

Summary
Human embryonic stem cells (hESC) show great promise for clinical and research applications, but their well-known proneness to genomic instability hampers the development to their full potential. Here, we demonstrate that medium acidification linked to culture density is the main cause of DNA damage and genomic alterations in hESC grown on feeder layers, and this even in the short time span of a single passage. In line with this, we show that increasing the frequency of the medium refreshments minimizes the levels of DNA damage and genetic instability. Also, we show that cells cultured on laminin-521 do not present this increase in DNA damage when grown at high density, although the (long-term) impact on their genomic stability remains to be elucidated. Our results explain the high levels of genome instability observed over the years by many laboratories worldwide, and show that the development of optimal culture conditions is key to solving this problem.
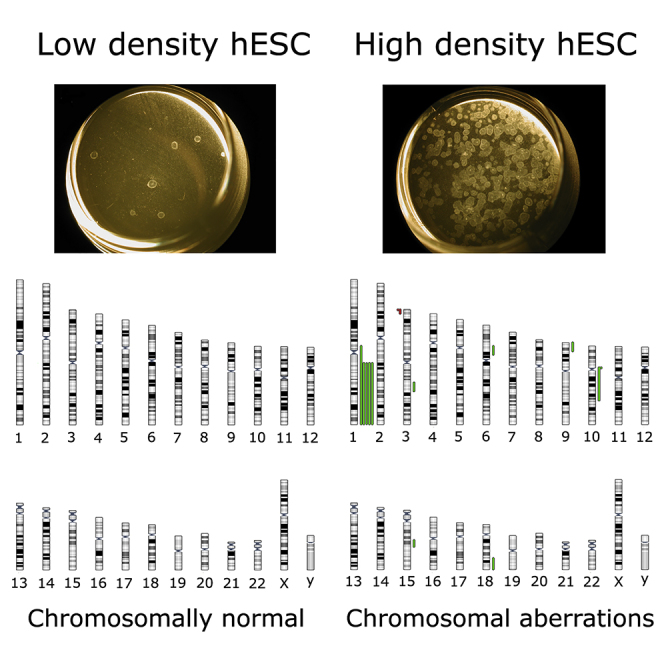
Graphical Abstract
Highlights
-
•
Increased culture density induces DNA damage and genomic alterations in hESC
-
•
Medium acidification due to lactic acid accumulation is the main driver
-
•
More frequent medium refreshments rescues genomic integrity in high-density culture
-
•
Laminin-521 reduces DNA damage but has no clear effect on genomic instability
In this article, Spits and colleagues show that the culture density of hESC is directly correlated to increased DNA damage and genomic instability. This is caused by medium acidification and can be countered by more frequent medium refreshments. When cultured on laminin-521, hESC show less DNA damage than in the feeder-based system, but no clear difference in genomic instability was found.
Introduction
The unique characteristics of human pluripotent stem cells (hPSC), such as human embryonic stem cells (hESC) and induced pluripotent stem cells (hiPSC), make them attractive not only as a potential source of cells in regenerative medicine, but also as a research tool to study early human developmental processes and human disorders (Ben-Nun and Benvenisty, 2006, Tabar and Studer, 2014). hPSC are kept in culture for long periods of time without this apparently affecting their self-renewal and pluripotent capacities. Nevertheless, these cultures are frequently taken over by cells carrying genetic abnormalities, some of which are highly recurrent (Amps et al., 2011, Lund et al., 2012, Nguyen et al., 2013, Spits et al., 2008). For instance, 20% of hPSC lines worldwide carry a gain of a small region of 20q11.21 (Amps et al., 2011). Recent work has shown that the recurrent takeover of cultures by cells carrying this mutation is due to the fact that the gain of 20q11.21 leads to decreased levels of apoptosis (Avery et al., 2013, Nguyen et al., 2014).
Conversely, not much is known about the origin of these mutations. Their high frequency, combined with the proficient DNA repair of hESC (Sokolov and Neumann, 2013), suggests that these cells undergo profuse DNA damage in culture. Studies on genetic mosaicism in hESC cultures, which reflect the spontaneous mutation rate of these cells, show that up to 35% of cells within one hESC culture have abnormal chromosome counts and structural aberrations (Dekel-Naftali et al., 2012, Jacobs et al., 2014, Lim et al., 2011).
This genetic heterogeneity of hPSC cultures presents an undeniable hurdle for their use in research and regenerative medicine. For instance, it is possible that experimental results are difficult to extrapolate from one cell line to another because of culture-induced variation in genetic or epigenetic content. There is evidence that genetically abnormal hPSC have aberrant differentiation capacity (Fazeli et al., 2011, Werbowetski-Ogilvie et al., 2009), and tend to produce immature teratomas containing a higher proportion of poorly differentiated or undifferentiated cells with an increased capacity for malignancy (Herszfeld et al., 2006, Werbowetski-Ogilvie et al., 2009, Yang et al., 2008). Furthermore, genetically abnormal hPSC display altered gene-expression profiles with an up-regulation of a number of oncogenes (Gopalakrishna-Pillai and Iverson, 2010, Werbowetski-Ogilvie et al., 2009, Yang et al., 2008). It is thus clear that understanding and, more importantly, controlling this genomic variability can significantly improve the value of hPSC and their derivatives for the clinic and as research models.
In this work, we focused on the study of hESC grown on mouse feeder layers. This culture system has been, over the years, the most commonly used worldwide (Fraga et al., 2011), and our aim was to identify the key factors behind the well-established proneness of these cultures to genetic instability. We hypothesized that suboptimal culture conditions lead to DNA damage in hESC, and that high-density culture in particular results in a nutrient deficit and/or detrimental concentration of waste products. These can interfere with the metabolism of the cells (Chen et al., 2010), cause replication stress, and increase the risk for DNA breakage and chromosomal abnormalities (Burrell et al., 2013).
Here, we show that during a single passage of 5 days, hESC growing on feeder layers in high densities show a significant increase in DNA fragmentation and genomic abnormalities at the single-cell level. These effects are largely caused by the accumulation of lactic acid in the culture medium and the associated medium acidification, and we show that this can be countered by refreshing the medium twice daily. Finally, our results demonstrate that hESC grown on laminin-521 show a decreased proneness to acquiring DNA damage.
