

Abstract
Background
Genome-wide association studies (GWAS) have identified thousands of genetic variants associated with complex traits and diseases. However, most of them are located in the non-protein coding regions, and therefore it is challenging to hypothesize the functions of these non-coding GWAS variants. Recent large efforts such as the ENCODE and Roadmap Epigenomics projects have predicted a large number of regulatory elements. However, the target genes of these regulatory elements remain largely unknown. Chromatin conformation capture based technologies such as Hi-C can directly measure the chromatin interactions and have generated an increasingly comprehensive catalog of the interactome between the distal regulatory elements and their potential target genes. Leveraging such information revealed by Hi-C holds the promise of elucidating the functions of genetic variants in human diseases.
Results
In this work, we present HiView, the first integrative genome browser to leverage Hi-C results for the interpretation of GWAS variants. HiView is able to display Hi-C data and statistical evidence for chromatin interactions in genomic regions surrounding any given GWAS variant, enabling straightforward visualization and interpretation.
Conclusions
We believe that as the first GWAS variants-centered Hi-C genome browser, HiView is a useful tool guiding post-GWAS functional genomics studies. HiView is freely accessible at: http://www.unc.edu/~yunmli/HiView.
Electronic supplementary material
The online version of this article (doi:10.1186/s13104-016-1947-0) contains supplementary material, which is available to authorized users.
Keywords: Integrative genome browser, Hi-C data, GWAS variants
Findings
The eukaryotic genome is organized at multiple levels ranging from chromosomal territories to topologically associated domains. Such hierarchical three-dimensional organization is closely related to genome function [1]. Historically, the study of genome organization has relied on microscopy-based techniques, which suffers from low resolution and low throughput. Recently, a series of technologies based on chromatin conformation capture (3C) [2], such as Hi-C [3] and in situ Hi-C [4], have been developed, enabling a high resolution genome-wide view of chromosomal architecture.
Data from 3C-based technologies can shed light on the structural and functional mechanisms, including non-coding variants identified for complex trait associations in genome-wide association studies (GWAS). GWAS has been resoundingly successful, identifying thousands of variants associated with complex traits. However, only a small proportion (7–12 %) of these variants fall into protein coding regions [5], making the interpretation of non-coding variants imperative. With the help of 3C-based technologies, a recent study [6] identified long-range (at megabase distances) interactions between the obesity-associated intronic variants in FTO gene and the promoter region of homeobox gene IRX3, demonstrating it is the expression of IRX3 rather than FTO that is directly linked to body mass and composition. This study showcased the power of 3C-based technologies for elucidating the functional mechanisms of genetic variants implicated by GWAS.
As 3C-derived technologies have been increasingly widely used, multiple visualization tools have been devised recently, such as Hi-C data browser [3] and 3D genome browser [7]. In addition, WashU EpiGenome browser is widely utilized for simultaneous visualization of Hi-C and other epigenetic data from the Roadmap Epigenomics project [8]. Most recently, Juicebox has been developed for visualizing the in situ Hi-C data [4]. Meanwhile, HiBrowse [9] has been developed to facilitate statistical analysis of Hi-C data.
Although many useful visualization tools have been developed, none of them is able to display 3C-based data with a focus on GWAS variants interpretation, preventing researchers from fully mining rich information, generating testable hypothesis, and visually validating biological findings. In addition, few of them incorporates peak calling results from 3C-based data or shows the magnitude of statistical evidence, making the interpretation of the statistical significance of 3C-based data extremely challenging.
To fill in the above gaps, we present HiView, the first genome browser for GWAS-variant centered visualization of Hi-C data. Additional file 1: Figure S1 shows the user interface of HiView. Users can select and extract genomic annotation of a GWAS variant by selecting the marker type and specifying the marker name. HiView displays raw and expected count data, and measures of statistical significance from several state-of-the-art Hi-C peak callers, such as AFC [10], Fit-Hi-C [11] and a hidden Markov random field (HMRF) based Hi-C peak caller [12]. By creating an ensemble of peak calling results from different approaches, users can have more robust data interpretations. For gene annotation, HiView incorporates three gene annotation tracks: (1) Ensembl genes, (2) UCSC genes and (3) RefSeq genes.
