT, suggesting that thymidylate kinase is the rate limiting enzyme in the synthesis of active AZT-TP2, 5. Most measurements of AZT are performed in plasma or serum, but because AZT is phosphorylated to its active form inside the cells, measurements of plasma levels of AZT may not directly correlate with antiviral activity or toxicity. Therefore, it is important to determine the intracellular levels of AZT and its metabolites.
Recently, several studies have shown that the intracellular concentration of the triphosphate moieties of NRTIs correlated better with the virologic response than did the levels of the parent compound in plasma6, 7. Long-term use of high-dosage AZT caused damage to many tissues, including a mitochondrial skeletal muscle myopathy, a dilated cardiomyopathy and hepatotoxicity associated with mitochondrial DNA depletion8. These conditions are related to AZT use and not to the progression of acquired immunodeficiency syndrome (AIDS) since when patients experiencing one or more of these adverse effects discontinued AZT therapy, the adverse effects would resolve9, 10. In modern therapeutic regimens, AZT is given at much lower doses and in combination with other drugs. Thus, these adverse effects have become fairly rare, but hematological toxicities, such as anemia and lipodystrophy are observed commonly in combination thera-py11, 12, 13.
Detection and quantitation of the NRTI-TP concentrations in hPBMCs will lead to a better understanding of the pharmacokinetic and pharmacodynamic characteristics of these agents, which will optimize treatment with AZT or combination therapy. The measurement of AZT-TP remains difficult because of the interference from endogenous nucleoside triphosphates. In this study, the pharmacokinetics of AZT and its phosphorylated metabolites was determined in hPBMCs.
2. Materials and methods
2.1. Materials and subjects
Zidovudine tablets (300 mg) were purchased from Northeast Pharmaceutical Group Shenyang No. 1 Pharmaceutical Co., Ltd. (Shenyang, China). Reference material of Zidovudine was purchased from Yidu HEC Pharma Co., Ltd. (Wuhan, China). Reference material of Zidovudine phosphate compounds was purchased from Moravek Biochemicals (CA, USA). All other reagents were commercially available and of analytical grade.
Twelve healthy Chinese male subjects were recruited and assessed for inclusion in the study. The subjects were underwent a standardized screening procedure to confirm their eligibility. The demographic characteristics of the study group are given in Table 1. All subjects gave their written informed consent to participate in the study after they had been well informed about the study objectives, method and possible risks. After a physical examination, subjects with any abnormality of the cardiovascular, respiratory, abdominal or central nervous system were excluded. Blood pressure (BP) and heart rate (HR) were measured. Additionally, an electrocardiogram was conducted and a general examination of the subject was performed to exclude any illness or abnormality.
Table 1.
Baseline demographic characteristics of volunteers.
| Parameter | Valuea | Range |
|---|---|---|
| Age (year) | 23.1±1.5 | 20–28 |
| Height (m) | 1.7±0.1 | 1.60–1.84 |
| Body weight (kg) | 64.8±3.0 | 49–78 |
Each value represents the Mean±SD, n=12.
2.2. Study design
This study was carried out in accordance with the International Conference on Harmonization (ICH) guidance on general considerations for clinical trials14. The dose in this study was selected based on the daily dosage regimen of AZT (600 mg) according to the terms of safety and therapeutic efficacy. The pharmacokinetics of AZT was determined following the administration of a single oral dose of 600 mg of AZT. An open-label design was used to evaluate the pharmacokinetics of AZT in the selected subjects after an overnight fast of at least 12 h. They received a single oral dose of 600 mg AZT followed by 240 mL of water. They received standardized meals 4 h and 10 h after drug administration.
2.3. Sample collection
For the pharmacokinetic assessments, blood samples (20 mL) were collected from an indwelling intravenous catheter inserted into the antecubital vein of the forearm of each subject prior to drug administration (blank), then at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8 and 10 h after drug administration in heparinized tubes. The blood samples were centrifuged and the plasma was harvested and transferred to polystyrene microcentrifuge tubes and stored at –20 °C until assay.