Results
Increased Culture Density Directly Correlates with Higher Levels of DNA Damage in hESC Grown on Feeder Layers
We studied the effect of culture density in hESC cultured on mouse embryonic fibroblasts (MEF) and in Knockout DMEM supplemented with 20% Knockout serum replacement, as commonly used by other laboratories worldwide (Fraga et al., 2011). We cultured hESC in increasing densities, with a 1:5 ratio between each condition, and all analyses were carried out 5 days after plating. We termed the conditions A to D (Figure 1A), considering condition A as the basal level to which all results were normalized (all absolute values can be found in Figure S1). Conditions B and C are regularly used for routine hESC culture. Condition D consisted of semi-confluent colonies. We carried out all experiments at least in triplicate; details on the setup, number of replicates, cell lines used, and methods for each experiment can be found in Table S1.
Figure 1.

Increased Culture Density Correlates with Increased DNA Damage
(A) Example of the hESC culture dishes at densities A to D on day 5 after plating.
(B) Alkaline COMET assay detects an increase in DNA fragmentation between lowest-density groups A and B and the two highest-density groups C and D (n = 6, p < 0.05; ANOVA).
(C) Neutral COMET assay does not detect a significant increase in double-stranded DNA damage (n = 3, p = 0.10; ANOVA).
(D) Quantification of immunocytochemical analysis of γH2AX foci detects a significant increase of double-stranded breaks in the two highest concentrations C and D, compared with A (n = 4, p < 0.05; ANOVA).
(E and F) Distribution of COMET assay results shown in (B) and (C), respectively, in four groups based on tail DNA per cell.
(G) Distribution of γH2AX foci shown in (D) in four groups based on number of nuclear foci.
All results are presented as mean ± SEM and are presented relative to condition A. Asterisk marks differences with p ≤ 0.05.
First we quantified DNA damage in the four culture conditions by COMET assay (single-cell gel electrophoresis, Figure S1A) and by immunocytochemistry for γH2AX foci, one of the earliest markers available for DNA damage (Figure S1B). We found a 75% increase in total DNA damage in the two highest culture densities (Figure 1B), a 37% increase in double-stranded breaks (Figure 1C), and a 60% increase in the mean number of γH2AX foci per nucleus (Figure 1D).
We then subdivided all analyzed cells in four groups based on their tail DNA content or number of γH2AX foci. We found that the relative increase in DNA damage was mainly due to a larger fraction of cells accumulating more DNA damage, rather than an overall increase in DNA breaks in all cells (Figures 1E–1G). Furthermore, it is worth noting a plateau effect of the culture density on DNA damage, with similar levels in conditions C and D.
High-Density Culture Induces Chromosomal Abnormalities in hESC
DNA repair has an intrinsic stochastic chance of causing mutations, presuming a correlation between DNA damage and mutation load. To test whether the increased DNA damage had an effect on the genomic stability of the hESC, we screened 29 individual cells from condition A and 30 from condition D for copy number variations (CNVs) using single-cell array-based comparative genomic hybridization (aCGH) as previously described (Jacobs et al., 2014). We detected abnormalities ranging from 0.6 to 134 Mb, mostly unique to one cell and forming a low-grade mosaic. All detected CNVs were segmental deletions or duplications, with predominantly duplications (85.7%), and the majority spanned the telomere (64.3%), as illustrated in the ideogram in Figure 2 (the breakpoints of all abnormalities are listed in Table S2). Interestingly, we found five cells with a gain of 1q, a chromosomal abnormality that recurrently takes over hESC cultures (Lund et al., 2012, Nguyen et al., 2013). While 33% of cells in condition D were genetically abnormal after only 5 days of high-density culture, all cells analyzed for condition A showed a normal genetic content. This provides an interesting parallel with the increase in DNA damage with increased culture density, suggesting that the cells cope with the DNA breaks by (mis-)repairing them. It appears that the more breaks the cells need to repair, the more likely it is that they generate genetic abnormalities. In addition to this, the subclone carrying the 1q duplication also suggests that the high culture density can lead to an environment with a higher selective pressure for the cells, favoring the culture takeover of hESC carrying mutations.
Figure 2.

High Culture Density Induces Genetic Mosaicism
Overview of all chromosomal imbalances found by single-cell aCGH in 30 hESC of VUB31 growing in the highest-density condition D. Cells grown in condition A showed no de novo chromosomal changes. Gains are marked in green and losses in red.
Culture Density Has No Impact on the Levels of Apoptosis and Correlates to G1-Phase Accumulation
We quantified the levels of apoptosis in the different conditions to test whether the DNA fragmentation observed at the higher densities was caused by cell death. AnnexinV/propidium iodide (PI) staining showed no differences in the fractions of early apoptotic cells (AnnexinV+/PI−) or total dead cells (AnnexinV+/PI+) in the different conditions (Figure 3A). This not only indicates that impaired cell survival is unlikely to bias the results of the DNA damage measurements, but also that hESC are capable of surviving, and probably repairing, the DNA damage.
Figure 3.

Culture Density Affects the Cells' Metabolism and Culture Medium
(A) AnnexinV/PI analysis did not detect any difference between the culture densities for the fractions of live, apoptotic, or dead cells (n = 4; two-way ANOVA).
(B) Cell cycle profile of hESC growing in the four culture densities. We detected a constant fraction of cells in S phase, while the fraction of cells in G2/M phase decreases and the G1 fraction increases significantly (n = 4; two-way ANOVA).
(C) ROS concentration was measured by flow cytometry after staining with 2′,7′-dichlorodihydrofluorescein diacetate. No difference could be detected between the four groups (n = 4; ANOVA). Concentration of aspartate (D), glutamine (E), glucose (F), and lactate (G) on day 5 after 24 hr incubation differs between the different culture densities and on mouse embryonic fibroblast feeders (MEF) only (n = 9, p < 0.05; ANOVA).
(H) pH of the culture medium on day 5 after 24 hr incubation on the different culture densities or on feeders only decreases significantly (n = 5, p < 0.05; ANOVA).
