

Abstract
Background
Schistosoma turkestanicum is a parasite of considerable veterinary importance as an agent of animal schistosomiasis in many countries, including China. The S. turkestanicum cercariae can also infect humans, causing cercarial dermatitis in many countries and regions of the world. In spite of its significance as a pathogen of animals and humans, there is little transcriptomic and genomic data in the public databases.
Methods
Herein, we performed the transcriptome Illumina RNA sequencing (RNA-seq) of adult males and females of S. turkestanicum and de novo transcriptome assembly.
Results
Approximately 81.1 (female) and 80.5 (male) million high-quality clean reads were obtained and then 29,526 (female) and 41,346 (male) unigenes were assembled. A total of 34,624 unigenes were produced from S. turkestanicum females and males, with an average length of 878 nucleotides (nt) and N50 of 1480 nt. Of these unigenes, 25,158 (72.7 %) were annotated by blast searches against the NCBI non-redundant protein database. Among these, 21,995 (63.5 %), 22,189 (64.1 %) and 13,754 (39.7 %) of the unigenes had significant similarity in the NCBI non-redundant protein (NR), non-redundant nucleotide (NT) and Swiss-Prot databases, respectively. In addition, 3150 unigenes were identified to be expressed specifically in females and 1014 unigenes were identified to be expressed specifically in males. Interestingly, several pathways associated with gonadal development and sex maintenance were found, including the Wnt signaling pathway (103; 2 %) and progesterone-mediated oocyte maturation (77; 1.5 %).
Conclusions
The present study characterized and compared the transcriptomes of adult female and male blood fluke, S. turkestanicum. These results will not only serve as valuable resources for future functional genomics studies to understand the molecular aspects of S. turkestanicum, but also will provide essential information for ongoing whole genome sequencing efforts on this pathogenic blood fluke.
Keywords: Schistosoma turkestanicum, Transcriptome, Differentially expressed genes, Next-generation sequencing
Background
Schistosomiasis is a neglected tropical disease caused by blood flukes (genus Schistosoma) that ranks second only to malaria among the parasitic diseases and affects more than 200 million people in a number of tropical and subtropical countries [1]. It is estimated that schistosomiasis causes an annual loss of between 1.7 and 4.5 million disability adjusted life years (DALYs) and contributes to several hundreds of thousands of deaths annually [2–4]. To date, no vaccines are available, and treatment relies on one drug, praziquantel [5].
S. turkestanicum (syn. Orientobilharzia turkestanicum) is a parasite of major veterinary importance as an agent of animal schistosomiasis, which infects a range of animals including cattle, sheep, goats, water buffaloes, horses, donkeys, mules, camels, and causes considerable economic losses [6]. Importantly, the cercariae of S. turkestanicum can also infect humans, in which they can cause cercarial dermatitis, and is considered the major pathogen of cercarial dermatitis in many countries and regions of the world, including China [6, 7]. In spite of its medical and veterinary importance, the genetics, epidemiology and biology of this parasite remain poorly understood.
Recent developments in deep sequencing or next-generation sequencing (NGS) provide an opportunity for rapid and cost-effective generation of genome-scale data [8, 9]. NGS technology, such as Solexa/Illumina platforms, has dramatically improved the efficiency of gene discovery, and is particularly suited to transcriptomics of distinct animal and plant taxa [10–13]. To date, many trematode transcriptomes have been sequenced using the NGS technology, such as Fascioloides magna [14]; Clonorchis sinensis [15]; Eurytrema pancreaticum [16], Schistosoma japonicum [17], Schistosoma mansoni [18], Schistosoma haematobium [19], Opisthorchis felineus [20], Paramphistomum cervi [21] and Fasciola hepatica [22]. In spite of the availability of advances in sequencing technologies and bioinformatic methods, there is still a paucity of knowledge of the complete transcriptome for this zoonotic fluke S. turkestanicum. Herein, a NGS platform and powerful de novo short-read assembly was employed to uncover a global view of the transcriptomes of adult male and female S. turkestanicum worms. The aim of the present study was to produce transcriptomic data to aid the better understanding of the biology of S. turkestanicum, which would facilitate the identification of intervention targets for S. turkestanicum and other medically and veterinary important trematodes.
