

Summary
Patients with an HNF1BS148L/+ mutation (MODY5) typically exhibit pancreatic hypoplasia. However, the molecular mechanisms are unknown due to inaccessibility of patient material and because mouse models do not fully recapitulate MODY5. Here, we differentiated MODY5 human-induced pluripotent stem cells (hiPSCs) into pancreatic progenitors, and show that the HNF1BS148L/+ mutation causes a compensatory increase in several pancreatic transcription factors, and surprisingly, a decrease in PAX6 pancreatic gene expression. The lack of suppression of PDX1, PTF1A, GATA4, and GATA6 indicates that MODY5-mediated pancreatic hypoplasia is mechanistically independent. Overexpression studies demonstrate that a compensatory increase in PDX1 gene expression is due to mutant HNF1BS148L/+ but not wild-type HNF1B or HNF1A. Furthermore, HNF1B does not appear to directly regulate PAX6 gene expression necessary for glucose tolerance. Our results demonstrate compensatory mechanisms in the pancreatic transcription factor network due to mutant HNF1BS148L/+ protein. Thus, patients typically develop MODY5 but not neonatal diabetes despite exhibiting pancreatic hypoplasia.
Graphical Abstract
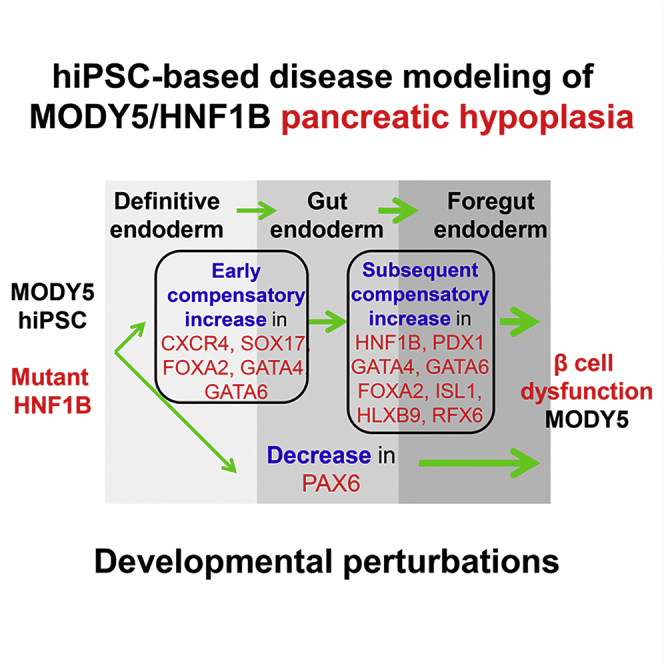
Highlights
-
•
HNF1BS148L/+ mutation elicits a compensatory increase in DE and pancreatic genes
-
•
MODY5-mediated pancreatic hypoplasia is independent of PDX1, PTF1A, GATA4, and GATA6
-
•
HNF1BS148L mutation directly causes a compensatory increase in PDX1 gene expression
-
•
HNF1BS148L/+ mutation limits PAX6 expression and consequently leads to MODY5
Kulkarni, Teo, and colleagues differentiated MODY5-hiPSCs into pancreatic progenitors and demonstrated that HNF1BS148L/+ mutation elicits a compensatory mechanism in the pancreatic transcription factor network. They report that MODY5-mediated pancreatic hypoplasia is independent of PDX1, PTF1A, GATA4, and GATA6. In addition, a decrease in PAX6 gene expression may lead to MODY5 development.
Introduction
Maturity-onset diabetes of the young (MODY) is a type of autosomal dominant monogenic diabetes classically characterized by non-ketotic, non-insulin-dependent diabetes occurring before the age of 25 years (Teo et al., 2013a). Many MODY genes are pancreatic developmental transcription factors, with the notable exception of GCK, CEL, and INS. Although MODY was discovered more than two decades ago, the molecular mechanisms underlying transcription factor MODY in humans is still largely unclear because mouse models do not fully recapitulate this disease (Maestro et al., 2007) and the lack of a suitable human model.
Patients with MODY5 commonly exhibit pancreatic hypoplasia due to an autosomal dominant mutation in the HNF1B gene (Edghill et al., 2006). HNF1B is a member of the complex pancreatic transcription factor network which includes HNF1A and HNF4A. HNF1B+ cells in the pancreatic trunk epithelium are multipotent pancreatic progenitors which play a role in endocrine and exocrine development (Haumaitre et al., 2005). Although MODY5 was discovered in 1997, to date the effects of an autosomal dominant mutation in HN1B on human pancreas development and the molecular mechanisms underlying pancreatic hypoplasia remain not fully understood. MODY5 phenotype in humans cannot be phenocopied by Hnf1β+/− mice since they do not develop diabetes (Haumaitre et al., 2005), highlighting the need for a suitable human model to study the perturbations in pancreas development (Teo et al., 2013a).
Human-induced pluripotent stem cell (hiPSC)-derived pancreatic cells now provide an excellent opportunity to study this monogenic diabetes phenotype. In this study, we established a well-controlled MODY5-hiPSC pancreatic differentiation model to elucidate the molecular mechanisms underlying MODY5 pancreatic hypoplasia. We differentiated four control and six mutant hiPSC lines, and observed that mutant HNF1BS148L/+ elicits a compensatory increase in definitive endoderm (DE) and pancreatic transcription factor gene expression. Mutant HNF1BS148L directly accounted for an increased PDX1 gene expression. These pancreatic transcription factor network perturbations could possibly explain the occurrence of maturity-onset diabetes rather than neonatal diabetes despite pancreatic hypoplasia. Importantly, pancreatic PAX6 gene expression, known to be important for pancreatic β-cell function, was distinctly down-regulated in MODY5 pancreatic progenitors, which in part explains the early-onset diabetes and pancreatic hypoplasia in MODY5 patients.