(I and J) Evolution of lactate concentration and pH of the culture medium over the 5 days, after overnight incubation on the four culture densities and on feeders only (n = 3).
Results are presented as mean ± SEM. Asterisk marks differences with p ≤ 0.05.
Next, we analyzed the cell cycle profiles of hESC in the different conditions. Okazaki fragments also can be misinterpreted as DNA damage using the COMET assay (Olive and Banáth, 1993), so an increase of cells in S phase could have influenced the interpretation of the results described above. However, the S-phase fraction remained constant for all conditions tested, ruling out this bias (Figure 3B; individual profiles are listed in Figure S2). In addition to this, the G2/M fraction in condition D almost halved while the G1-phase fraction increased. A decrease of the number of dividing cells (G2/M phase) should normally imply a comparable decrease in the number of cells replicating their DNA (S phase). In our results, however, the S-phase fraction does not follow this pattern, staying relatively constant over all conditions, which indicates that the cells in the highest density proceed abnormally slow through S phase. We ruled out that this was due to differentiation, causing the cells to leave the pluripotent cell cycle with its characteristic short G1 phase (Becker et al., 2006) (Figure S3). Rather, the accumulation of cells in G1 is suggestive of genotoxic stress (Bárta et al., 2010, Sokolov and Neumann, 2013) while the stalling of the S phase may indicate replication stress.
Medium Acidification Is the Main Driver of DNA Damage and Genomic Instability in High-Density hESC Cultures
To identify the cause of the increased DNA damage, we quantified the levels of cellular reactive oxygen species (ROS) and analyzed the culture medium for differences in nutrients and metabolites. We found no differences in the levels of ROS (Figure 3C) and found that the medium still contained an excess of aspartate, glutamine, glucose, and folic acid (>20 μg/l) in all conditions (Figures 3D–3F). On the other hand, the lactate concentration increased to above 14 mM in condition D. This accumulation of lactic acid was directly correlated to a significant drop in pH, to below 6.9 in the highest density (Figures 3G and 3H; Table S3). These levels are known to affect cell growth and metabolism (Chen et al., 2010). Furthermore, daily measurements showed that the lactic acid concentration was significantly different between conditions A and D from day 1 onward, and the pH from day 2 onward (Figures 3I and 3J).
To determine the contribution of lactic acid accumulation on DNA damage, we cultured hESC at the density of condition B, and on day 5 applied medium with concentrations of lactic acid corresponding to conditions A to D (termed conditions L1–L4; details in Table S3). We found a relative increase of 88.4% in total DNA damage in condition L4 compared with L1 (Figures 4A and 4B). Next, we tested whether medium acidification was causing these effects by artificially mimicking the pH of the different conditions in the same experimental setup as above (conditions pHA–pHD, details in Table S3). In pHD we found an increase of 72.6% in total DNA fragmentation (Figures 4C and 4D). Combined, these results show that medium acidification due to lactic acid accumulation is the key factor, although possibly not the only factor, responsible for the negative effects of culture density on the DNA integrity (summarized in Figure 4E).
Figure 4.

Lactate-Induced Medium Acidification Induces DNA Damage
(A–D) Alkaline COMET assay shows that the DNA damage increases in hESC growing in medium with increasing lactic acid concentration (A, B) and decreasing pH (C, D) (n = 3, p < 0.05; ANOVA). The results are presented relative to condition L1 and pH1, respectively. L1–L4 and pHA–pHD contain lactic acid concentrations or are acidified to the equivalents of densities A–D, on a cell density equivalent to B. (B) and (D) present the distribution of COMET assay results shown in (A) and (C), respectively, in four groups based on tail DNA per cell.
(E) Summary of the relative increase of DNA fragmentation as measured with alkaline COMET assay in the culture density series (Figure 1B), lactic acid series (A), and pH series (C). The different increases in DNA damage (comparing D, L4, and pH4) are not statistically significantly different (p = 0.86; ANOVA).
All results are presented as mean ± SEM, and presented relative to condition A. Asterisk marks differences with p ≤ 0.05.
More Frequent Medium Replacements Prevent Impact of Culture Density on DNA Integrity
As more frequent medium replacements may overcome this effect, we refreshed the medium 8 hr before harvesting and analysis, on day 5 after plating (“prevented” conditions pA–pD, Figure 5A and Table S3). Strikingly, this resulted in a complete restoration of the DNA damage to a basal level (Figure 5B). We then tested whether we could also prevent the increase in genomic instability. Given that the pH and lactic acid were already critically abnormal before day 5, we performed a second round of experiments in which we refreshed the medium twice daily. On day 5, we carried out fluorescence in situ hybridization (FISH) for the telomeres and centromere of two chromosomes, and found that double medium changes in condition D resulted in a decrease in abnormal cells of 45%, reaching a level comparable with that of condition A (Figure 5C and Table S4). As with the measurements carried out by aCGH, here we found predominantly gains and losses of the telomeric signals, with only a few aneuploidies in condition D. Also, flow cytometry analysis showed that the decrease in G2/M cells was completely countered (Figure 5D). Thus, in line with the DNA damage, more frequent medium changes prevented an increase in chromosomal aberrations in high-density cultures.
Figure 5.

Extra Medium Replacements Can Counter Culture Density-Induced DNA Damage
(A) Schematic overview of the medium refreshments (represented by arrows) of the three experimental setups. Prev. 1 (Prevention 1) was used for the rescue of DNA damage, with the results shown in (B). Prev. 2 (Prevention 2) was used for the rescue of chromosomal abnormalities, with the results shown in (C).
(B) The DNA damage induced by growing hESC in different culture densities (A, B, C, D) is countered by refreshing the medium 8 hr before measurement (condition pA, pB, pC, and pD, respectively, n = 3, p < 0.05; ANOVA). DNA damage is measured by alkaline COMET assay, and the results are presented relative to condition A.
(C) Relative percentage of chromosomal abnormal cells as measured by FISH for telomeres and centromere of chromosome 18, in cells growing in culture densities A and D, and following the Prevention 2 protocol (condition pA and pD, respectively). The results are presented relative to condition A, and pD is significantly lower than D (n = 3, p < 0.05, Student's t test).
