

Abstract
Obesity is a well-known risk factor for various cardiovascular diseases. Recent clinical data showed that overweight and obese patients have higher incidence of atrial fibrillation (AF) compared with individuals with normal body weights, but the underlying mechanisms remain to be elucidated. In this study, we investigated the effects of a high fat diet on atrial activities in mice. ICR male mice were fed a regular diet (RD) or a high fat diet (HFD) for 8 weeks. Mice fed HFD showed significantly greater body weight gains and visceral fat accumulation compared with RD mice. Under anesthetic condition, baseline arterial blood pressure and heart rate were not significantly different between RD and HFD groups. Although no spontaneous or atrial stimulation-induced atrial fibrillation was observed, this study revealed several alterations in the activities and protein levels in the atria in HFD mice. Surface electrocardiogram (ECG) revealed significantly shortened PR interval in HFD mice. In the atrial stimulation experiments, the sinoatrial (SA) node recovery time was significantly prolonged whereas the atrial effective refractory period was significantly reduced in HFD mice as compared with RD mice. Western blot showed protein levels of two major potassium channels, Kv1.5 and Kv4.2/3, were significantly increased in atria of HFD mice. These data indicate that HFD induces atrial electrophysiological remodeling in mice, which may be a potential mechanism underlying the increased risk for atrial arrhythmias in obesity and metabolic disorders.
Keywords: High fat diet, obesity, atrial arrhythmia, potassium channels, mice
Introduction
The obese population has been persistently growing in the last three decades in the United States and around the world [1,2]. High fat, high sugar, and high calorie Western style diets, sedentary life style, and increased social pressure are among the major contributors to the obesity pandemic. Obesity has become a severe public health issue and economic burden in the US and worldwide [3].
Obesity is a major risk factor for a variety of cardiovascular diseases [4-6]. Particularly, clinical data indicate that obesity is an independent risk factor for cardiac arrhythmias [7]. Atrial fibrillation (AF) is the most common atrial arrhythmia [8,9], which can compromise cardiac pump function and increase the risk for thrombosis and stroke. Recent clinical studies revealed that the incidence of AF was significantly higher in obese patients [10-12]. The precise underlying mechanisms, however, remain to be fully studied. Electrical and structural remodeling in atria is known to contribute to AF [13,14]. In this study, we examined and compared the surface electrocardiography (ECG), atrial pacing responses, and some key proteins that may contribute to cardiac remodeling in mice fed a regular diet or high fat diet.
Methods
Animal model
All protocols using animals in the experiments of this study were in full compliance with NIH guidelines and were approved by the University of South Dakota IACUC. Male ICR mice with a body weight range of 22 g to 26 g were purchased from Harlan Laboratories, Inc (Indianapolis, IN). The mice were housed in the USD Animal Resource Center with food and water ad-libitum and 12:12 light-dark cycle. Mice were randomly separated into two groups: RD group mice were fed a regular diet chow (RD, TD2920X, Harlan), which has an energy density of 3.1 Kcal/g, with 24% from protein, 60% from carbohydrate, and 16% from fat of the total calories; HFD group mice were fed a high fat diet chow (HFD, TD. 06414, Harlan), which has an energy density of 5.1 Kcal/g, with 18.4% from protein, 21.3% from carbohydrate, and 60.3% from fat of the total calories. The mice were fed these diets for 8 weeks. The food intake and body weight of the animals were weighed weekly. The body weight gain of each mouse was determined by the body weight at each week minus the initial body weight at the beginning of the diet schedule. Two independent batches of animals, containing both diet groups, were used for acute functional experiments or for tissue collection.
Hemodynamics and ECG
Anesthetized by intraperitoneal injection with saline containing urethane (2 g/kg) and α-chloralose (50 mg/kg), animals were placed on a warming pad. Tracheotomy and tracheal intubation were performed to facilitate breathing. The Lead II surface electrocardiography (ECG) was taken via a pair of electrodes that were connected to a bio-amplifier (AD Instruments Inc. Colorado Springs, CO) with Low- and High-pass filters at 1 kHz and 10 Hz, respectively. The left carotid artery was catheterized for measuring blood pressure via a pressure transducer that was coupled to a bridge amplifier (AD Instruments Inc). The signals were then recorded by a Powerlab data acquisition system (Model 8/sp, AD Instruments Inc) at the sampling rate 2 kHz.