The remaining blood cells without plasma are used for the separation of mononuclear cells by the Ficoll-Hypaque density-gradient centrifugation method. First, the blood cells were diluted (1:1 blood cells/diluted solution). Second, a volume of 3–4 mL Ficoll-Hypaque solution was added into 15-mL conical centrifuge tubes using a sterile pipet, then the diluted blood cells (6–8 mL) were slowly layered by placing the tip of the pipet containing the diluted blood cells 0.7–1.5 cm above the level of Ficoll-Hypaque solution. Third, the tube was centrifuged at 20 °C in a rotor at 2000 rpm (900×g) for 15 min with no break. Last, the mononuclear lymphocyte cell layer was transferred to a centrifuge tube. The layer will appear as a white, cloudy band between the blood cells and the Ficoll-Hypaque layers. Then the cells were washed by adding Hanks' balanced salt solution (3 times the volume of the mononuclear cell layer) and centrifuged for 20 min at 1800 rpm. The precipitate is the mononuclear cells. After removing the supernatant, the mononuclear cells were re-suspended in 5 mL Hanks' balanced salt solution. The cells were counted by using 100 μL cell resuspension solution, and the remaining cell resuspension solution was used to recover the mononuclear cells by centrifugation. To store the cells, methyl alcohol–water (7:3, –20 °C) was added to lyse the cells and 50 mmol/L sodium orthovanadate was added as a phosphatase inhibitor. The cells were stored at –80 °C until use. The mononuclear cells were used for concentration determination of AZT, AZT-MP, AZT-DP and AZT-TP by a validated LC-MS/MS method.
2.4. Bioanalytic assessments
The level of AZT and its phosphorylated metabolites in plasma and mononuclear cells was determined by a validated LC–MS/MS method15. Briefly, the separation of the analytes was achieved on a Phenomenex Gemini-C18 column with 0.2% formic acid solution and methanol as the mobile phase. AZT was detected by electrospray ionization-tandem mass spectroscopy in the multiple-reaction-monitoring positive mode. The precursor-product ion transitions monitored were m/z 267.9→126.8 for zidovudine and m/z 247.9→126.8 for the internal standard tinidazole. The calibration curve of AZT-TP was established over the range of 0.1125–82.05 pg/μL (r2=0.9941). The lower limit of quantification (LLOQ) was 0.1125 pg/μL. The intra- and inter-day precision (RSD) was less than 8.0%, the average extraction recoveries and the matrix effects were satisfied with the requirements of biological sample measurement. The concentration of the quality control samples was 1.013, 9.117 and 82.05 pg/μL. The absence of interference of AZT with tinidazole was verified.
3. Results and discussion
The study enrolled 12 subjects and all of them completed the study. Safety profiles and pharmacokinetic characteristics were assessed in all subjects who completed the study. In the present study, measures were taken to protect the safety of the subjects, including prolonging the time of lying in bed and remaining in the clinical research unit, and enhancing the clinical monitoring and nursing. All subjects were negative for hepatitis and HIV. None of the 12 subjects had hypersensitivity to AZT.
None of the enrolled subjects showed any signs of serious adverse drug reactions (ADRs) during or after the study. AZT was well tolerated in the participating subjects. The most commonly reported ADRs associated with AZT administration were headache (one report) and nausea (one report), which are well-known ADRs associated with AZT. Physical examination, electrocardiograms and laboratory tests revealed no clinically significant changes.
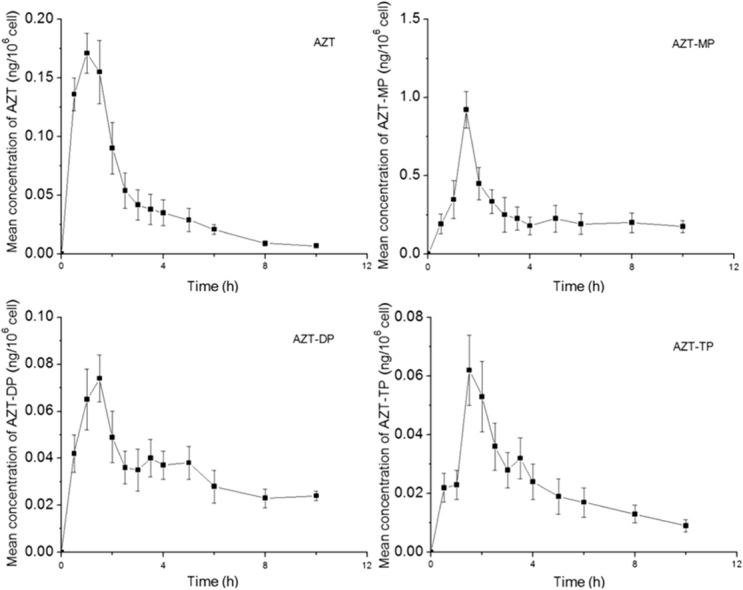
The mean linear and plasma concentration-time curve of AZT administered orally as a single dose are presented in Fig. 1. The mean linear and intracellular concentration-time curve of AZT, AZT-MP, AZT-DP and AZT-TP are presented in Fig. 2. The average non-compartmental pharmacokinetic parameters of AZT, AZT-MP, AZT-DP and AZT-TP in hPBMCs are summarized in Table 2. Pharmacokinetic parameters of AZT in plasma are summarized in Table 3.