(D) Average percentages of cells in G1, S, or G2/M phase for hESC grown in the Prevention 2 series. All fractions stay constant in the four culture conditions (p > 0.05; two-way ANOVA).
(E) Alkaline COMET assay for hESC growing in HEPES-enriched medium did not show significant differences in DNA damage compared with the normal culture density series (n = 3, p > 0.05; ANOVA).
(F) Average percentages of cells in G1, S, or G2/M phase for hESC grown in the HEPES density series. The results show that while there is a constant fraction of cells in S phase in all conditions, in condition D there is a decrease of cells in G2/M (n = 3, p < 0.05; two-way ANOVA).
(G) An example of the hESC culture dishes at densities LaA to LaD on day 5 after plating on laminin-521. Scale bar, 250 μm.
(H) The DNA damage induced by growing hESC in different culture densities in feeder-free conditions (LaA–LaD), showing no increase in DNA damage, as measured by alkaline COMET assay, and less DNA damage than hESC growing in high density on MEFs (n = 3, p < 0.05, ANOVA).
(I) Relative percentage of genetically abnormal cells as measured by FISH for the telomeres and centromere of chromosome 18, in cells grown in conditions LaA and LaD relative to condition A (n = 3, p = 0.11 for A and LaA, p = 0.60 for LaA and LaD; Student’s t test).
(J) Average percentages of cells in G1, S, or G2/M phase for hESC grown in the laminin density series (n = 3). The results suggest a decrease in proliferation in the highest-density condition LaD, as shown by a decrease in S and G2/M phases.
All results are presented as mean ± SEM, and presented relative to condition A. Asterisk marks differences with p ≤ 0.05.
Next, we tested whether increasing the pH-buffer capacity of the bicarbonate-buffered culture medium by adding 25 mM HEPES could yield the same effect (series HA to HD). We found that the pH in condition D still dropped to 6.9 and that the DNA damage was not significantly different compared with the normal culture density series (Figure 5E and Table S3). The decrease in G2/M cells in the highest-density group was also still present, though less pronounced (Figure 5F).
The Impact of Culture Density on Cells Grown on Laminin-521
Finally, we tested the effect of hESC growing in different culture densities on laminin-521, a feeder-free system (Rodin et al., 2014), using a similar experimental setup, in which condition LaD became confluent between days 4 and 5 (Figure 5G). While LaA showed a degree of DNA fragmentation similar to that of A, LaD showed significantly less DNA fragmentation than D (Figure 5H) despite the pH in the highest-density condition LaD still dropping to 6.9 (Table S3). In line with this, FISH analysis showed that condition LaD did not contain more chromosomally abnormal cells than LaA (p = 0.60; Student’s t test), and although the basal level of abnormal cells in laminin-521 cultures appeared higher than on feeder layers, this difference did not reach statistical significance (p = 0.11, Student’s t test; Figure 5I and Table S3). The cell cycle profiles of the cells in the higher densities showed that the cells are slowing down their proliferation (Figure 5J), likely due to contact inhibition. This way the cells might be indirectly protected from acquiring DNA damage in higher-density cultures.
Discussion
In this study we demonstrate a direct correlation between culture density and the occurrence of DNA damage and genomic alterations in hESC. When grown in higher densities, hESC are exposed to higher lactate concentrations, leading to a lower medium pH. This, in turn, results in an increase of cells in G1 and a stalling of the S phase, without an increase in cell death or a loss of pluripotency. The accumulation of cells in G1 indicates that the hESC are under genotoxic stress, while the stalling of the S phase suggests that the cells are undergoing replication stress. The latter may lead to DNA damage and mediate the occurrence of de novo chromosomal rearrangements. Finally, we show that medium acidification is the main causal factor of these effects on the hESC DNA in high-density cultures, and that by increasing the frequency of the medium refreshments the levels of DNA damage and genetic instability can be restored to those found in the lowest culture density. Conversely, our modification of the culture medium did not result in a satisfactory increase in its buffering capacity, and consequently did not affect the effects of the high-density culture. In short, we describe a direct correlation between culture conditions on the one hand and genomic integrity and instability on the other, and provide evidence of the key factors involved: a drop in pH caused by lactic acid accumulation.
The exact mechanism by which the low pH induces DNA damage remains unanswered so far, and is an intriguing question. Whether it is a direct effect of the pH, or is rather caused by other aberrations of the cells' metabolism, is unclear. Notably, by adding lactate or lowering the pH of the medium we could only mimic the effects of condition D, while the effect of C was only partially replicated. This indicates that other medium components also have a role in this, although much less pronounced. In cancer research, a correlation has been found between hypoxia combined with a low pH and a lower capacity to repair DNA damage (Yuan et al., 2000). The authors suggest that this defective DNA repair may be caused by abnormal protein levels or folding. In the light of this, it is likely that the final outcome of an increased genomic instability is the result of a multi-step process, in which medium acidification through lactate accumulation is the trigger.
Our study highlights the importance of optimizing the current culture conditions used for hPSC, to prevent, or at least limit, genetic drift of the cells during long-term culture. As we describe herein a significant increase in genomic instability after only 5 days of culture, the long-term effect of suboptimal culture conditions on the genomic integrity of cell lines could be substantial. Our work shows that there is still much room for improvement regarding culture systems. One of the possible optimizations could be to ensure frequent medium refreshment in high-density cultures. This could be achieved using, for instance, perfusion culture systems, which provide a continuous flow of medium and allow for an optimal monitoring and control of nutrient and metabolite concentrations. For mouse ESC, for example, it has been shown that perfusion cultures are able to limit the metabolic toxicity on the growth rate and pluripotent state (Yeo et al., 2013).
Since the production of lactic acid seems to be the cause of the genotoxic stress, modifying the culture medium could control its production and its effects. Because hESC do not metabolize carbon sources such as fructose or galactose, which generate less lactate, an option could be to use lower concentrations of glucose in the medium. This seems to lead to lower levels of lactate in hESC cultures, without significantly decreasing cell growth (Chen et al., 2010). A second possibility is to provide a better pH buffer, as we tried to achieve by the addition of HEPES. In this sense, the lack of detailed information of the composition of the KnockOut DMEM culture medium is probably the main reason for our lack of success.