Atrial catheterization and stimulation
In the anesthetized mice, the right jugular vein was isolated and catheterized with an octapolar electrodes catheter (CIB’er, NuMED Inc. Hopkinton, NY) [15,16]. The first two pairs of electrodes in the catheter were connected with two pre-amplifiers at Low- 10 kHz and High-pass at 30 Hz. The signals were recorded and monitored via the PowerLab system. The electrical signals were monitored while the catheter was advanced toward the right atrium. The dominant atrial electrical signals, which corresponded to the P wave in the surface ECG, indicated that both electrodes pairs were correctly placed in the right atrium. The first pair of electrodes was then switched to connect with a stimulator for atrial stimulation. The stimuli were rectangular pulses with 1 ms in duration, 1 V in amplitude with various pulse-pulse intervals (p-p), depending on experimental needs. In an attempt to induce AF, the right atrium was stimulated with p-p bursts of 50 ms, 40 ms, and 30 ms for 10 seconds each. To assess the sino-atrial node recovery time (SNRT), atrial stimulations with 100 ms p-p were applied for 30 seconds. The SNRT was measured as the time between the last stimulus and the first sinus P wave. For testing Wenckebach AV block (WC), continuous atrial stimulation was applied with decreasing p-p from 90 ms to 70 ms with 1 ms decrements. For testing atrial effect refractory period (AERP), continuous stimulation was applied with decreasing p-p from 50 ms to 30 ms at 1 ms decrements.
Western blot
Under deep anesthesia with isoflurane, animals were sacrificed by cervical dislocation. The heart and other tissues were collected and were quickly frozen on dry ice and stored at -70°C. Frozen tissues were homogenized in RIPA buffer containing 1% protease inhibitor cocktail (Pierce Inc.) and 0.1% SDS by a tissue homogenizer (BBX24, Next Advance, Inc. Averill Park, NY) in a 1.5 ml tube at 4°C. Following centrifugation, BCA assay, and adjusting protein concentration, the supernatants were mixed with 4x loading buffer containing beta mercaptoethanol (BME), boiled for 10 minutes, and subjected to standard acrylamide SDS gel electrophoresis and membrane transfer. The proteins on the membrane were blotted with appropriate primary and then secondary antibodies conjugated with fluorescent dye. The bands on the membranes were visualized by the Odyssey Imaging System (LiCor Biosciences, Lincoln, NE). The band intensity was measured using NIH ImageJ software. The quantifications were carried out using the ratio of the intensity of the protein of interest versus a loading control protein (pan-actin or GAPDH).
Data analysis
All quantified data were expressed as mean ± standard deviation (SD). Data of body weight gain over time were analyzed by ANOVA followed by the student’s t-test. Other measurements were compared between the two groups using single factor ANOVA. The statistical significances between the two groups were determined by p value equal or less than 0.05.
Results
HFD induced obesity
Compared with RD mice, mice fed HFD gained more body weight over time, which reached statistical significance at 4 weeks, which continued through the end the experiment at 8 weeks (Figure 1A and 1B). At the end of the feeding period, the HFD mice had significantly greater visceral fat compared with the RD mice (Figure 1A and 1C). The average daily food intake per mouse was 4.9 ± 0.1 g in RD group and 3.9 ± 0.2 g in HFD group. Since the calorie density of HFD (5.1 kcal/g) is greater than that of RD (3.1 kcal/g), the average daily calorie intake per mouse was significantly greater in HFD mice compared with RD mice (Figure 1E).
Figure 1.