Users can configure HiView for customized visualization in many ways (detailed in the online tutorial) including but not limited to (1) selecting tracks to display, (2) specifying the order of displayed tracks, (3) moving the viewing window upstream and downstream, zooming in and out, and specifying the range of the viewing window, (4) specifying the genomic regions to highlight, (5) specifying the text and color used for each track and (6) specifying the picture size and width. HiView also provides a table of numerical values of Hi-C data and peak calling results that can be downloaded by users. Figures 1 and Additional file 1: Figure S2 show an example of HiView figure and HiView table, respectively. A detailed tutorial to generate Fig. 1 can be found in the Additional file 1: Section S1.
Fig. 1.
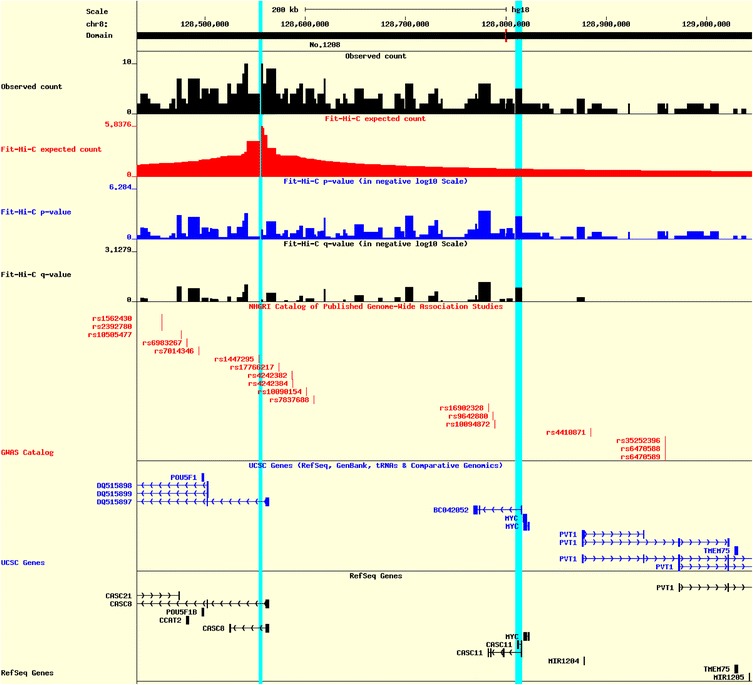
HiView snapshot of GWAS variant rs1447295. The left and right light blue bars highlight the location of GWAS variant rs1447295 and gene MYC, respectively. Using Hi-C data from human IMR90 cells, we observe five paired-end reads spanning between rs1447295 and the transcription start site of gene MYC, while the expected contact frequency is 0.8281. Such long-range chromatin interaction is statistically significant, with p-value 0.0016. Therefore, we hypothesize that gene MYC is a potential target of this likely regulatory GWAS variant rs1447295
Here is an example of using HiView to leverage Hi-C results for the interpretation of GWAS variants. Multiple studies [13, 14] have identified rs1447295 to be associated with the risk of prostate cancer. Although rs1447295 was mapped as an intronic variant in CASC8 lncRNA, its functional mechanisms are still unknown. Both RgulomeDB [15] and HaploReg [16] identify this variant as an enhancer for multiple cell lines, indicating its potential regulatory role. Using the high resolution fragment level Hi-C data from human IMR90 lung fibroblastic cells [10], we observed statistically significant long-range chromatin interactions between rs1447295 and the transcription start site of the MYC gene with p value 0.0016 (Fig. 1). Therefore, we hypothesized that MYC gene is a potential target of this likely regulatory GWAS variant rs1447295 [17]. In this work, the Hi-C data and GWAS variant were collected from different cell types. It would be more informative to integrative Hi-C data and GWAS variants from the same cancer cell line, to fully understand the mechanistic relationship. As Hi-C data from more tissue and cell types are generated, we will have a more comprehensive understanding of tissue or cell type specific target genes.
The HiView interface is implemented using PHP, HTML and cascading styling sheets (CSS) languages. Hi-C and GWAS data are stored in a MySQL database in the UNC Linux server. HiView is compatible with Internet Explorer, Chrome and Firefox. HiView also allows users to upload their own Hi-C dataset for customized comparison and visualization.
In summary, we present HiView, a visualization tool that integrates raw Hi-C data and chromatin interactions identified by various peak callers for the interpretation of GWAS variants. HiView is the first genetic GWAS-variant centered visualization tool for Hi-C data. The resulting one-dimensional view allows close examination of interactions between each GWAS variant and all genes in the region the variant resides. We believe that HiView will facilitate the interpretation of GWAS variants, particularly the identification of their potential target genes.