Alternatively, we show that during culture in the feeder-free system on laminin-521, the cells are significantly less prone to DNA damage linked to increased culture density, despite similar decreases in the pH of the culture medium. The exact reason for this is unclear, but the different medium used for this system (Nutristem) could have an impact on the differences between the laminin-521 and the MEF-based system. Indeed, cells grown on a matrix differ significantly in morphology and culture dynamics to cells on feeders. Furthermore, cultures on laminin-521 show other remarkable differences, such as their tolerance to single-cell passaging, which causes massive cell death in cells on feeders. It is likely that these significant biological differences lead to the cells slowing down their cell cycle progression by the time that the culture medium is becoming critically acidified, mainly because of the levels of confluence in the dish, and in this way indirectly protecting the cells from DNA damage. Notably, despite this decrease in DNA damage, we could not detect any significant differences in the number of genomic aberrations between the lowest density on feeders and the cells grown on laminin-521. Conversely, despite the differences being not statistically significant, our results suggest that cells on laminin-521 may carry a higher percentage of low-grade mosaicism.
In light of this, the capacity of this culture system to preserve genomic stability, especially during long-term culture, still remains to be assessed. There is currently little information in this regard (Garitaonandia et al., 2015, Rodin et al., 2014); future studies will tell whether these environments are indeed capable of providing the pristine cell cultures necessary for cell-based therapies.
Experimental Procedures
hESC Culture
Human ESC lines (VUB07, VUB14, and VUB31) were derived and characterized at our institute (Mateizel et al., 2006, Mateizel et al., 2010). Undifferentiated cells were maintained on mitomycin C-inactivated CF1-MEF (Millipore), in Knockout DMEM (Invitrogen) supplemented with 20% Knockout serum replacement (Invitrogen), 2 mM L-glutamine (Invitrogen), 1% non-essential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 4 ng/ml human recombinant basic fibroblast growth factor (hrbFGF; Invitrogen), penicillin (10 units/ml), and streptomycin (10 μg/ml) (Gibco), and were manually passaged every 5–6 days.
For culture density experiments, cells were plated in a 1:5 dilution series (illustrated in Figure 1A). The lowest-density condition (A) contained 1.99 ± 0.14 colonies per cm2 (the equivalent of 1.73 ± 0.27 × 104 cells), condition B 8.21 ± 0.47 colonies per cm2 (5.32 ± 1.97 × 104 cells), condition C 31.61 ± 2.15 colonies per cm2 (35.53 ± 15.86 × 104 cells), and the highest-density condition (D) consisted of semi-confluent colonies (133.77 ± 29.50 × 104 cells). For analysis, undifferentiated hESC were isolated after 5 min incubation with non-enzymatic cell dissociation solution (Sigma-Aldrich). For each experiment, two 12-well plates were used, with three wells grown in density D, three in density C, six in density B, and 12 in density A. For analysis, all cells of each condition were collected and pooled to minimize possible stochastic variation among wells.
For the pH and lactate concentration experiments, cells were plated in 12-well plates at a density of 8.97 ± 0.44 colonies per cm2. Here, lactic acid was added to standard hESC medium to create a dilution series of 0, 5, 10, and 15 mM lactate (condition L1, L2, L3, and L4, respectively). For the pH experiments, HCl was added to the standard hESC culture medium, creating culture medium with pH 7.37, 7.25, 7.07 and 6.79 (condition pHA, pHB, pHC, and pHD, respectively).
For feeder-free culture, hESC were cultured on dishes coated with 1 μg/cm2 laminin-521 (Biolamina) in Nutristem hESC XF culture medium (Biological Industries) supplemented with 10 mM penicillin and streptomycin (Life Technologies). Cells were cultured at 37°C, 5% CO2, and atmospheric O2 conditions, and the medium was changed daily. Cells were passaged enzymatically as single cells using TrypLE Select Enzyme (Life Technologies). After centrifugation at 120 × g for 5 min, cells were plated in a ratio ranging from 1:10 to 1:50 depending on the growth of the cell line during normal routine culture. For the density series, cell were plated in a 1:5 dilution series, resulting in 0.59 ± 0.16 × 104 cells per cm2 for the lowest-density condition LaA, 4.05 ± 1.3 × 104 cells per cm2 for LaB, 17.46 ± 1.19 × 104 cells per cm2 for LaC, and 54.13 ± 11.2 × 104 cells per cm2 for LaD.
Medium Analysis
Medium incubated overnight from days 4 to 5 was analyzed by high-performance liquid chromatography for L-glutamine and L-aspartate as previously described (Van Hemelrijck et al., 2005). Glucose, lactate, and folic acid concentrations were measured on a VITROS 5.1 FS (Ortho Clinical Diagnostics; Laboratory for Clinical Chemistry, UZ Brussels). The pH was measured with a Cobas b221 (Roche; Laboratory for Clinical Chemistry, UZ Brussels).
Single-Cell Gel Electrophoresis Assay
Alkaline and neutral COMET assays were performed as previously described (De Boeck et al., 2003, Wojewódzka et al., 2002). In brief, cells embedded in agarose on glass slides were lysed overnight in lysis buffer (2.5 M NaCl, 10 mM Tris, 100 mM EDTA salt [pH 10], supplemented with 10% DMSO and 1% Triton X-100 before use). For the alkaline COMET assay, slides were incubated in denaturation buffer for 40 min, followed by electrophoresis for 20 min at 25 V and 300 mA, and washed with neutralization buffer (360 mM Tris [pH 7.5]). For the neutral COMET assay, slides were kept for 1 hr in electrophoresis buffer (300 mM sodium acetate, 100 mM Tris [pH 8.3]) before electrophoresis for 1 hr at 14 V and 12 mA. Analysis was performed using a Leitz Dialux 20 microscope and Komet 5.5 software. The percentage of labeled DNA migrating out of the nucleus, and thus forming the tail of the “comet,” is indicative of the fragmentation of the cell DNA and is here termed “tail DNA.”