Results
Establishment of a Human Stem Cell Model for MODY5
We recently reported the derivation of several hiPSC lines from a MODY5 family (Haldorsen et al., 2008) for in vitro disease modeling of monogenic diabetes (Teo et al., 2013b). Our experimental design included a “node” comprising a healthy family member (N805-6) and two members of the family with an autosomal dominant (S148L) mutation in the HNF1B gene, of which one of them has developed diabetes (N805-2) whereas the other has not (N805-1) (Figure 1A). Three independent hiPSC lines from each subject were established (iN805-6A/B/C, iN805-1A/B/C, and iN805-2A/B/C) and verified for the absence or presence of the S148L (C443T in exon 2) mutation in the HNF1B gene (Figures 1B and S1A).
Figure 1.

Establishment of a Human Stem Cell Model for MODY5
(A) N805 family node pedigree for MODY5 family. Square denotes male, circles denote females, solid symbol denotes diabetes, NN denotes no mutation, and NM denotes mutation. N805-6 is a family control, N805-1 has HNF1BS148L/+ phenotype but no diabetes, whereas N805-2 has HNF1BS148L/+ phenotype and is diabetic.
(B) S148L (C443T in exon 2) mutation (or lack thereof) in HNF1B has been verified in iN805 hiPSCs at least three times.
(C–E) Schematic representation of a human pancreatic differentiation protocol for disease modeling of MODY5 in vitro. (C) CHIR99021 (CHIR) is a GSK-3 inhibitor whereas LY294002 (LY) is a PI3K inhibitor. Expression of (D) pluripotency (OCT4 and NANOG), mesendoderm (EOMES and MIXL1), definitive endoderm (CXCR4, SOX17, GATA4, and GATA6) and (E) foregut endoderm (ISL1, HLXB9, HNF1B, PAX6, SOX9, PDX1, PTF1A, and RFX6) markers in control hiPSCs differentiated for 17 days. All error bars indicate SD of three biological replicates in an independent experiment.
(F) Gene-expression heatmap of representative pluripotency, mesendoderm, definitive endoderm, and pancreas-related genes in undifferentiated (first two columns) and day-10 differentiated (last two columns) control hiPSCs (up-regulation in red, down-regulation in blue). The colors in the heatmap depict gene expression in units of SD from the mean across all samples. Asterisks indicate genes for which mutations are known to result in pancreatic hypoplasia/agenesis.
See also Figures S1 and S2.
We then set out to establish a human pancreatic differentiation protocol for disease modeling of MODY5 in vitro (Figure 1C) based on a chemically defined medium (no serum) (Teo et al., 2014), which is a modified version of our recently reported protocol (Teo et al., 2012, Teo et al., 2015). Careful time-course analyses of differentiated control hiPSCs (derived from AG16102) (Teo et al., 2013b) indicated that pluripotency factors OCT4 and NANOG plummet by day 3, and that hiPSCs transit through EOMES+ MIXL1+ mesendoderm before differentiating into DE marked by CXCR4, SOX17, GATA4, and GATA6 (Figure 1D).
Further differentiation toward foregut endoderm and pancreatic progenitors by day 17 revealed an up-regulation of numerous key pancreatic progenitor markers such as ISL1, HLXB9, HNF1B, PAX6, SOX9, PDX1, PTF1A, RFX6, NEUROD1, HNF6, DLK1, SOX4, and MAFB, indicating that our pancreatic differentiation protocol is suitable for studying the impact of HNF1B S148L/+ mutation on early pancreatic development and its transcriptional network (Figures 1E and S1B). Immunostaining on day 12 and fluorescence-activated cell sorting (FACS) analyses on day 17 further confirmed the protein expression of key pancreatic developmental genes (Figures S1C and S1D). Since HNF1B transcripts exhibit peak expression between days 7 and 10, we performed genome-wide microarray analyses on day-10 differentiated control hiPSCs, and confirmed the up-regulation of pancreas-related genes such as PDX1, HNF1B, ISL1, RFX6, PAX6, GATA4, GATA6, SOX9, and PTF1A, and a reciprocal down-regulation of numerous pluripotency-related genes (Figures 1F and Table S1). Gene ontology (GO) analyses on up- and down-regulated genes (fold change >2) indicated general changes in developmental processes and possibly a down-regulation of vascular development (Figure S2A).
MODY5-hiPSC-Derived Pancreatic Progenitors Exhibit a Compensatory Increase in DE and Pancreatic Markers but Down-Regulation of PAX6 Gene Expression
We first differentiated the MODY5-hiPSCs into DE (day 3), gut endoderm (days 5–7), and early foregut progenitors (day 10) (Figure 2). Hnf1β gene is expressed as early as embryonic day 8.75 (E8.75) in the mouse primitive gut/foregut (Ott et al., 1991), corresponding to ∼days 3–7 of the hiPSC in vitro differentiation. Interestingly, we observed an early compensatory increase in DE markers CXCR4, SOX17, FOXA2, GATA4, and GATA6 on day 5 and beyond (Figure 2) in the mutant hiPSCs, just when HNF1B gene is beginning to be expressed (Figure 1E; days 3–5). This suggests that the early (low) expression of mutant HNF1B S148L/+ during gut endoderm development is already causing an early compensatory increase in DE and gut endoderm markers.
Figure 2.

Transcriptional Perturbations in MODY5-hiPSC-Derived Early Pancreatic Progenitors
Gene expression of CXCR4, SOX17, FOXA2, GATA4, GATA6, and HNF1B in a non-family control and in iN805 family hiPSCs differentiated for 0, 5, 7 and 10 days. Data are representative of at least two independent hiPSC lines per subject. Replicate experiments are presented. The experiments were replicated at least twice. All error bars indicate SD of three biological replicates in an independent experiment. See also Figures S2 and S3.