Methods
Ethics statement
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Permit code. LVRIAEC2013-006). The sheep, from which S. turkestanicum was collected, were handled in accordance with good animal practices required by the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Sample preparation
Adult blood flukes representing female and male S. turkestanicum (codes: OutF and OutM) were obtained from the portal and mesenteric veins of naturally infected sheep at a local slaughter house in Heilongjiang province, China. One blood fluke was collected from each of the five sheep, which were randomly selected for slaughter and sale, following the perfusion of the mesenteric and intestinal vessels using physiological saline (37 °C). Five blood flukes (three females and two males, respectively) were washed in physiological saline six times to remove any contamination with bacterial and host DNA, identified morphologically as S. turkestanicum according to existing keys and descriptions [23], and their identity was further ascertained by direct amplification and sequencing of the internal transcribed spacer (ITS) region, as previously described [24]. The male and female S. turkestanicum worms were immediately frozen in liquid nitrogen and stored at −80 °C until use.
RNA isolation and Illumina sequencing
Total RNA was extracted from pooled females (n = 3) and pooled males (n = 2) of S. turkestanicum, respectively, using Trizol reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol, and was stored at −80 °C until use. The Oligo (dT) was used to isolate poly (A) mRNA from total RNA. Mixed with the fragmentation buffer, the mRNA was fragmented into short fragments. Then cDNA was synthesized using the mRNA fragments as templates. Short fragments were purified and resolved with EB buffer for end reparation and single nucleotide A (adenine) addition. After that, the short fragments were connected with adapters. The suitable fragments were selected as templates for the PCR amplification. During the Quality Control (QC) steps, an Agilent 2100 Bioanaylzer and an ABI StepOnePlus Real-Time PCR System were used in quantification and qualification of the sample library. Illumina HiSeq™ 2000 was applied for sequencing at BGI-Shenzhen, Shenzhen, China according to the manufacturer’s instructions (Illumina, San Diego, CA, USA).
De novo assembly
Prior to assembly, the high-quality clean reads were obtained from raw reads by removing adaptor sequences, highly redundant sequences, reads that contained more than 10 % ‘N’ rate (the ‘N’ character representing ambiguous bases in reads), and low quality reads containing more than 50 % bases with Quality (Q) value (Q-value ≤ 10). De novo assembly of the clean reads was performed by using the Trinity software, which was designed specifically for transcriptome assembly [25]. Briefly, Trinity (this is in Trinity software) first combines reads of a certain length of overlap to form longer fragments, which are called contigs. Then the reads are mapped back to contigs. Trinity connects the contigs and identifies sequences that cannot be extended on either end. Such sequences are defined as unigenes. Unigenes from each sample’s assembly can be used in further processes of sequence splicing and redundancy removal with sequence clustering software TGICL [26] in order to acquire non-redundant unigenes that are as long as possible.
Bioinformatics analysis
Unigene sequences were first aligned to the NCBI non-redundant proteins (NR), non-redundant nucleotide (NT), Swiss-Prot, Cluster of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases by BLASTx, using an e-value <0.00001. The unigenes were tentatively annotated according to the known sequences with the highest sequence similarity. The annotated unigenes direction and predicted protein coding sequences (CDS) were identified by the best alignment results. ESTScan [27] was used to predict the CDS and the sequence direction when unigenes were unaligned to any of the databases. With NR annotation, the Blast2GO program was used to classify unigenes to Gene Ontology (GO) terms such as molecular function, biological processes, and cellular components [28]. After obtaining GO annotations for all unigenes, WEGO software [29] was used to perform GO function classification for all unigenes and to analyse the distribution of S. turkestanicum gene functions at the macro-level. Simple sequence repeats (SSRs) in the nucleotide sequences were identified using the MIcroSAtellite (MISA) identification tool [30]. The poly-A and poly-T sequences at the terminal regions of the UTs were removed before SSR identification. SOAPsnp [31] was used (with parameters -u t -Q i -L 90) on pileup files to output lists of single nucleotide polymorphisms (SNPs) and their locations.
Identification of female and male differentially expressed genes
The calculation of Unigene expression used the FPKM method (Fragments Per kb per Million reads) [32]. RPKM was directly used to compare the difference of gene expression levels between females and males. False Discovery Rate (FDR) control is a statistical method used in multiple hypothesis testing to correct for p-value. In our analysis, we chose those with FDR ≤ 1 × 10−3 and |log2 ratio| ≥2 to identify sex-biased genes. Differentially expressed genes were then subjected to GO functional analysis and KEGG Pathway analysis.