Immunocytochemistry
Cells were dissociated using non-enzymatic cell dissociation solution for 5–8 min and washed in PBS with 1% BSA (all from Sigma-Aldrich). Cytospins were made on glass slides and samples were frozen. After 10 min fixation with 4% formaldehyde, cells were permeabilized with PBS-T (0.1% Triton X-100 in PBS) and blocking was done with 3% BSA. Anti-γH2AX antibody (ab11175, Abcam) diluted in PBS-T with 3% BSA was incubated overnight at 4°C; Alexa Fluor 488-labeled secondary antibody (Invitrogen) was incubated for 1 hr at room temperature. Subsequently, anti-mouse laminin (ab30320, Abcam) diluted in PBS-T with 3% BSA was incubated overnight at 4°C; Alexa Fluor 546-labeled secondary antibody was incubated for 1 hr at room temperature. DNA was counterstained with Hoechst 33342 and the slides were mounted with ProLong Gold Antifade Reagent (Invitrogen). Analysis was done with an Olympus IX81 microscope and CellSens software.
Cell Cycle and ROS Analysis
Cells were dissociated using non-enzymatic cell dissociation solution for 5–8 min, washed in PBS with 1% BSA, and fixed in ice-cold methanol (all from Sigma-Aldrich). For cell cycle analysis, cells were rehydrated in PBS and treated with RNase A (Sigma) for 30 min at 37°C. After staining with 2 μg/ml PI (Sigma), cells were analyzed for DNA content using a BDFACSCanto flow cytometer. Forward and side scatter gates were chosen to select for undifferentiated cells. Cell cycle profile was calculated with the Dean-Jett-Fox model using Flowjo software.
For ROS determination, cells were incubated with 30 μM 2′,7′-dichlorodihydrofluorescein diacetate (Invitrogen) for 1 hr, followed by 40 min incubation in conditioned medium. Afterward, cells were dissociated using non-enzymatic cell dissociation solution, washed in PBS with 1% BSA, counterstained with PI, and analyzed using a BDFACSCanto flow cytometer.
Cell Viability Assay
Cell viability was measured by Annexin V and PI staining. Cells were dissociated using non-enzymatic cell dissociation solution and incubated for 20 min with Annexin V Alexa Fluor 488 at room temperature. After washing with Annexin binding buffer, cells were resuspended in 1 μg/ml PI (all from Invitrogen). Analysis was done using the Tali Image-Based Cytometer (Invitrogen).
Quantitative Real-Time PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) following the manufacturer's instructions. Reverse transcription was performed using the First-Strand cDNA Synthesis Kit (GE Healthcare) following the manufacturer's protocol. For the gene-expression studies using multiple endogenous controls (UBC, GAPDH, and GUSB), the real-time PCR was carried out using the ViiA 7 thermocycler (Life Technologies) and analyzed using ViiA 7 software version 1.2 (Life Technologies). Reactions were performed under the fast mode, in 20 μl total volume, comprising 10 μl of 2× TaqMan Fast Universal Master Mix (Life Technologies), 40 ng of cDNA, 1 μl of TaqMan gene-expression assays for GUSB (Hs99999908_m1), or 900 nM primer mix and 250 nM probes for NANOG, POU5F1 (OCT4), UBC, and GAPDH. The sequences for the probe and the reverse and forward primers were: NANOG-Probe: 6-FAM-CAG AGA CTG TCT CTC CTC-MGB; NANOG-F: 5′-TGCAAATGTCTTCTGCTGGATG-3′; NANOG-R: 5′-TCCTGAATAAGCAGATCCATGGA-3′; UBC-Probe: 6-FAM-TCG CAG TTC TTG TTT GTG-MGB; UBC-F: 5′-CGC AGC CGG ATT TG-3′; UBC-R: 5′-TCAAGTGACGATACAGCGA-3′, GAPDH-Probe: 6-FAM-CAG GAG CGA GAT CC-MGB; GAPDH-F: 5′-ATGGAAATCCCATCACCATCTT-3′; GAPDH-R: 5′-CGCCCCACTTGATTTTGG-3′. hESC were differentiated into osteogenic progenitor-like cells as described earlier (Mateizel et al., 2008) and used as control sample.
Fluorescence In Situ Hybridization
FISH probing was performed following the manufacturer's instructions using Telvysion 18p, Telvysion 18q, and Vysis CEP 18 probes (Abbott Molecular). In short, cell fixation was performed using Carnoy fixative (3:1 methanol/acetic acid), and slides were subsequently dehydrated in a dilution series of ethanol in water. Probe co-denaturation was done on a 75°C hot plate for 3 min, followed by hybridization in a humidified chamber at 37°C overnight. After post-hybridization washing in saline sodium citrate (SSC) buffer (Gibco) and Igepal CA-630 (Sigma), slides were mounted with Vectashield with DAPI (Vector Laboratories). Analysis was done using a Zeiss axyoplan-2 microscope.
Single-Cell Array-Based Comparative Genomic Hybridization
Single hESC were washed and collected into sterile 0.2-ml PCR tubes containing 2 μl of PBS (Cell Signaling Technologies) with 0.1% polyvinylpyrrolidone as previously described (Mertzanidou et al., 2013). Cells were lysed and the genomic DNA was amplified using the Sureplex DNA Amplification System (BlueGnome) following the manufacturer's instructions. Male and female amplified genomic DNA (SureRef Reference DNA, BlueGnome, Illumina) were used as reference DNA. The BlueGnome 24sure platform was used for aCGH. In brief, the amplified DNA was labeled following the standard labeling plan, with a PCR reaction at 37°C for 4 hr. After ethanol precipitation at −80°C for 20 min, the DNA was resuspended and denatured in hybridization buffer for 10 min at 75°C combined with regular vortexing. The samples were incubated overnight at 47°C in humidified chambers (50% formamide/2% SSC), followed by the standard 24sure washing protocol. An Agilent dual-laser DNA microarray scanner G2566AA (Agilent Technologies) was used for scanning the arrays.