Given the phenotypic similarities in pancreatic agenesis/hypoplasia caused by HNF1B, PDX1, PTF1A, GATA4, and GATA6 mutations, we hypothesized that the dorsal pancreatic agenesis in MODY5 (HNF1B S148L/+) (Haldorsen et al., 2008) could be directly linked to the down-regulation of downstream pancreatic genes GATA4, GATA6, PDX1, and PTF1A. Surprisingly, time-course transcriptional analyses of MODY5-hiPSC-derived pancreatic progenitors indicate higher gene expression of HNF1B, PDX1, GATA4, and GATA6 in mutant hiPSCs (iN805-1A/B/C and iN805-2A/B/C; three independent lines; each in biological triplicate) compared with two control hiPSCs (non-family-related control iAG16102 and family control iN805-6A/B; two independent lines; each in biological triplicate) (Figure 3A). These findings were also substantiated by immunostaining of HNF1B and PDX1 on day 12 (Figure 3B; representative of two to three hiPSC lines) and FACS analyses of SOX9 and PDX1 on day 17 (Figure 3C; two to three hiPSC lines per subject were pooled together) in MODY5-hiPSC-derived pancreatic progenitors.
Figure 3.

Transcriptional Perturbations in MODY5-hiPSC-Derived Pancreatic Progenitors
(A) Gene expression of HNF1B, PAX6, PDX1, PTF1A, GATA4, and GATA6 in a non-family control and in iN805 family hiPSCs differentiated for 0, 12, 14, and 17 days. Data are representative of at least two independent hiPSC lines per subject. Replicate experiments are presented. The experiments were replicated at least twice. All error bars indicate SD of three biological replicates in an independent experiment.
(B) Immunostaining for HNF1B and PDX1 on iN805 hiPSCs differentiated for 12 days. Data are representative of at least two independent hiPSC lines per subject. The experiment was replicated at least twice. Scale bar, 200 μm.
(C) The percentage of PDX1+ and SOX9+ cells after differentiation of iN805 hiPSCs (two to three hiPSC lines per subject were pooled together) for 17 days.
See also Figures S2 and S3.
Subsequently, to address the hypothesis that mutant HNF1B S148L/+ results in its decreased expression and thus leads to pancreatic hypoplasia and MODY, we analyzed the transcriptional profile to potentially identify a gene(s) which is down-regulated in the mutant hiPSC-derived pancreatic progenitors. Contrary to expectations, we found a compensatory increase in FOXA2, ISL1, HLXB9, and RFX6 (Figures S2B and S2C), suggesting that this set of genes (including HNF1B, PDX1, GATA4, and GATA6) play an important role in alleviating the negative effects of mutant HNF1BS148L/+ so as to delay the overall impact of pancreatic hypoplasia and/or mutant HNF1BS148L/+ on diabetes onset. Surprisingly, PAX6 was the singular gene among the many examined to exhibit down-regulation in mutant hiPSC-derived pancreatic progenitors (Figure 3A). Following this candidate gene approach, we performed microarray analyses for cells obtained from days 0, 12, and 17 (Figure S3A and Table S2). Reassuringly, our genome-wide analyses indicated that our candidate gene approach has captured all the key changes in pancreatic genes: increase in PDX1, ISL1, HNF1B, TCF2, RFX6, FOXA2, GATA4, GATA6, and HLXB9 (MNX1) expression in the mutant hiPSC-derived pancreatic progenitors (Figure S3A). In addition, PAX6 was also the most relevant pancreatic gene (expressed in our differentiation) to be down-regulated in the mutant hiPSC-derived pancreatic progenitors (Figure S3A). Further GO analyses on the up- and down-regulated genes (fold change >2) on days 12 and 17 mostly reflected changes in developmental processes (Figures S3B and S3C). Uniquely, neural development appeared to be more down-regulated in MODY5 pancreatic progenitors on day 17 (Figure S3C).
We then performed chromatin immunoprecipitation (ChIP) analyses and confirmed that HNF1B binds onto the genomic loci of HLXB9 and HNF1B (Figure S3D), suggesting that these two genes could be the earliest mediators of pancreatic transcription factor network control. To evaluate whether the HNF1B mutation affected pancreatic progenitor cell growth, we performed a cell counting assay and, interestingly, observed that there is a huge retardation of cell growth from day 12 onward, when the mutant HNF1B protein exhibits peak expression (Figure S3E). This strongly suggests that the mutant HNF1B protein affects pancreatic cell growth, thereby accounting for the pancreatic hypoplasia phenotype in MODY5.
Wild-Type HNF1B Suppresses, While Mutant HNF1B Increases, PDX1 Gene Expression
The compensatory increase in both HNF1B and PDX1 gene expression in the mutant hiPSC-derived pancreatic progenitors prompted us to further investigate this molecular relationship. FACS analyses performed on day-12 differentiated control hiPSCs confirmed that 36.2% are PDX1+ (Figure 4A) whereas 91.4% are HNF1B+ (Figure 5A), corroborating their transcriptional profile (Figure 1E). We first performed luciferase assays to study the transcriptional regulation of cardinal pancreatic gene PDX1 (Stoffers et al., 1997) by HNF1B. Overexpression of HNF1B from days 10–12 in differentiated control hiPSCs surprisingly suppressed PDX1 transcriptional activity, in both the ∼7 kb full-length PDX1 promoter construct and a construct containing areas I–III of PDX1 promoter (∼2.4 kb) known to be the principal control region of PDX1 gene expression (Gerrish et al., 2000) (Figure 4B). This indicates a repressive pattern of HNF1B whereby HNF1B gene expression is decreasing when PDX1 is beginning to be expressed from days 10–12 (Figure 1E).
Figure 4.

Mutant HNF1B Is Responsible for the Increase in PDX1 Gene Expression
(A) The percentage of PDX1+ cells after differentiation of hiPSCs for 12 days.
(B and C) Luciferase assay showing the effect of (B) HNF1B and/or (C) HNF1A overexpression on PDX1 transcriptional activity in hiPSCs differentiated for 12 days.
(D and E) Expression of pancreas-related (HNF1B, mutant HNF1BS148L/+, PDX1, GATA4, and GATA6) markers in hiPSCs differentiated for 10 days and overexpressing increasing amounts of (D) mutant HNF1BS148L or (E) HNF1B from days 7–10. The replicate experiment for mutant HNF1BS148L is presented.