Availability and requirements
Project name: HiView.
Project home page: http://www.unc.edu/~yunmli/HiView.
Operating system(s): Platform independent.
Programming language: PHP, HTML and cascading styling sheets (CSS) languages.
Other requirements: browser such as Internet Explorer, Chrome and Firefox.
License: GNU GPL (version 3, 06/29/2007).
Any restriction to use by non-academics: none.
Availability of supporting data
Original raw data used in Fig. 1, Additional file 1: Figures S1 and S2 were retrieved from the NCBI Gene Expression Omnibus repository (GSE43070: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43070).
Authors’ contributions
ZX developed and implemented the software, performed the data analysis, constructed the database, and prepared the manuscript. GZ performed the data analysis and constructed the database. QD and SC performed the data analysis and wrote the online tutorial. BZ, CW and FJ performed the data analysis and constructed the database. FY prepared the manuscript. YL, MH conceived and coordinated the project, prepared the manuscript. All authors read and final approved the final manuscript.
Acknowledgements
We thank Drs. Karen Mohlke, Terrance S. Furey and their lab members for providing feedback on our web browser. This research was supported by the National Institute of Health grants R01-HG006292 and R01-HG006703 (awarded to YL), and 1U54DK107977-01 (awarded to MH).
Competing interests
The authors declare that they have no competing interests.
Additional file
10.1186/s13104-016-1947-0 HiView graphic user interface. Users can input the genomic location of a GWAS variant by (1) select the marker type, (2) type the marker name and then (3) click the Run button (red-colored). Users can configure HiView in many ways (refer to online tutorial) to obtain customized figures. HiView is freely accessible at http://www.unc.edu/~yunmli/HiView. Figure S2. HiView Table for GWAS variant rs1600249.
Contributor Information
Zheng Xu, xuzheng@email.unc.edu.
Guosheng Zhang, gszhang@email.unc.edu.
Qing Duan, qduan@email.unc.edu.
Shengjie Chai, shengjie@email.unc.edu.
Baqun Zhang, Email: zhangbaqun@ruc.edu.cn.
Cong Wu, Email: wucong0451@outlook.com.
Fulai Jin, Email: fulai.jin@case.edu.
Feng Yue, Email: fyue@hmc.psu.edu.
Yun Li, Email: yunli@med.unc.edu.
Ming Hu, Email: ming.hu@nyumc.org.
References
- 1.Dekker J. Gene regulation in the third dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao SSP, Huntley MH, Durand NC, Stamenova EK. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, Lee JH, Puviindran V, Tam D, Shen M, Son JE, Vakili NA, Sung H-K, Naranjo S, Acemel RD, Manzanares M, Nagy A, Cox NJ, Hui C-C, Gomez-Skarmeta JL, Nóbrega MA. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, Madden PAF, Smirnov I, Costello JF, Wang T. Epigenomic annotation of genetic variants using the roadmap epigenome browser. Nat Biotechnol. 2015;33(4):345–346. doi: 10.1038/nbt.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulsen J, Sandve GK, Gundersen S, Lien TG, Trengereid K, Hovig E. HiBrowse: multi-purpose statistical analysis of genome-wide chromatin 3D organization. Bioinformatics. 2014;30:1620–1622. doi: 10.1093/bioinformatics/btu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen C-A, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ay F, Bailey TL, Noble WS. Analysis of genome architecture data reveals regulatory chromatin contacts in human and mouse cell lines spline fitting corrects for binning artifacts. 2014; 1136996.
- 12.Xu Z, Zhang G, Jin F, Chen M, Furey TS, Patrick F, Qin Z, Hu M, Li Y. A hidden Markov random field based Bayesian method for the detection of long-range chromosomal interactions in Hi-C data. Bioinformatics. 2016;32(5):650–656. doi: 10.1093/bioinformatics/btv650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Tres A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 14.Knipe DW, Evans DM, Kemp JP, Eeles R, Easton DF, Kote-Jarai Z, Al Olama AA, Benlloch S, Donovan JL, Hamdy FC, Neal DE, Davey Smith G, Lathrop M, Martin RM. Genetic variation in prostate-specific antigen-detected prostate cancer and the effect of control selection on genetic association studies. Cancer Epidemiol Biomark Prev. 2014;23:1356–1365. doi: 10.1158/1055-9965.EPI-13-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward LD, Kellis M. Haplorreg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]