TIFF files were imported and processed using BlueFuse Multi 3.1 (BlueGnome). Cutoff values for our quality control were SD <0.2, included clones >80%, signal-to-background ratio >3, and derivative log ratio <0. Log2 ratios per clone were exported and analyzed using the online “circular binary segmentation” logarithm (http://compbio.med.harvard.edu/CGHweb). As described previously (Jacobs et al., 2014); the resolution of our system was set at a minimum of seven consecutive clones, with a log2-ratio of ≥0.3 for chromosomal gains and ≤−0.45 for chromosomal loss.
Statistics
All results are presented as mean ± SEM. The COMET and γH2AX foci groups of Figures 1E–1G, cell viability analysis (Figure 3A), cell cycle profiles (Figures 3B, 5C, and 5E), and the pH and lactic acid time series presented in Figures 3I and 3J were analyzed using two-way ANOVA with a Bonferroni post hoc test. The aCGH data were analyzed using a Fisher exact test. For comparison of the mean number of aberrant cells after FISH analysis, we used Student’s t test because of the small number of replicates (n = 3). For all other datasets, we used ANOVA variation analysis with a Bonferroni post hoc test for statistical analysis.
Author Contributions
K.J. co-wrote the manuscript and performed all experiments except when stated otherwise; F.Z. assisted with COMET assays; A.M. performed single-cell tubing for aCGH; I.S. performed high-performance liquid chromatography measurements; M.G. supervised hESC culture; H.T.N. performed gene-expression studies; L.B. assisted with COMET assays; K.S. and C.S. supervised the study and co-wrote the manuscript. All co-authors proofread the manuscript.
Acknowledgments
The authors acknowledge Laetitia Gonzalez for her help with the COMET assay, Pedro Couck and Ilse Weets for their assistance in measuring pH and lactic acid concentrations, and their colleagues from the hESC laboratory for the derivation and culture of the cell lines. This work was supported by the Fund for Scientific Research Flanders (Fonds voor Wetenschappelijk Onderzoek [FWO] Vlaanderen) and the Methusalem grant to Karen Sermon of the Research Council of the Vrije Universiteit Brussel. Claudia Spits is a postdoctoral fellow at the FWO Vlaanderen. Lise Barbé is supported by the Leerstoel Mireille Aerens (Vrije Universiteit Brussel).
Published: February 25, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes three figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.01.015.
Accession Numbers
The GEO accession number for the aCGH data reported in this paper is GEO: GSE58281.
Supplemental Information
References
- Amps K., Andrews P.W., Anyfantis G., Armstrong L., Avery S., Baharvand H., Baker J., Baker D., Munoz M.B., Beil S. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery S., Hirst A.J., Baker D., Lim C.Y., Alagaratnam S., Skotheim R.I., Lothe R.A., Pera M.F., Colman A., Robson P. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Rep. 2013;1:379–386. doi: 10.1016/j.stemcr.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárta T., Vinarský V., Holubcová Z., Dolezalová D., Verner J., Pospísilová S., Dvorák P., Hampl A. Human embryonic stem cells are capable of executing G1/S checkpoint activation. Stem Cells. 2010;28:1143–1152. doi: 10.1002/stem.451. [DOI] [PubMed] [Google Scholar]
- Becker K.A., Ghule P.N., Therrien J.A., Lian J.B., Stein J.L., van Wijnen A.J., Stein G.S. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell. Physiol. 2006;209:883–893. doi: 10.1002/jcp.20776. [DOI] [PubMed] [Google Scholar]
- Ben-Nun I.F., Benvenisty N. Human embryonic stem cells as a cellular model for human disorders. Mol. Cell. Endocrinol. 2006;252:154–159. doi: 10.1016/j.mce.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Burrell R.A., McClelland S.E., Endesfelder D., Groth P., Weller M.-C., Shaikh N., Domingo E., Kanu N., Dewhurst S.M., Gronroos E. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Chen A., Woo T.L., Choo A.B.H., Reuveny S., Oh S.K.W. Investigations into the metabolism of two-dimensional colony and suspended microcarrier cultures of human embryonic stem cells in serum-free media. Stem Cells Dev. 2010;19:1781–1792. doi: 10.1089/scd.2010.0077. [DOI] [PubMed] [Google Scholar]
- De Boeck M., Lombaert N., De Backer S., Finsy R., Lison D., Kirsch-Volders M. In vitro genotoxic effects of different combinations of cobalt and metallic carbide particles. Mutagenesis. 2003;18:177–186. doi: 10.1093/mutage/18.2.177. [DOI] [PubMed] [Google Scholar]
- Dekel-Naftali M., Aviram-Goldring A., Litmanovitch T., Shamash J., Reznik-Wolf H., Laevsky I., Amit M., Itskovitz-Eldor J., Yung Y., Hourvitz A. Screening of human pluripotent stem cells using CGH and FISH reveals low-grade mosaic aneuploidy and a recurrent amplification of chromosome 1q. Eur. J. Hum. Genet. 2012;20:1248–1255. doi: 10.1038/ejhg.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli A., Liew C.G., Matin M.M., Elliott S., Jeanmeure L.F.C., Wright P.C., Moore H., Andrews P.W. Altered patterns of differentiation in karyotypically abnormal human embryonic stem cells. Int. J. Dev. Biol. 2011;55:175–180. doi: 10.1387/ijdb.103177af. [DOI] [PubMed] [Google Scholar]
- Fraga A.M., Souza de Araújo É.S., Stabellini R., Vergani N., Pereira L.V. A survey of parameters involved in the establishment of new lines of human embryonic stem cells. Stem Cell Rev. 2011;7:775–781. doi: 10.1007/s12015-011-9250-x. [DOI] [PubMed] [Google Scholar]