The experiments were replicated at least twice. All error bars indicate SD of three biological replicates in an independent experiment. See also Figure S4.
Figure 5.

HNF1B Is Not Directly Involved in Gene Regulation of PAX6
(A) The percentage of HNF1B+ and PAX6+ cells after differentiation of hiPSCs for 12 days.
(B and C) Luciferase assay showing (B) basal PAX6 promoter activity and (C) the effect of HNF1B and/or HNF1A overexpression on PAX6 promoter activity in hiPSCs differentiated for 10 days.
(D and E) Expression of PAX6 in hiPSCs differentiated for 10 days and overexpressing increasing amounts of (D) HNF1B or (E) mutant HNF1BS148L from days 7–10. The experiments in (B) to (E) were replicated at least twice. All error bars indicate SD of three biological replicates in an independent experiment. ∗p < 0.05 by two-sided Student's t test on three independent experiments.
(F) Clinical characteristics of MODY5 patients including HbA1c (%), blood glucose (glu), insulin (INS), C-peptide (C-pep), and plasma glucagon (P-GCG) concentrations.
(G) Model depicting the impact of mutant HNF1BS148L/+ on early human pancreas development. Mutant HNF1BS148L/+ evokes an early increase in definitive endoderm genes followed by a subsequent increase in pancreas-related foregut endoderm genes. Thus, although MODY5 patients typically develop pancreatic hypoplasia there is a low occurrence of neonatal diabetes. The decrease in early PAX6 gene expression in pancreatic progenitors may partly account for the subsequent β-cell dysfunction in MODY5 patients.
HNF1B protein functions either as a homodimer or a heterodimer with the structurally related HNF1A (Mendel et al., 1991, Rey-Campos et al., 1991). We thus overexpressed both HNF1B and HNF1A from days 10–12 in differentiated control hiPSCs, only to discover that HNF1A does not have any transcriptional regulatory effect on PDX1, at least during this early pancreatic progenitor stage when PDX1 is beginning to be expressed (Figure 4C). This is consistent with the observation that HNF1A is expressed very late during pancreatic differentiation (Figure S1D). Thus, the suppressive effect of HNF1B on PDX1 transcriptional activity is likely due to homodimers in action.
Very little is known about the target genes of HNF1B in a developing pancreas. HNF4A is one potential candidate (Thomas et al., 2001). HNF1 binding sites have been found to be present in pancreas-/islet-specific P2 (Thomas et al., 2001) and P1 promoters (Taraviras et al., 1994) of the Hnf4α gene (Eeckhoute et al., 2003). Thus, we cloned both P1 and P2 promoters of HNF4A, and performed similar luciferase assays. Dismally, HNF1B overexpression did not have any effect on P1 promoter and only a very marginal effect on P2 promoter activity in our stem cell model on day 12 (Figure S4A), suggesting that the regulation of HNF4A by HNF1B (or HNF1A) is likely apparent only at later stages of pancreatic development.
To delineate whether the wild-type HNF1B or mutant HNF1BS148L allele is responsible for the compensatory increase in pancreatic gene expression (Figure 3A), we next overexpressed wild-type HNF1B or mutant HNF1BS148L from days 7–10 in differentiated control hiPSCs. Interestingly, overexpression of mutant HNF1BS148L resulted in an increase in PDX1 gene expression but was insufficient to up-regulate other pancreatic transcription factors such as GATA4, GATA6, or SOX9 (Figures 4D and S4B). On the contrary, overexpression of wild-type HNF1B from days 7–10 did not alter the expression level of PDX1 or other genes we investigated, including GATA4, GATA6, and SOX9 (Figures 4E and S4C). ChIP analyses performed on HNF1B also did not reveal binding on PDX1 genomic loci (Cebola et al., 2015 and data not shown). Last but not least, qPCR and western blot analyses confirmed that the HNF1B and HNF1A genes/proteins were overexpressed successfully (Figures S4D and S4E). Together, these data indicate that mutant HNF1BS148L (and not HNF1B or HNF1A) gene expression is responsible for the compensatory increase in PDX1 gene expression in mutant hiPSC-derived pancreatic progenitors. The molecular mediator(s) of increased GATA4 and GATA6 gene expression in mutant hiPSC-derived pancreatic progenitors is unclear and requires further investigation.
HNF1B Is Not Directly Involved in Gene Regulation of PAX6
Pax6 is known to be expressed during early pancreatic development (E9–9.5) in cells committed exclusively to the endocrine cell fate (Ashery-Padan et al., 2004, Sander et al., 1997). Among the transcription factors we analyzed, only PAX6 transcripts were clearly down-regulated (Figure 3A). Since PAX6 exhibits a similar (but right-shifted) gene-expression profile to that of HNF1B, we sought to investigate whether HNF1B or HNF1BS148L is involved in the regulation of PAX6 gene expression. FACS analyses on day-12 differentiated control hiPSCs (PAX6 peak transcript expression) (Figure 1E), indicated that 27.2% are PAX6+, of which 22.0% are also HNF1B+ (Figure 5A). This percentage of PAX6+ cells is consistent with the report that PAX6 protein is detected only in a small subset of cells in the pancreatic endoderm at E9–9.5 (Sander et al., 1997). Luciferase assays performed on day-10 differentiated control hiPSCs indicated that PAX6 promoter is active (Figure 5B). However, overexpression of HNF1B and/or HNF1A had no impact, whereas overexpression of mutant HNF1BS148L only resulted in a marginal suppression of PAX6 promoter activity (Figure 5C). Overexpression of wild-type HNF1B from days 7–10 in differentiated control hiPSCs (Figure 4E) did not alter PAX6 gene-expression levels (Figure 5D). Overexpression of mutant HNF1BS148L alone (Figure 4D) was not sufficient to suppress PAX6 gene-expression levels (Figure 5E). Furthermore, HNF1B was not found to bind onto the PAX6 promoter region (Cebola et al., 2015 and data not shown). This indicates that the decrease in PAX6 gene expression is an indirect effect of the mutant HNF1BS148L/+ and is possibly a result of the altered pancreatic transcription factor network.