Results
Sequencing and assembly
Using Illumina (paired-end) sequencing technology, we obtained a total of 86,697,820 and 84,578,440 raw reads from female and male adults of S. turkestanicum, respectively. After quality filtration, 81,086,908 (total clean nucleotides: 7,297,821,720 nt) and 80,469,244 (total clean nucleotides: 7,242,231,960 nt) high quality clean reads were generated for assembly from females and males, respectively. All clean reads were submitted to the Sequence Read Archive database at NCBI (accession nos: SRP068812 and SRP068813). The Q20 percentage (Q20 percentage is the proportion of sequencing nucleotides error rate with quality value <1 %) and GC percentage of females were 97.69 % and 40.62 %, respectively. The Q20 and GC percentages of males were 97.86 % % and 41.26 %, respectively. Based on the clean reads, a total of 46,041 (>200 nt) and 69,543 (>200 nt) contiguous sequences (contigs) without gaps were produced by Trinity software for S. turkestanicum females and males, respectively (Table 1). Short unigenes (less than 200 nt) were removed. A total of 34,624 unigenes (total length: 30,412,123 nt) were produced from all samples (females and males), with an average length of 878 nt and N50 (A measure of the contig or scaffold length. It is the maximum length L such that 50 % of all nucleotides lie in contigs or scaffolds of size at least L) of 1480 nt. A total of 29,526 and 41,346 unigenes were produced by the clustering of S. turkestanicum sequences from females and males, respectively (Table 1). In females, 5356 unigenes (18.1 %) were longer than 500–1000 nt, and 3160 unigenes (10.7 %) were longer than 1000-1500 nt. In males, 6458 unigenes (15.6 %) were longer than 500–1,000 nt, and 3356 unigenes (8.1 %) were longer than 1000-1500 nt. All unigenes showed no gaps, thus demonstrating the high quality of Trinity assembly. The length distribution of these unigenes is shown in Fig. 1.
Table 1.
Summary of transcriptome data for adult Schistosoma turkestanicum
| Data generation and filtering | Female | Male |
|---|---|---|
| Raw reads | 86,697,820 | 84,578,440 |
| Clean reads | 81,086,908 | 80,469,244 |
| GC content | 40.62 % | 41.26 % |
| Q20 percentage | 97.69 % | 97.86 % |
| Contigs (mean length; N50) | 46,041 (419 nt; 977 nt) | 69,543 (355 nt; 699 nt) |
| Unigenes (mean length; N50) | 29,526 (744 nt; 1363 nt) | 41,346 (673 nt; 1324 nt) |
Fig. 1.
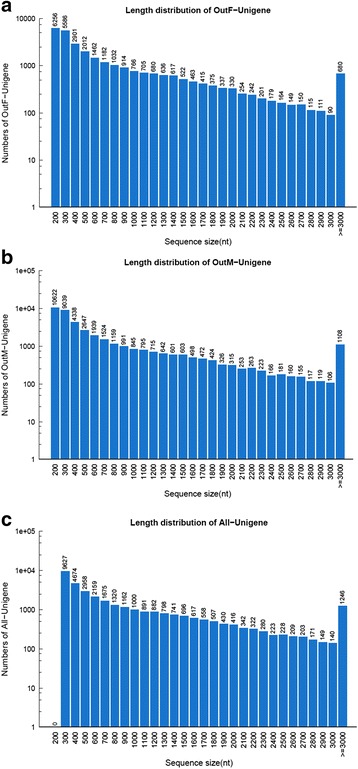
Length distribution of unigenes of adult Schistosoma turkestanicum transcriptome. a: Female- unigene; b: Male- unigene; c: All-unigene
Functional annotation
In order to obtain and validate sequence-based annotations for all assembled unigenes, we employed Blastx for a sequence similarity search against the NR, NT, Swiss-Prot, GO, COG and KEGG databases, with an e-value <0.00001. Of these unigenes, 25,158 (72.7 %) were annotated by BLAST searches against the public databases. Among these, 21,995 (63.5 %), 22,189 (64.1 %), 13,754 (39.7 %), 11,816 (34.1 %), 5,889 (17 %) and 3,764 (10.9 %) unigenes had homologous sequences in NR, NT, Swiss-Prot, KEGG, COG and GO databases, respectively. Furthermore, 9,466 (27.34 %) unigenes showed no homology to known sequences deposited in these databases.
A total of 3,764 unigenes were assigned 3,652 GO term annotations, which could be classified into three categories: biological process, molecular function, and cellular component. The biological process category consisted of 2,328 GO terms, which were assigned to 2,728 (7.9 %) unigenes. The cellular component category consisted of 495 GO terms, which were assigned to 2,108 (6.1 %) unigenes, and the molecular function category consisted of 829 GO terms, which were assigned to 2,965 (8.6 %) unigenes (Fig. 2). Within the biological process category, most unigenes (2,296) were assigned to “cellular processes”. In the cellular component category, most unigenes (1,844) were assigned to “cell”. In the molecular function category, the majority of unigenes (1,775) were associated with “binding” (Fig. 2).