Figure 1.

Plasma concentration-time curve of AZT in healthy Chinese subjects after a single-dose oral administration of 600 mg AZT. Values are Mean±SD, n=12.
Figure 2.

Concentration-time curves of AZT and its phosphate metabolites in hPBMCs after a single-dose oral administration of 600 mg AZT. Values are Mean±SD, n=12.
Table 2.
Pharmacokinetic parameters of AZT and its phosphorylated metabolites in HPBMCs after a single-dose oral administration of 600 mg AZT.
| Parameter | AZT | AZT-MP | AZT-DP | AZT-TP |
|---|---|---|---|---|
| Cmax (ng/106 cell) | 0.175±0.018 | 0.922±0.115 | 0.076±0.010 | 0.062±0.012 |
| Tmax (h) | 1.083±0.289 | 1.500±1.500 | 1.417±0.195 | 1.583±0.289 |
| AUC0-t (ng⋅h/106 cell) | 0.371±0.193 | 2.548±0.656 | 0.345±0.055 | 0.223±0.043 |
| AUC0-∞ (ng⋅h/106 cell) | 0.396±0.189 | 6.343±2.964 | 0.615±0.161 | 0.279±0.069 |
| t1/2 (h) | 2.493±0.638 | 13.428±9.611 | 8.285±2.840 | 4.240±0.977 |
| MRT0-t (h) | 4.941±19.287 | 4.190±0.347 | 0.792±12.028 | 3.872±0.235 |
| MRT0-∞ (h) | 14.853±32.375 | 18.715±12.856 | 14.270±6.350 | 6.328±1.137 |
Each value represents the mean±SD, unless otherwise specified; n=12.
Table 3.
Pharmacokinetic parameters of AZT in plasma after a single-dose administration of 600 mg AZT.
| Parameter | AZT |
|---|---|
| Cmax (μg/L) | 3505.176±362.932 |
| Tmax (h) | 0.583±0.195 |
| AUC0-t (μg·h/L) | 7050.142±1275.517 |
| AUC0-∞ (μg·h/L) | 7167.961±1267.624 |
| t1/2 (h) | 2.022±0.946 |
| MRT0-t (h) | 2.197±0.253 |
| MRT0-∞ (h) | 2.404±0.216 |
| Vd/F (L) | 263.908±161.840 |
| CL/F (L/h) | 86.092±14.962 |
Each value represents the mean±SD, unless otherwise specified, n=12.