The loss of Pax6 expression early on during pancreatic development reduces pancreatic insulin content (Ashery-Padan et al., 2004, Sander et al., 1997) and affects postnatal pancreatic β-cell function, which results in early-onset diabetes (Ashery-Padan et al., 2004). In this study, we found that PAX6 gene expression is decreased in mutant (HNF1BS148L/+) hiPSC-derived pancreatic progenitors. To translate our findings to the MODY5 patients, we tracked the clinical phenotype of the N805 patients over time. N805-2 (HNF1BS148L/+) exhibited high fasting glucose levels (16.3 mmol/l) and required insulin (58–68 units/24 hr), whereas N805-1 (HNF1BS148L/+), who was not diabetic, presented with elevated fasting glucose levels (normal range: 4.0–6.0 mmol/l) and elevated 2-hr glucose levels after a standard oral glucose challenge (75 g) (impaired glucose tolerance range: 7.8–11.1 mmol/l) after the age of 8 years (Figure 5F). N805-2 presented low fasting insulin (2.4 mIE/l) whereas N805-1 exhibited a transition from normal to subnormal levels of fasting insulin, at 5.4 mIE/l (normal range: 6.0–27.0 mIE/l) (Figure 5F). As diabetes develops, plasma glucagon levels usually rise due to impaired α-cell glucose-sensing function and lack of appropriate suppression (Dunning et al., 2005). This scenario fits with the substantially elevated plasma glucagon levels (72.2 pmol/l) in the diabetic individual N805-2 and the moderately elevated plasma glucagon levels in the pre-diabetic individual N805-1 (46.9 pmol/l; normal range: 14.3–43 pmol/l) (Figure 5F).
Discussion
We have successfully established a human stem cell model to study the molecular mechanisms underlying MODY5. Our experimental design, which includes a non-family control hiPSC line and three independent hiPSC lines from each of the three subjects in a MODY5 family, is a well-controlled experimental “node” which minimizes potential genetic background influence and line-to-line variation in hiPSC differentiation. While we observe HNF1B gene expression in the family control data to resemble that in the MODY5-hiPSC-derived pancreatic progenitors on rare occasions (Figure 2), suggesting initial genetic background effects (difference between control lines becomes attenuated later), the use of non-family control samples provides confidence in the overall distinct phenotypes we observed in our in vitro stem cell model. In addition, more than 90% of day-12 differentiated cells are HNF1B+, making our pancreatic differentiation protocol well suited for studying MODY5.
HNF1B has been reported to be a transcriptional activator (Rey-Campos et al., 1991), but any positive regulation of PDX1 gene expression by HNF1A and/or HNF1B could be specific to pancreatic β cells (Ben-Shushan et al., 2001) and not pancreatic progenitors, implying that HNF1B and/or HNF1A switch their binding partners during pancreatic development to regulate specificity in gene regulation.
We observed that mutant HNF1BS148L (and not HNF1B or HNF1A) gene expression is responsible for the compensatory increase in PDX1 gene expression in mutant hiPSC-derived pancreatic progenitors. This could, in part, be acting to counter the increased transcriptional repression by wild-type HNF1B (Figures 3B and 3C), although increasing HNF1B gene expression for 3 days from days 7 to 10 appears to be insufficient to significantly suppress PDX1 gene expression (Figure 4E). This gene regulation is likely to be indirect, since we did not find HNF1B protein to be bound onto the genomic loci of PDX1 (Cebola et al., 2015). It is interesting to note that β-cell-specific knockout of Hnf1β similarly results in an increase in Pdx1 gene expression (Wang et al., 2004), indicating that perturbations in HNF1B gene expression, either early during pancreatic development or in mature β cells, both prompt compensatory PDX1 gene expression.
The presence of a wild-type HNF1B allele and the combinatorial effects of wild-type-mutant HNF1B dimers in MODY5 (HNF1BS148L/+) patients partly explains the compensatory up-regulation of numerous pancreatic transcription factors. The lack of down-regulation of critical pancreatic genes PDX1, PTF1A, GATA4, and GATA6 implies that the mechanism underlying dorsal pancreatic agenesis in MODY5 is independent of these genes, which are also known to result in pancreatic agenesis/hypoplasia when mutated (D'Amato et al., 2010, Lango Allen et al., 2012, Sellick et al., 2004, Stoffers et al., 1997). This compensation in pancreatic transcriptional network due to an HNF1B autosomal dominant mutation may also account for the diabetes onset relatively later in life seen in MODY5 compared with the early onset observed in genetic forms of neonatal diabetes (Teo et al., 2013a) (Figure 5G). In this context, it is interesting that the MODY5 subjects with dorsal pancreatic agenesis also exhibit compensatory increase in physiological acinar function with hypersecretion from the remaining ventral portions of the pancreas (Tjora et al., 2013).
Hnf1β−/− mice with pancreatic agenesis exhibit loss of numerous pancreatic genes, including Pax6 expression (Haumaitre et al., 2005). Pax6-deficient pancreatic progenitors that are unable to mature and reach terminal differentiation later in life cannot be rescued by postnatal neogenesis (Ashery-Padan et al., 2004). Thus, the reduction in pancreatic endocrine cell number in Pax6−/− embryos (Sander et al., 1997) and altered acinar structure (Hart et al., 2013) could partly account for the pancreatic hypoplasia phenotype in MODY5 patients. Given that PAX6 directly regulates genes that regulate β-cell function (Ashery-Padan et al., 2004, Wen et al., 2009), we speculate that the loss of PAX6 gene expression early on in the MODY5 patients also partly accounts for the β and α islet cell dysfunction, and contributes to their diabetic phenotype (Figure 5G). This concept is concordant with the observations that patients with PAX6 heterozygous mutations also develop glucose intolerance (Wen et al., 2009, Yasuda et al., 2002). It is interesting to note that administration of exendin-4, a glucagon-like peptide 1 receptor (GLP-1R) agonist, to Pax6+/− mice can rescue the metabolic abnormalities observed, either via increased insulin secretion or β-cell regeneration (Ding et al., 2009), suggesting a potential for treatment for patients lacking pancreatic PAX6 gene expression, and the MODY5 patients in this study.