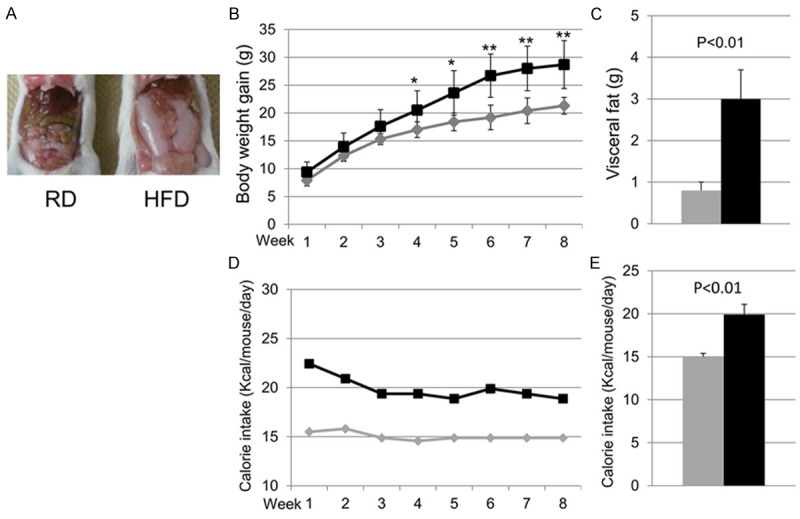
The animal models. A. Raw image of a mouse body fed regular diet (RD, left) and high fat diet (HFD, right); B. Quantification of body weight gain (the current body weight minus that at the beginning of the diet); C. Quantification of the weight (g) of visceral fat tissue at the end of 8 weeks for each diet group; D. The time course of calorie intake per mouse per day at each week; E. The average calorie intake per mouse per day over 8 weeks. In all diagrams, the gray lines or bars represent RD group and the black lines or bars represent HFD group. Data were presented as mean ± SD. For both groups, n=5. *p<0.05, **p<0.01.
General surface ECG and hemodynamics
There were no significant differences between RD and HFD groups in heart rate (HR) and arterial blood pressure (BP) (Table 1). Both groups showed regular surface ECG. No significant atrial or ventricular arrhythmias were detected (Figure 2). However, the ECG PR interval was significantly shortened, whereas the duration of the QRS complex was significantly prolonged in HFD mice as compared with RD mice (Figure 2 and Table 1).
Table 1.
Summary of hemodynamics and ECG
| RD (n=5) | HFD (n=5) | P value | |
|---|---|---|---|
| sBP (mmHg) | 112 ± 21 | 126 ± 23 | 0.38 |
| dBP (mmHg) | 67 ± 16 | 75 ± 24 | 0.57 |
| HR (bpm) | 556 ± 45 | 544 ± 26 | 0.64 |
| BCL (ms) | 108.3 ± 8.9 | 110.4 ± 5.4 | 0.67 |
| P wave width (ms) | 11.7 ± 1.9 | 10.6 ± 0.9 | 0.27 |
| PR interval (ms) | 39.5 ± 3.7 | 35.4 ± 1.7 | 0.05* |
| QRS interval (ms) | 23.8 ± 0.4 | 26.4 ± 2.1 | 0.03* |
Summary of hemodynamics and ECG measurements in RD and HFD groups. Abbreviations: sBP: systolic blood pressure; dBP: diastolic blood pressure; HR: heart rate; BCL: basal cyclic length. Sample size n=5 for each group.
Statistical significance as compared between RD and HFD groups.
Figure 2.
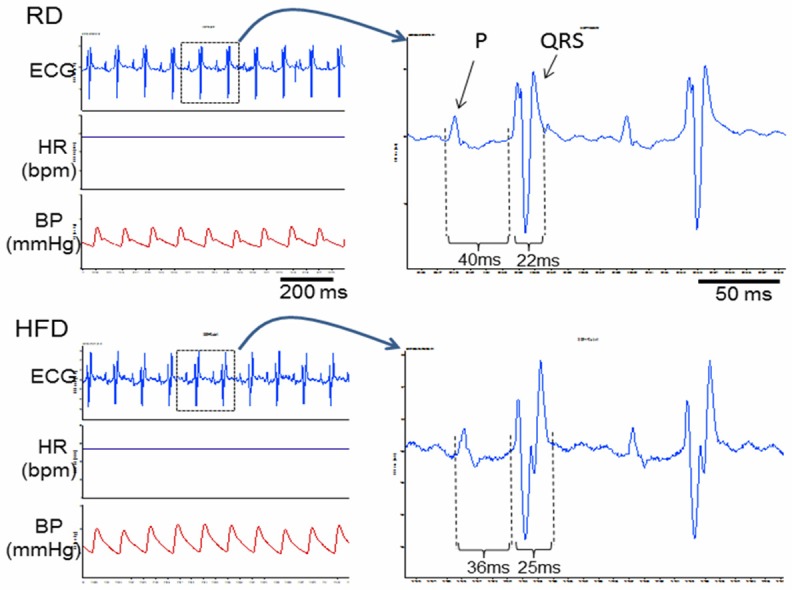
Raw tracings of ECG, HR and BP. The left panel contains raw recording tracings of surface ECG, heart rate (HR), and arterial blood pressure (BP) in a RD mouse (top) and a HFD mouse (bottom). Two cycles of ECG from the tracings of RD (top) and HFD (bottom) were enlarged in the right panel, showing the shortened PR interval and widened QRS interval in the HFD mouse as compared with that in the RD mouse.