The present study evaluated the pharmacokinetics of AZT in plasma and pharmacokinetics of AZT, AZT-MP, AZT-DP and AZT-TP in hPBMCs. After oral administration of AZT, its appearance in plasma was rapid and its plasma concentration peaked within 0.583 h; the intracellular concentration of AZT, AZT-MP, AZT-DP and AZT-TP peaked within 1.083, 1.500, 1.417 and 1.583 h, respectively, and these differences result from the distribution and transformation of AZT in vivo. AZT was eliminated with an apparent t1/2 of 2.493 h in hPBMCs, and was longer than that of plasma (2.022 h) due to transfer of AZT from plasma to cells. AZT-MP, AZT-DP and AZT-TP were eliminated with t1/2 of 13.428, 8.285 and 4.24 h respectively, indicating that AZT in plasma was eliminated rapidly, and metabolized to AZT-MP, AZT-DP and AZT-TP in hPBMCs rapidly after oral administration. The AUC value of AZT-MP was 2.548 (ng⋅h)/106cell, more than that of other phosphate metabolites and the parent drug in hPBMCs after oral administration. On the other hand, the plasma concentration of the phosphate metabolites was not quantifiable, indicating AZT was metabolized in hPBMCs instead of plasma. All NRTI antiretroviral agents, while differing in pharmacokinetics, toxicity and efficacy, require conversion to the triphosphate form in order to inhibit viral replication. Efficacy of this class of compounds depends on many factors including parent drug pharmacokinetics (absorbance, clearance, etc.), intracellular metabolism factors such as transport to the cell, multi-step phosphorylation and de-phosphorylation or other enzymatic processes, and the activation status of cells16. AZT was converted to AZT-MP in cells by thymidine kinase, then to AZT-DP by thymidylate kinase, and further to its active form of AZT-TP presumably by pyrimidine nucleoside diphosphate kinase. Previous studies have shown that intracellular concentrations of AZT-TP correlated with anti-viral activity and immunological response to therapy17. No AZT-TP was detected during the incubation in either liver or heart mitochondria18. This may be due to the limited time frame in which isolated mitochondria may be studied and to the poor reactivity of AZT-MP with thymidylate kinase. In non-mitotic cells, like those found in heart and liver, no thymidine kinase 1 is expressed, and the only route available for these cells to phosphorylate thymidine is by the mitochondrial thymidine kinase 2 pathways19. AZT, by acting to inhibit thymidine kinase 2, may greatly reduce the conversion of thymidine to AZT-MP. Reduction in the production of AZT-MP directly results in a reduction in AZT-TP production, as AZT-MP is a necessary precursor to AZT-TP. As AZT-TP becomes limiting, mitochondrial DNA replication slows, and eventually mitochondrial DNA depletion is evident. Further evaluation of this potential mechanism will require studies to correlate the depletion of thymidine triphosphate pools with cytotoxicity, mitochondrial toxicity and mitochondrial DNA depletion, perhaps in hepatocyte or cardiomyocyte cell culture. These pharmacokinetic studies on the metabolism of AZT to AZT-TP in hPBMCs should lead to a better understanding of the pharmacokinetic and pharmacodynamic characteristics of this drug class, and better treatment of viral infections.
4. Conclusions
This study demonstrated that AZT phosphate metabolites (AZT-MP, AZT-DP and AZT-TP) are not present in measurable levels in plasma, and AZT was eliminated rapidly, which may lead to an inaccurate understanding of its pharmacodynamic characteristics. These results suggest that determination of AZT-TP in hPBMCs is more accurate in determining pharmacokinetic and pharmacodynamics profiles.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Beijing, China) for Project No. 81102499 and the Fundamental Research Funds for the Central Universities of Central South University (No. 2015zzts286).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Kakuda T.N. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther. 2000;22:685–708. doi: 10.1016/S0149-2918(00)90004-3. [DOI] [PubMed] [Google Scholar]
- 2.Furman P.A., Fyfe J.A., St Clair M.H., Weinhold K., Rideout J.L., Freeman G.A. Phosphorylation of 3'-azido-3'-deoxythymidine and selective interaction of the 5'-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A. 1986;83:8333–8337. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsuya H., Broder S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2',3'-dideoxynucleosides. Proc Natl Acad Sci U S A. 1986;83:1911–1915. doi: 10.1073/pnas.83.6.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sungkanuparph S., Anekthananon T., Hiransuthikul N., Bowonwatanuwong C., Supparatpinyo K., Mootsikapun P. Guidelines for antiretroviral therapy in HIV-1 infected adults and adolescents: the recommendations of the Thai AIDS Society (TAS) 2008. J Med Assoc Thai. 2008;91:1925–1935. [PubMed] [Google Scholar]
- 5.Balzarini J., Baba M., Pauwels R., Herdewijn P., de Clerq E. Anti-retrovirus activity of 3'-fluoro- and 3'-azido-substituted pyrimidine 2',3'-dideoxynucleoside analogues. Biochem Pharmacol. 1988;37:2847–2856. doi: 10.1016/0006-2952(88)90049-4. [DOI] [PubMed] [Google Scholar]
- 6.Sommadossi JP, Valentin MA, Zhou XJ, Xie MY, Moore J, Calvez V, et al. Intracellular phosphorylation of staduvine (d4T) and 3TC with their antiviral activity in naive and zidovudine (ZDV)-experienced HIV-infected patients, abstr 262. In: Proceedings of the program and abstracts of the 5th conference on retroviruses and opportunistic infections; 1998. p. 146.