Collectively, we have evaluated the impact of a MODY5-causing point mutation (S148L) in the HNF1B gene on human pancreas development using a unique human stem cell model. We report molecular phenotypes (up- and down-regulation of pancreatic genes) when HNF1B is increasingly expressed from days 7 to 10, and observed that these phenotypes extend beyond the expression window of HNF1B. These data provide insights into the long-term impact of the MODY5 mutation on the pancreatic transcriptional network and the subsequent development of human pancreatic progenitors into mature functional endocrine cells. This is in agreement with Haumaitre et al. (2006), who indicated that MODY5 is certainly due to defective morphogenesis of the pancreas. Future efforts to further differentiate these hiPSCs into pancreatic β cells will reveal the impact of transcriptional perturbations on β-cell formation and function.
Experimental Procedures
Cell Culture
Informed consent was obtained from MODY5 patients. This study was reviewed and approved by the Institutional Review Boards at Haukeland University Hospital and Joslin Diabetes Center, and in accordance with the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. hiPSCs used were tested mycoplasma negative and were cultured as described previously (Teo et al., 2013b, Teo et al., 2014). hiPSCs were differentiated into pancreatic progenitors as described previously (Teo et al., 2014). In general, 2–3 independent hiPSC lines per subject were used, and biological triplicates were used for each hiPSC line.
Sequencing of MODY5-hiPSCs
The method for sequencing has been described previously (Teo et al., 2013b).
qRT-PCR, Immunostaining, FACS, Western Blot, and ChIP Analyses
Methods for qRT-PCR, immunostaining, FACS, western blot (Teo et al., 2014), and ChIP analyses (Cebola et al., 2015, Teo et al., 2011, Teo et al., 2015) have been described previously. p < 0.05 indicates statistical significance by Student's t test (two-sided; equal variance). In general, qRT-PCR and immunostaining data of MODY5-hiPSC-derived cells are representative of one non-family control hiPSC line (iAG16102), two family control hiPSC lines (iN805-6A/C), and three lines each for the two MODY5 patients (iN805-1A/B/C and iN805-2A/B/C). All error bars indicate SD of three biological replicates. Two to three hiPSC lines per subject were pooled together before being set up in triplicate for FACS analyses. Primers and antibodies used are provided in Table S3.
Microarray
Microarray was performed by the Molecular Genetics Core Facility at Children's Hospital, Boston. Two biological replicates for undifferentiated and day-10 differentiated control hiPSCs were analyzed. One sample from iAG16102, iN805-6, iN805-1, and iN805-2 each was used for day-0, -12 and -17 analyses.
Gene Ontology Overrepresentation Analysis
The overrepresentation of GO biological process categories was assessed using DAVID.
Overexpression Studies
HNF1B gene was amplified from cDNA obtained from differentiated hPSCs using PfuUltra II Fusion HS DNA Polymerase with a melting temperature (Tm) of 50°C and extension at 72°C for 2 min. HNF1B cDNA was then subcloned into pCDH-FlagV5 vector using EcoRI and XhoI RE sites to obtain pCDH-hHNF1B. HNF1B cDNA was also subcloned into PCR-Blunt II-TOPO vector, and hHNF1BC443TF and hHNF1BC443TR primers were used to amplify HNF1BS148L using PfuUltra II Fusion HS DNA Polymerase with an extension at 72°C for 5 min. HNF1BS148L cDNA was then subcloned into pCDH-FlagV5 vector using EcoRI and XhoI RE sites to obtain pCDH-hHNF1BS148L. pcDNA3.1-hHNF1A was a gift from Y.-I. Chi. Primers used are provided in Table S3. Day-6 differentiated control hiPSCs were split and replated onto 12-well plates. These cells were transduced with lentiviruses (multiplicity of infection 10, 20, 30 or 50, 100, 200) containing pCDH-GFP, pCDH-HNF1B, or pCDH-HNF1BS148L on day 7, washed with PBS on day 8, and harvested on day 10.
Luciferase Assays
hPDX1 promoter (∼7 kb) was cloned into pGL4.10 using NheI and XhoI RE sites. hPDX1 promoter/enhancer (areas I–III) was cloned into pGL4.23 using XhoI and BglII RE sites. hHNF4A P1 and P2 promoters were cloned into pGL4.10 using KpnI and XhoI RE sites. hPAX6 promoter (346 bp) containing HNF1B and HNF1A binding sites was a gift from Y.-H. Zhou. Primers used are provided in Table S3. Day-9 differentiated control hiPSCs were split and replated onto 12-well plates. These cells were transfected with hPDX1 promoter, hHNF4A P1 or P2 promoter in the absence and presence of pCDH-HNF1B, and/or pcDNA3.1-hHNF1A on day 10 and harvested on day 12. Day-6 differentiated control hiPSCs were split and replated onto 12-well plates. These cells were transfected with hPAX6 promoter in the absence and presence of pCDH-GFP, pCDH-HNF1B, pCDH-HNF1BS148L, and/or pcDNA3.1-hHNF1A on day 7 and harvested on day 10. The method for luciferase assay has been described previously (Teo et al., 2011).
Hormone Assays
Glucagon was assessed with a radioimmunoassay from Millipore. This assay reports a precision (coefficient of variation) for high levels (72.4 pmol/l) of 10.1%, for intermediate levels (52.2 pmol/l) of 12.6%, and for low levels (28.8 pmol/l) of 9.2%. Prior to analysis, we collected blood samples in plasma tubes containing 250 kIU Trasvlol (Aprotinin) per ml of whole blood. This resulted in a final concentration of approximately 500 kIU Trasvlol per ml of serum or plasma, and the aliquot was frozen at −70°C.