Atrial stimulation
To test the inducibility of AF, stimulations of the right atrium with stimuli bursts were applied. No sustained AF was induced in either group. The HFD mice, however, exhibited increased atrial activities and longer time to resume the normal sinus rhythm (example tracings on the left of Figure 3). Consistent with this phenomenon, the SNRT was significantly prolonged in HFD mice as compared with RD mice (Figure 3A and 3B). Additionally, the AERP was significantly shortened in HFD mice (Figure 3D). The p-p intervals of stimuli for inducing Wenckebach AV block were not different between the two groups (Figure 3C).
Figure 3.
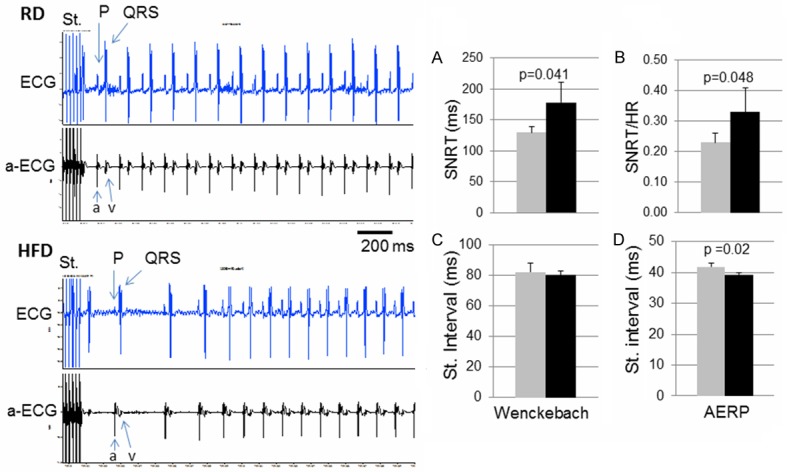
Responses to atrial stimulations. The left panel: Raw recording tracings of responses in surface ECG and atrial ECG following atrial stimulation (St.) bursts, in a RD mouse (top) and a HFD mouse (bottom). In the atrial ECG, “a” indicates atrial activity and “v” indicates ventricular activity. (A and B) the SA node recovery time (SNRT) (A) or normalized SNRT by heart rate (B) following atrial stimulation. (C and D) the stimulation p-p interval value that induced a Wenckebach AV block (C) or that revealed the atrial effective refractory period (AERP) (D). Data were presented as mean ± SD. In the diagrams, grey bars represent RD group and black bars represent HFD group. In each group, n=5.
Western blot
To explore the potential mechanisms underlying the electrophysiological changes, protein levels of some critical ion channels were determined in the heart samples. In atrial samples, the antibody against Kv1.5, the critical potassium channel involved in atrial repolarization, detected two major bands at the molecular weights of ~55 kDa and ~75 kDa, which were both significantly increased in HFD as compared with RD (Figure 4A). In the ventricular samples, the ~55 kDa band of Kv1.5 was undetectable and the ~75 kDa band was significantly increased in HFD as compared with RD (Figure 4B). In skeletal muscle samples, both ~55 kDa and ~75 kDa Kv1.5 bands were detected but there was no significant change between the two groups (Figure 4C).
Figure 4.