- Garitaonandia I., Amir H., Boscolo F.S., Wambua G.K., Schultheisz H.L., Sabatini K., Morey R., Waltz S., Wang Y.-C., Tran H. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS One. 2015;10:e0118307. doi: 10.1371/journal.pone.0118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishna-Pillai S., Iverson L.E. Astrocytes derived from trisomic human embryonic stem cells express markers of astrocytic cancer cells and premalignant stem-like progenitors. BMC Med. Genomics. 2010;3:12. doi: 10.1186/1755-8794-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herszfeld D., Wolvetang E., Langton-Bunker E., Chung T.-L., Filipczyk A.A., Houssami S., Jamshidi P., Koh K., Laslett A.L., Michalska A. CD30 is a survival factor and a biomarker for transformed human pluripotent stem cells. Nat. Biotechnol. 2006;24:351–357. doi: 10.1038/nbt1197. [DOI] [PubMed] [Google Scholar]
- Jacobs K., Mertzanidou A., Geens M., Thi Nguyen H., Staessen C., Spits C. Low-grade chromosomal mosaicism in human somatic and embryonic stem cell populations. Nat. Commun. 2014;5:4227. doi: 10.1038/ncomms5227. [DOI] [PubMed] [Google Scholar]
- Lim H., Han J., Woo D., Kim S., Kim S. Biochemical and morphological effects of hypoxic environment on human embryonic stem cells in long-term culture and differentiating embryoid bodies. Mol. Cells. 2011;31:123–132. doi: 10.1007/s10059-011-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund R.J., Närvä E., Lahesmaa R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012;13:732–744. doi: 10.1038/nrg3271. [DOI] [PubMed] [Google Scholar]
- Mateizel I., De Temmerman N., Ullmann U., Cauffman G., Sermon K., Van de Velde H., De Rycke M., Degreef E., Devroey P., Liebaers I., Van Steirteghem A. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum. Reprod. 2006;21:503–511. doi: 10.1093/humrep/dei345. [DOI] [PubMed] [Google Scholar]
- Mateizel I., De Becker A., Van de Velde H., De Rycke M., Van Steirteghem A., Cornelissen R., Van der Elst J., Liebaers I., Van Riet I., Sermon K. Efficient differentiation of human embryonic stem cells into a homogeneous population of osteoprogenitor-like cells. Reprod. Biomed. Online. 2008;16:741–753. doi: 10.1016/s1472-6483(10)60490-7. [DOI] [PubMed] [Google Scholar]
- Mateizel I., Spits C., De Rycke M., Liebaers I., Sermon K. Derivation, culture, and characterization of VUB hESC lines. In Vitro Cell. Dev. Biol. Anim. 2010;46:300–308. doi: 10.1007/s11626-010-9284-4. [DOI] [PubMed] [Google Scholar]
- Mertzanidou A., Spits C., Nguyen H.T., Van de Velde H., Sermon K. Evolution of aneuploidy up to day 4 of human preimplantation development. Hum. Reprod. 2013;28:1716–1724. doi: 10.1093/humrep/det079. [DOI] [PubMed] [Google Scholar]
- Nguyen H.T., Geens M., Spits C. Genetic and epigenetic instability in human pluripotent stem cells. Hum. Reprod. Update. 2013;19:187–205. doi: 10.1093/humupd/dms048. [DOI] [PubMed] [Google Scholar]
- Nguyen H.T., Geens M., Mertzanidou A., Jacobs K., Heirman C., Breckpot K., Spits C. Gain of 20q11.21 in human embryonic stem cells improves cell survival by increased expression of Bcl-xL. Mol. Hum. Reprod. 2014;20:168–177. doi: 10.1093/molehr/gat077. [DOI] [PubMed] [Google Scholar]
- Olive P.L., Banáth J.P. Induction and rejoining of radiation-induced DNA single-strand breaks: “tail moment” as a function of position in the cell cycle. Mutat. Res. 1993;294:275–283. doi: 10.1016/0921-8777(93)90010-e. [DOI] [PubMed] [Google Scholar]
- Rodin S., Antonsson L., Niaudet C., Simonson O.E., Salmela E., Hansson E.M., Domogatskaya A., Xiao Z., Damdimopoulou P., Sheikhi M. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nat. Commun. 2014;5:3195. doi: 10.1038/ncomms4195. [DOI] [PubMed] [Google Scholar]
- Sokolov M., Neumann R. Lessons learned about human stem cell responses to ionizing radiation exposures: a long road still ahead of us. Int. J. Mol. Sci. 2013;14:15695–15723. doi: 10.3390/ijms140815695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits C., Mateizel I., Geens M., Mertzanidou A., Staessen C., Vandeskelde Y., Van der Elst J., Liebaers I., Sermon K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 2008;26:1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- Tabar V., Studer L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat. Rev. Genet. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hemelrijck A., Sarre S., Smolders I., Michotte Y. Determination of amino acids associated with cerebral ischaemia in rat brain microdialysates using narrowbore liquid chromatography and fluorescence detection. J. Neurosci. Methods. 2005;144:63–71. doi: 10.1016/j.jneumeth.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Werbowetski-Ogilvie T.E., Bossé M., Stewart M., Schnerch A., Ramos-Mejia V., Rouleau A., Wynder T., Smith M.-J., Dingwall S., Carter T. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009;27:91–97. doi: 10.1038/nbt.1516. [DOI] [PubMed] [Google Scholar]
- Wojewódzka M., Buraczewska I., Kruszewski M. A modified neutral comet assay: elimination of lysis at high temperature and validation of the assay with anti-single-stranded DNA antibody. Mutat. Res. 2002;518:9–20. doi: 10.1016/s1383-5718(02)00070-0. [DOI] [PubMed] [Google Scholar]
- Yang S., Lin G., Tan Y., Zhou D., Deng L., Cheng D., Luo S., Liu T., Zhou X., Sun Z. Tumor progression of culture-adapted human embryonic stem cells during long-term culture. Genes Chromosomes Cancer. 2008;47:665–679. doi: 10.1002/gcc.20574. [DOI] [PubMed] [Google Scholar]
- Yeo D., Kiparissides A., Cha J.M., Aguilar-Gallardo C., Polak J.M., Tsiridis E., Pistikopoulos E.N., Mantalaris A. Improving embryonic stem cell expansion through the combination of perfusion and Bioprocess model design. PLoS One. 2013;8:e81728. doi: 10.1371/journal.pone.0081728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Narayanan L., Rockwell S., Glazer M.P. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000;60:4372–4376. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.