Author Contributions
A.K.K.T. designed the study, performed most of the experiments, collected and analyzed data, and wrote the paper; H.H.L. performed experiments for manuscript revision; I.A.V. performed FACS analyses; E.D. contributed to discussions; E.T. collected patient data; H.R. contributed to the design of the study and to data interpretation; R.N.K. contributed to conceptual discussions, supervised the studies, and edited and approved the paper. All authors discussed the results and commented on the manuscript.
Acknowledgments
The hiPSC lines used were derived with support from the Joslin DRC iPS Core Facility (NIH 5 P30 DK036836-27). FACS analysis was supported by the HSCI/DRC Flow Core (NIH P30DK036836). Microarray studies were performed by the Molecular Genetics Core Facility at Children's Hospital Boston, supported by NIH-P50-NS40828 and NIH-P30-HD18655. We thank B. Wagner for assistance with microarray analyses; and K. Tomita, E.K. Tan, N.R. Dunn, Y.-H. Zhou, and Y.-I. Chi for providing plasmids. A.K.K.T. was supported by a Juvenile Diabetes Research Foundation (JDRF) Postdoctoral Fellowship and is currently supported by the Institute of Molecular and Cell Biology (IMCB), A∗STAR, NHG-KTPH SIG/14033, the NUHS-CG Metabolic In-Vitro Core Seed Funding, and the JCO Career Development Award (CDA) 15302FG148, A∗STAR. I.A.V. is supported by an HSCI Sternlicht Director's Fund Award and a NIH F31DK098931 award. E.D. is supported by an Advanced JDRF Postdoctoral Fellowship. H.R. is supported by grants from Bergen Forskningsstiftelse (BFS), the Western Norway Regional Health Authority and the Novo Nordisk Foundation. R.N.K. is supported by the HSCI, NIH grants RO1 DK 67536, RO1 DK 055523, and R01103215, and a grant from AstraZeneca.
Published: February 11, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes four figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.01.007.
Contributor Information
Adrian Kee Keong Teo, Email: ateo@imcb.a-star.edu.sg.
Rohit N. Kulkarni, Email: rohit.kulkarni@joslin.harvard.edu.
Accession Numbers
Microarray data have been uploaded to GEO with accession number GEO: GSE74885.
Supplemental Information
References
- Ashery-Padan R., Zhou X., Marquardt T., Herrera P., Toube L., Berry A., Gruss P. Conditional inactivation of Pax6 in the pancreas causes early onset of diabetes. Dev. Biol. 2004;269:479–488. doi: 10.1016/j.ydbio.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Ben-Shushan E., Marshak S., Shoshkes M., Cerasi E., Melloul D. A pancreatic beta-cell-specific enhancer in the human PDX-1 gene is regulated by hepatocyte nuclear factor 3beta (HNF-3beta ), HNF-1alpha, and SPs transcription factors. J. Biol. Chem. 2001;276:17533–17540. doi: 10.1074/jbc.M009088200. [DOI] [PubMed] [Google Scholar]
- Cebola I., Rodriguez-Segui S.A., Cho C.H., Bessa J., Rovira M., Luengo M., Chhatriwala M., Berry A., Ponsa-Cobas J., Maestro M.A. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol. 2015;17:615–626. doi: 10.1038/ncb3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amato E., Giacopelli F., Giannattasio A., D'Annunzio G., Bocciardi R., Musso M., Lorini R., Ravazzolo R. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet. Med. 2010;27:1195–1200. doi: 10.1111/j.1464-5491.2010.03046.x. [DOI] [PubMed] [Google Scholar]
- Ding J., Gao Y., Zhao J., Yan H., Guo S.Y., Zhang Q.X., Li L.S., Gao X. Pax6 haploinsufficiency causes abnormal metabolic homeostasis by down-regulating glucagon-like peptide 1 in mice. Endocrinology. 2009;150:2136–2144. doi: 10.1210/en.2008-1006. [DOI] [PubMed] [Google Scholar]
- Dunning B.E., Foley J.E., Ahren B. Alpha cell function in health and disease: influence of glucagon-like peptide-1. Diabetologia. 2005;48:1700–1713. doi: 10.1007/s00125-005-1878-0. [DOI] [PubMed] [Google Scholar]
- Edghill E.L., Bingham C., Ellard S., Hattersley A.T. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J. Med. Genet. 2006;43:84–90. doi: 10.1136/jmg.2005.032854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckhoute J., Moerman E., Bouckenooghe T., Lukoviak B., Pattou F., Formstecher P., Kerr-Conte J., Vandewalle B., Laine B. Hepatocyte nuclear factor 4 alpha isoforms originated from the P1 promoter are expressed in human pancreatic beta-cells and exhibit stronger transcriptional potentials than P2 promoter-driven isoforms. Endocrinology. 2003;144:1686–1694. doi: 10.1210/en.2002-0024. [DOI] [PubMed] [Google Scholar]
- Gerrish K., Gannon M., Shih D., Henderson E., Stoffel M., Wright C.V., Stein R. Pancreatic beta cell-specific transcription of the pdx-1 gene. The role of conserved upstream control regions and their hepatic nuclear factor 3beta sites. J. Biol. Chem. 2000;275:3485–3492. doi: 10.1074/jbc.275.5.3485. [DOI] [PubMed] [Google Scholar]
- Haldorsen I.S., Vesterhus M., Raeder H., Jensen D.K., Sovik O., Molven A., Njolstad P.R. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet. Med. 2008;25:782–787. doi: 10.1111/j.1464-5491.2008.02460.x. [DOI] [PubMed] [Google Scholar]
- Hart A.W., Mella S., Mendrychowski J., van Heyningen V., Kleinjan D.A. The developmental regulator Pax6 is essential for maintenance of islet cell function in the adult mouse pancreas. PLoS One. 2013;8:e54173. doi: 10.1371/journal.pone.0054173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumaitre C., Barbacci E., Jenny M., Ott M.O., Gradwohl G., Cereghini S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc. Natl. Acad. Sci. USA. 2005;102:1490–1495. doi: 10.1073/pnas.0405776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumaitre C., Fabre M., Cormier S., Baumann C., Delezoide A.L., Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum. Mol. Genet. 2006;15:2363–2375. doi: 10.1093/hmg/ddl161. [DOI] [PubMed] [Google Scholar]