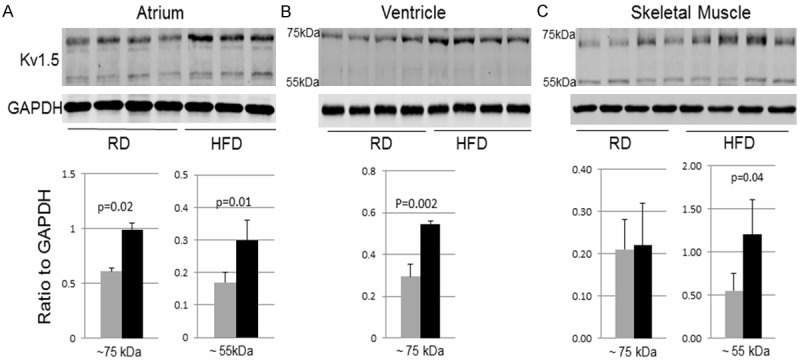
Western blot of Kv1.5. The raw images (top) and quantifications (bottom) of Western blot detecting Kv1.5 in atrial, ventricular, or skeletal muscle at the ~75 kDa (left graph for each tissue) or ~55 kDa band (right graph for each tissue). Data were presented as mean ± SD. In the diagrams, the grey bars represent the RD group and the black bars represent the HFD group. In the atrial diagram, n=4 in RD group and n=3 in HFD group. In the ventricular and skeletal muscle diagrams, n=4 in both RD and HFD groups.
Kv4.2/3 potassium channels, which are involved in the early repolarization in the heart, were detected as two major bands at ~60 kDa and ~80 kDa by Western blot. While there was no significant difference in ventricular and skeletal muscle samples, the ~80 kDa band was significantly increased in atrial samples in the HFD group as compared with the RD group (Figure 5A).
Figure 5.
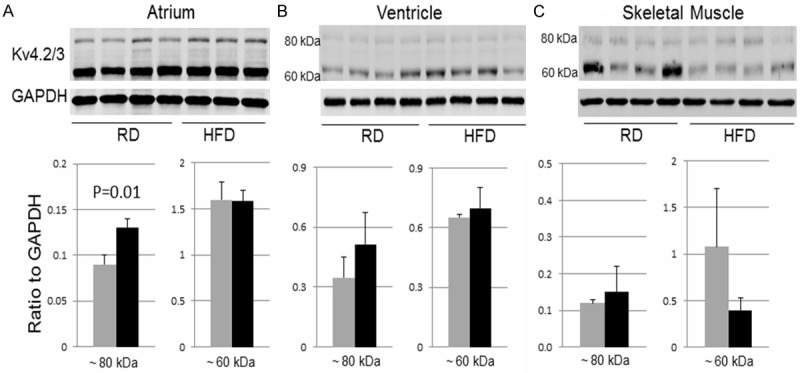
Western blot of Kv4.2/3. The raw images (top) and quantifications (bottom) of Western blot for Kv4.2/3 in atrial, ventricular, or skeletal muscle samples at ~80 kDa (left graph in each tissue) or ~60 kDa band (right graph in each tissue). Data were presented as mean ± SD. In the diagrams, the grey bars represent the RD group and the black bars represent the HFD group. For atrial samples, n=4 in RD group and n=3 in HFD group. For ventricular and skeletal muscle samples, n=4 in both RD and HFD groups.
TGFβ plays a critical role in cardiac remodeling and arrhythmia [17]. In addition, altered connexin43, a components of gap junction critical for cardiac electrical conduction, is known to contribute to cardiac arrhythmia [18]. In this study, however, Western blot did not detect any significant changes in protein levels of TGFβ or connexin43 in atrial and ventricular samples between RD and HFD groups (Figure 6).
Figure 6.
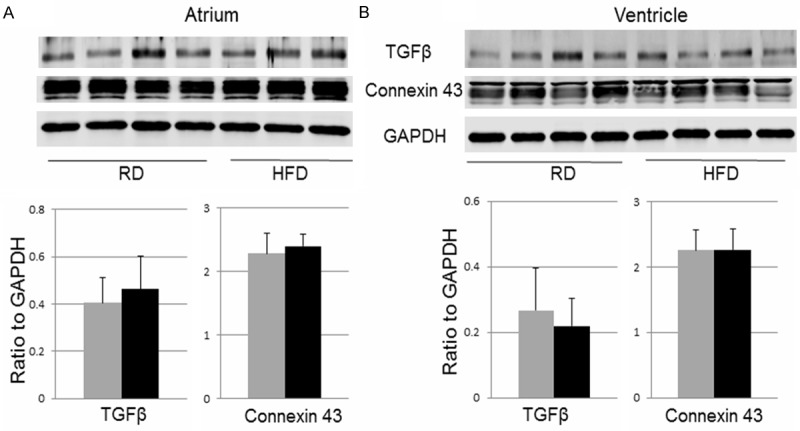
Western blot of TGFβ and connexin43. The raw images (top) and quantifications (bottom) of Western blot for TGFβ and connexin43 in atrial (left) and ventricular (right) samples. Data were presented as mean ± SD. In the diagrams, the grey bars represent the RD group and the black bars represent the HFD group. For atrial samples, n=4 in RD group and n=3 in HFD group. For ventricular and skeletal muscle samples, n=4 in both RD and HFD groups.