- 7.Sommadossi JP, Zhou XJ, Moore J, Havlir DV, Friedland G, Tierney C, et al. Impairment of stavudine (d4T) phosphorylation in patients receiving a combination of zidovudine (ZDV) and d4T (ACTG 290). In: Proceedings of the program and abstracts of the 5th conference on retroviruses and opportunistic infections; 1998. p. 1–5.
- 8.Benbrik E., Chariot P., Bonavaud S., Ammi-Saïd M., Frisdal E., Rey C. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. J Neurol Sci. 1997;149:19–25. doi: 10.1016/s0022-510x(97)05376-8. [DOI] [PubMed] [Google Scholar]
- 9.Mhiri C., Baudrimont M., Bonne G., Geny C., Degoul F., Marsac C. Zidovudine myopathy: a distinctive disorder associated with mitochondrial dysfunction. Ann Neurol. 1991;29:606–614. doi: 10.1002/ana.410290607. [DOI] [PubMed] [Google Scholar]
- 10.Dalakas M.C., Illa I., Pezeshkpour G.H., Laukaitis J.P., Cohen B., Griffin J.L. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med. 1990;322:1098–1105. doi: 10.1056/NEJM199004193221602. [DOI] [PubMed] [Google Scholar]
- 11.Barile M., Valenti D., Quagliariello E., Passarella S. Mitochondria as cell targets of AZT (zidovudine) Gen Pharmacol. 1998;31:531–538. doi: 10.1016/s0306-3623(98)00041-x. [DOI] [PubMed] [Google Scholar]
- 12.Shah I. Adverse effects of antiretroviral therapy in HIV-1 infected children. J Trop Pediatr. 2006;52:244–248. doi: 10.1093/tropej/fmi086. [DOI] [PubMed] [Google Scholar]
- 13.Moh R., Danel C., Sorho S., Sauvageot D., Anzian A., Minga A. Haematological changes in adults receiving a zidovudine-containing HAART regimen in combination with cotrimoxazole in Côte d'Ivoire. Antivir Ther. 2005;10:615–624. doi: 10.1177/135965350501000510. [DOI] [PubMed] [Google Scholar]
- 14.Baber N. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH) Br J Clin Pharmacol. 1994;37:401–404. doi: 10.1111/j.1365-2125.1994.tb05705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu P., Gu Z.K., Guo X., Ran L.L., Cheng H., Zhang X.Y. LC–APCI-MS–MS analysis of zidovudine in human plasma, and application to a bioequivalence study. Chromatographia. 2010;72:151–155. [Google Scholar]
- 16.Gao W.Y., Agbaria R., Driscoll J.S., Mitsuya H. Divergent anti-human immunodeficiency virus activity and anabolic phosphorylation of 2',3'-dideoxynucleoside analogs in resting and activated human cells. J Biol Chem. 1994;269:12633–12638. [PubMed] [Google Scholar]
- 17.Rodman J.H., Robbins B., Flynn P.M., Fridland A. A systemic and cellular model for zidovudine plasma concentrations and intracellular phosphorylation in patients. J Infect Dis. 1996;174:490–499. doi: 10.1093/infdis/174.3.490. [DOI] [PubMed] [Google Scholar]
- 18.McKee E.E., Bentley A.T., Hatch M., Gingerich J., Susan-Resiga D. Phosphorylation of thymidine and AZT in heart mitochondria: elucidation of a novel mechanism of AZT cardiotoxicity. Cardiovasc Toxicol. 2004;4:155–167. doi: 10.1385/ct:4:2:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johansson M., van Rompay A.R., Degrève B., Balzarini J., Karlsson A. Cloning and characterization of the multisubstrate deoxyribonucleoside kinase of Drosophila melanogaster. J Biol Chem. 1999;274:23814–23819. doi: 10.1074/jbc.274.34.23814. [DOI] [PubMed] [Google Scholar]