- Lango Allen H., Flanagan S.E., Shaw-Smith C., De Franco E., Akerman I., Caswell R., Ferrer J., Hattersley A.T., Ellard S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2012;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro M.A., Cardalda C., Boj S.F., Luco R.F., Servitja J.M., Ferrer J. Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocr. Dev. 2007;12:33–45. doi: 10.1159/000109603. [DOI] [PubMed] [Google Scholar]
- Mendel D.B., Hansen L.P., Graves M.K., Conley P.B., Crabtree G.R. HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes Dev. 1991;5:1042–1056. doi: 10.1101/gad.5.6.1042. [DOI] [PubMed] [Google Scholar]
- Ott M.O., Rey-Campos J., Cereghini S., Yaniv M. vHNF1 is expressed in epithelial cells of distinct embryonic origin during development and precedes HNF1 expression. Mech. Dev. 1991;36:47–58. doi: 10.1016/0925-4773(91)90071-d. [DOI] [PubMed] [Google Scholar]
- Rey-Campos J., Chouard T., Yaniv M., Cereghini S. vHNF1 is a homeoprotein that activates transcription and forms heterodimers with HNF1. EMBO J. 1991;10:1445–1457. doi: 10.1002/j.1460-2075.1991.tb07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M., Neubuser A., Kalamaras J., Ee H.C., Martin G.R., German M.S. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Sellick G.S., Barker K.T., Stolte-Dijkstra I., Fleischmann C., Coleman R.J., Garrett C., Gloyn A.L., Edghill E.L., Hattersley A.T., Wellauer P.K. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat. Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- Stoffers D.A., Zinkin N.T., Stanojevic V., Clarke W.L., Habener J.F. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Taraviras S., Monaghan A.P., Schutz G., Kelsey G. Characterization of the mouse HNF-4 gene and its expression during mouse embryogenesis. Mech. Dev. 1994;48:67–79. doi: 10.1016/0925-4773(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Teo A.K., Arnold S.J., Trotter M.W., Brown S., Ang L.T., Chng Z., Robertson E.J., Dunn N.R., Vallier L. Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes Dev. 2011;25:238–250. doi: 10.1101/gad.607311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A.K., Ali Y., Wong K.Y., Chipperfield H., Sadasivam A., Poobalan Y., Tan E.K., Wang S.T., Abraham S., Tsuneyoshi N. Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells. 2012;30:631–642. doi: 10.1002/stem.1022. [DOI] [PubMed] [Google Scholar]
- Teo A.K., Wagers A.J., Kulkarni R.N. New opportunities: harnessing induced pluripotency for discovery in diabetes and metabolism. Cell Metab. 2013;18:775–791. doi: 10.1016/j.cmet.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A.K., Windmueller R., Johansson B.B., Dirice E., Njolstad P.R., Tjora E., Raeder H., Kulkarni R.N. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J. Biol. Chem. 2013;288:5353–5356. doi: 10.1074/jbc.C112.428979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A.K., Valdez I.A., Dirice E., Kulkarni R.N. Comparable generation of activin-induced definitive endoderm via additive Wnt or BMP signaling in absence of serum. Stem Cell Rep. 2014;3:5–14. doi: 10.1016/j.stemcr.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A.K., Tsuneyoshi N., Hoon S., Tan E.K., Stanton L.W., Wright C.V., Dunn N.R. PDX1 binds and represses hepatic genes to ensure robust pancreatic commitment in differentiating human embryonic stem cells. Stem Cell Rep. 2015;4:578–590. doi: 10.1016/j.stemcr.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas H., Jaschkowitz K., Bulman M., Frayling T.M., Mitchell S.M., Roosen S., Lingott-Frieg A., Tack C.J., Ellard S., Ryffel G.U. A distant upstream promoter of the HNF-4alpha gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum. Mol. Genet. 2001;10:2089–2097. doi: 10.1093/hmg/10.19.2089. [DOI] [PubMed] [Google Scholar]
- Tjora E., Wathle G., Erchinger F., Engjom T., Molven A., Aksnes L., Haldorsen I.S., Dimcevski G., Raeder H., Njolstad P.R. Exocrine pancreatic function in hepatocyte nuclear factor 1beta-maturity-onset diabetes of the young (HNF1B-MODY) is only moderately reduced: compensatory hypersecretion from a hypoplastic pancreas. Diabet. Med. 2013;30:946–955. doi: 10.1111/dme.12190. [DOI] [PubMed] [Google Scholar]
- Wang L., Coffinier C., Thomas M.K., Gresh L., Eddu G., Manor T., Levitsky L.L., Yaniv M., Rhoads D.B. Selective deletion of the Hnf1beta (MODY5) gene in beta-cells leads to altered gene expression and defective insulin release. Endocrinology. 2004;145:3941–3949. doi: 10.1210/en.2004-0281. [DOI] [PubMed] [Google Scholar]
- Wen J.H., Chen Y.Y., Song S.J., Ding J., Gao Y., Hu Q.K., Feng R.P., Liu Y.Z., Ren G.C., Zhang C.Y. Paired box 6 (PAX6) regulates glucose metabolism via proinsulin processing mediated by prohormone convertase 1/3 (PC1/3) Diabetologia. 2009;52:504–513. doi: 10.1007/s00125-008-1210-x. [DOI] [PubMed] [Google Scholar]
- Yasuda T., Kajimoto Y., Fujitani Y., Watada H., Yamamoto S., Watarai T., Umayahara Y., Matsuhisa M., Gorogawa S., Kuwayama Y. PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes. 2002;51:224–230. doi: 10.2337/diabetes.51.1.224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.