Discussion
Clinical evidence has indicated an increased risk for atrial fibrillation in obese patients but the underlying mechanisms are elusive. The attempt of this study was to examine the potential effect of HFD on electrophysiological activities in mice. Although there was neither spontaneous nor induced AF in mice fed a HFD for 8 weeks, the results indeed showed several significant changes in cardiac electrophysiological activities in HFD mice. In particular, the atrial stimulation experiments showed an increase in atrial activities, a prolonged SNRT, and a shortened AERP in HFD mice as compared with RD mice. As for the potential underlying mechanisms of these alterations, data show the significant increases in protein levels of Kv1.5 and Kv4.2/3 in atria of HFD mice. These observations provide novel information lending to a better understanding and further investigation of the potential link between high fat diet, obesity, and atrial arrhythmia.
One of the well-accepted mechanisms underlying AF is the formation of re-entry circuits, mainly caused by an altered electrical refractory period and/or conduction velocity [8,9]. Potassium channels play a critical role in determining the myocardial repolarization and refractory periods. Enhancing the function of potassium channels accelerates repolarization and consequently shortens the refractory period leading to AF, while the blockade of potassium channels prolongs the refractory period and thus exerts antiarrhythmic effects [8,19]. As shown in our study, the protein levels of Kv1.5 and Kv4.2/3, crucial potassium channels that determine the repolarization process in the heart, were significantly increased in atria of HFD mice. As a functional result of this remodeling, the AERP was reduced in the HFD. Consistent with these pro-atrial arrhythmic changes, we indeed observed the increased atrial activities following atrial stimulation in HFD mice. For the potential reasons why the spontaneous and inducible AF were not observed in this study, one determining variable may be the period of HFD feed. In this study, mice were fed HFD for 8 weeks, shorter than other studies with timelines up to 20 weeks on HFD [20,21]. It is possible that in the period of 8 weeks on HFD, the animals may be at an early stage of obesity, in which the electrical remodeling is underway but not yet to capable of developing sustained or inducible AF.
Kv1.5 is considered an atrial selective potassium channel that mediates a major part of repolarization in an action potential (AP) in atrial cardiomyocytes in humans [22]. Blockage of Kv1.5 is a potential selective strategy for treatment of AF without compromising ventricular function [22]. In this study, Western blot detected two major bands of Kv1.5. The ~55 kDa band, which was only present in atrial samples but not in ventricular samples, was significantly increased in HFD as compared with RD. The ~75 kDa band of Kv1.5 was detected and shown increases in both atrial and ventricular samples. Kv1.5 is also expressed in ventricles in mice [23,24] but the significance of the altered Kv1.5 in ventricles in HFD mice remains to be defined. Our Western blot detected two major bands for Kv4.2/3, the potassium channels that contribute to the phase 1 repolarization of AP both in atria and ventricles [25]. Interestingly, the ~80 kDa band of Kv4.2/3 was significantly increased only in HFD atrial samples but not in ventricular samples as compared with RD samples. These data suggest the Kv1.5 and Kv4.2/3 could be potential therapeutic targets for obesity related atrial arrhythmia.
It should be pointed out that the role of potassium currents in the development of arrhythmia is rather complicated. Evidence has shown that in tissues from patients with disease related AF, protein levels of potassium channels, including Kv1.5, were decreased [26]. For example, although Kv1.5 is considered a potential selective target for AF, experimental and clinical trial data are not consistent [22]. There is evidence that Kv1.5 loss of function by genetic mutation can cause AF [27]. All these data indicate the complexity of potassium channels in the pathophysiology of atrial arrhythmia. Interestingly, a recent study reported a decrease in protein and mRNA levels of Kv1.5 in ventricles with a prolonged QT interval in mice fed HFD for 20 weeks [28]. It would be interesting to further compare whether Kv1.5 protein levels were increased early and then decreased later in the heart in HFD regimen. Nonetheless, these data indicate that Kv1.5 potassium channels are sensitive molecules in responding to metabolic disorders.
An interesting question to be addressed is why HFD causes the atria specific remodeling of these potassium channels. A recent study using HFD fed rats showed an atrial specific alteration in autonomic innervations [29], which may well be a contributor to the atria specific remodeling. However, the question remains: why does HFD induce these alterations specifically in atria but not in ventricles? We hypothesize that HFD and obesity increase body mass and alter renal function, which consequently increases in volume load. As atria are more sensitive to the volume stress, they therefore undergo electrophysiological and structural remodeling prior to the changes in ventricles. Our ongoing study is designed to test this potential mechanism underlying the HFD-induced atria-specific remodeling.
Cardiac electrical properties and function are determined by many molecular and structural factors. In addition to the ion channel remodeling, tissue fibrosis and altered gap junction are also implicated in AF [30]. In addition to potassium channels, we also measured the protein levels of TGFβ and Connexin43, two major factors impacted in cardiac fibrosis and gap junction remodeling [17,31-33], but found no significant change in atrial or ventricular samples in HFD mice at 8 weeks. Alterations in other ion channels, including sodium channels, calcium channels, and other types of potassium channels, will also be investigated in HFD mice as compared to RD in our future study. In addition, it is also important to further observe and compare the atrial activities and responses in RD and HFD regimens in a conscious condition.
Collectively, the results of this study provide novel and potentially important information regarding the increased risk for AF in obesity. Mounting clinical studies are implicating the link of overweight and obesity to AF [10,11]. Further investigation of the underlying mechanisms involved in the altered atrial electrical activities indicated by this study may provide a better understanding of these issues leading to new strategies for prevention and treatments of obesity related cardiac arrhythmia.
Acknowledgements
This study was supported by NIH grant No. 1R15HL118696, NIH SD BRIN grant No P20 RR016479, and USD Sanford School of Medicine BBS bridge grants and the graduate program.
References
- 1.Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- 2.McLellan F. Obesity rising to alarming levels around the world. Lancet. 2002;359:1412. doi: 10.1016/S0140-6736(02)08397-6. [DOI] [PubMed] [Google Scholar]
- 3.Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378:815–825. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- 4.Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, Lang CC, Rumboldt Z, Onen CL, Lisheng L, Tanomsup S, Wangai P Jr, Razak F, Sharma AM, Anand SS INTERHEART Study Investigators. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366:1640–1649. doi: 10.1016/S0140-6736(05)67663-5. [DOI] [PubMed] [Google Scholar]
- 5.Powell BD, Redfield MM, Bybee KA, Freeman WK, Rihal CS. Association of obesity with left ventricular remodeling and diastolic dysfunction in patients without coronary artery disease. Am J Cardiol. 2006;98:116–120. doi: 10.1016/j.amjcard.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 6.Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, Sowers J. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment--a position paper of the The Obesity Society and The American Society of Hypertension. Obesity (Silver Spring) 2013;21:8–24. doi: 10.1002/oby.20181. [DOI] [PubMed] [Google Scholar]
- 7.Anand RG, Peters RW, Donahue TP. Obesity and dysrhythmias. J Cardiometab Syndr. 2008;3:149–154. doi: 10.1111/j.1559-4572.2008.00003.x. [DOI] [PubMed] [Google Scholar]
- 8.Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation. 2011;124:2264–2274. doi: 10.1161/CIRCULATIONAHA.111.019893. [DOI] [PubMed] [Google Scholar]
- 9.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 10.Wanahita N, Messerli FH, Bangalore S, Gami AS, Somers VK, Steinberg JS. Atrial fibrillation and obesity--results of a meta-analysis. Am Heart J. 2008;155:310–315. doi: 10.1016/j.ahj.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Wang TJ, Parise H, Levy D, D’Agostino RB Sr, Wolf PA, Vasan RS, Benjamin EJ. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292:2471–2477. doi: 10.1001/jama.292.20.2471. [DOI] [PubMed] [Google Scholar]
- 12.Magnani JW, Hylek EM, Apovian CM. Obesity begets atrial fibrillation: a contemporary summary. Circulation. 2013;128:401–405. doi: 10.1161/CIRCULATIONAHA.113.001840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nattel S, Li D, Yue L. Basic mechanisms of atrial fibrillation--very new insights into very old ideas. Annu Rev Physiol. 2000;62:51–77. doi: 10.1146/annurev.physiol.62.1.51. [DOI] [PubMed] [Google Scholar]
- 14.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nature Reviews Drug Discovery. 2012;11:275–291. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 15.Wakimoto H, Maguire CT, Kovoor P, Hammer PE, Gehrmann J, Triedman JK, Berul CI. Induction of atrial tachycardia and fibrillation in the mouse heart. Cardiovasc Res. 2001;50:463–473. doi: 10.1016/s0008-6363(01)00264-4. [DOI] [PubMed] [Google Scholar]
- 16.Stockigt F, Brixius K, Lickfett L, Andrie R, Linhart M, Nickenig G, Schrickel JW. Total betaadrenoceptor knockout slows conduction and reduces inducible arrhythmias in the mouse heart. PLoS One. 2012;7:e49203. doi: 10.1371/journal.pone.0049203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verheule S, Sato T, Everett Tt, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ, Gollob MH. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236–244. doi: 10.1161/CIRCULATIONAHA.110.961227. [DOI] [PubMed] [Google Scholar]
- 19.Dobrev D, Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010;375:1212–1223. doi: 10.1016/S0140-6736(10)60096-7. [DOI] [PubMed] [Google Scholar]
- 20.Gronros J, Wikstrom J, Brandt-Eliasson U, Forsberg GB, Behrendt M, Hansson GI, Gan LM. Effects of rosuvastatin on cardiovascular morphology and function in an ApoE-knockout mouse model of atherosclerosis. Am J Physiol Heart Circ Physiol. 2008;295:H2046–2053. doi: 10.1152/ajpheart.00133.2008. [DOI] [PubMed] [Google Scholar]
- 21.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–2828. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrlich JR, Nattel S. Atrial-selective pharmacological therapy for atrial fibrillation: hype or hope? Curr Opin Cardiol. 2009;24:50–55. doi: 10.1097/HCO.0b013e32831bc336. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Guo W, Xu H, Hood R, Benedict AT, Nerbonne JM. Functional expression of a GFPtagged Kv1.5 alpha-subunit in mouse ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H1955–1967. doi: 10.1152/ajpheart.2001.281.5.H1955. [DOI] [PubMed] [Google Scholar]
- 24.London B, Guo W, Pan X, Lee JS, Shusterman V, Rocco CJ, Logothetis DA, Nerbonne JM, Hill JA. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridinesensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001;88:940–946. doi: 10.1161/hh0901.090929. [DOI] [PubMed] [Google Scholar]
- 25.Grant AO. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2:185–194. doi: 10.1161/CIRCEP.108.789081. [DOI] [PubMed] [Google Scholar]
- 26.Nattel S, Frelin Y, Gaborit N, Louault C, Demolombe S. Ion-channel mRNA-expression profiling: Insights into cardiac remodeling and arrhythmic substrates. J Mol Cell Cardiol. 2010;48:96–105. doi: 10.1016/j.yjmcc.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 27.Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776–785. doi: 10.1093/cvr/cvq398. [DOI] [PubMed] [Google Scholar]
- 28.Huang H, Amin V, Gurin M, Wan E, Thorp E, Homma S, Morrow JP. Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J Mol Cell Cardiol. 2013;59:151–158. doi: 10.1016/j.yjmcc.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCully BH, Hasan W, Streiff CT, Houle JC, Woodward WR, Giraud GD, Brooks VL, Habecker BA. Sympathetic cardiac hyperinnervation and atrial autonomic imbalance in dietinduced obesity promote cardiac arrhythmias. American journal of physiology Heart and circulatory physiology. 2013;305:H1530–1537. doi: 10.1152/ajpheart.00196.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 31.Burstein B, Nattel S. Atrial structural remodeling as an antiarrhythmic target. J Cardiovasc Pharmacol. 2008;52:4–10. doi: 10.1097/FJC.0b013e3181668057. [DOI] [PubMed] [Google Scholar]
- 32.Van Norstrand DW, Asimaki A, Rubinos C, Dolmatova E, Srinivas M, Tester DJ, Saffitz JE, Duffy HS, Ackerman MJ. Connexin43 mutation causes heterogeneous gap junction loss and sudden infant death. Circulation. 2012;125:474–481. doi: 10.1161/CIRCULATIONAHA.111.057224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover: implications for cardiac excitability. Circ Res. 2000;86:723–728. doi: 10.1161/01.res.86.7.723. [DOI] [PubMed] [Google Scholar]