Fig. 2.
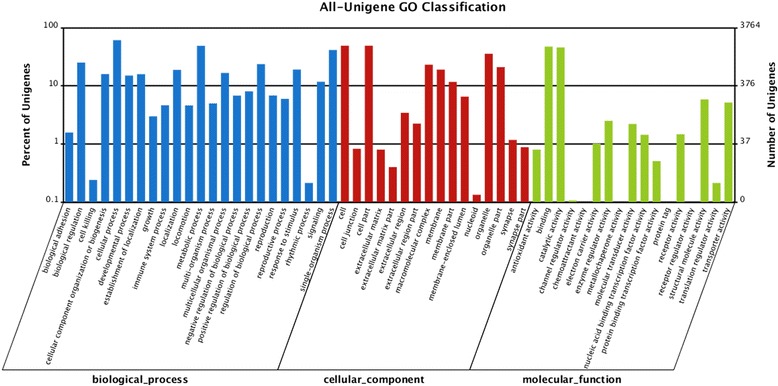
GO annotations of unigenes in Schistosoma turkestanicum transcriptome
All assembled unigenes were further annotated based on COG category. A total of 5,889 unigenes were further annotated, which could be grouped into 25 functional categories (Fig. 3). Among these functional categories, R (general function prediction only) included 5.9 % (2,036/34,624) unigenes, followed by L (replication, recombination and repair), K (transcription), J (translation, ribosomal structure and biogenesis), O (post-translational modification) and T (Signal transduction mechanisms) had 2.7 % (947/34,624), 2.7 % (923/34,624), 2.3 % (811/34,624), 2.1 % (739/34,624) and 1.9 % (666/34,624) unigenes, respectively. Furthermore, Y (Nuclear structure) had only six unigenes (Fig. 3).
Fig. 3.
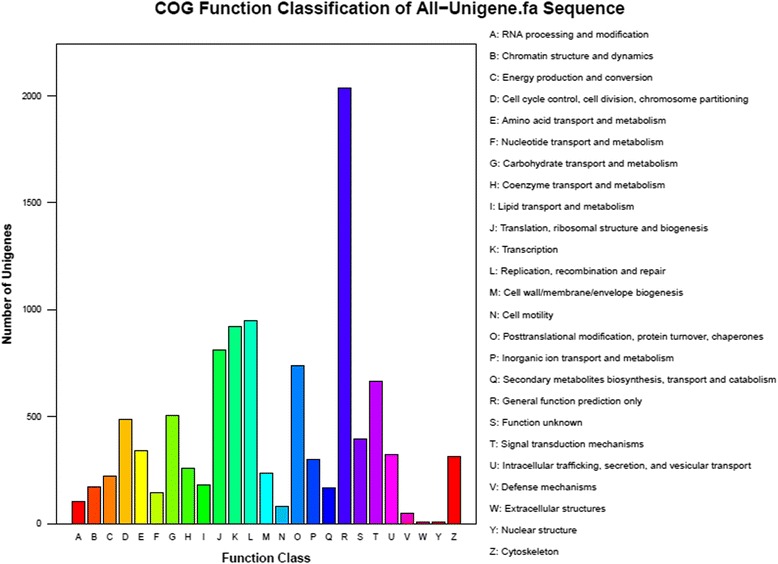
COG functional annotations of unigenes in Schistosoma turkestanicum transcriptome
Functional classification and pathway assignments were based on the KKEGG analysis. A total of 11,816 (34.1 %) uningenes were classified into six categories (Metabolism, Genetic information processing, Environmental information processing, Cellular processes, Organismal systems and Human disease) that mapped to 256 KEGG pathways. The top five KEGG pathways were metabolic pathways (1,545; 13.08 %), pathways in cancer (472; 3.99 %), regulation of actin cytoskeleton (444; 3.76 %), focal adhesion (443; 3.75 %) and spliceosome (401; 3.39 %) pathway. Interestingly, the ‘Immune system or Immune diseases’ sub-category, including 22 pathways, was associated with 1,802 (1,802/11,816; 15.3 %) KEGG annotated unigenes.
SNP identification and microsatellite
In S. turkestanicum females, a total of 124,242 SNPs were also identified with the frequency of 1/58,738 nt, including transition 83,574 (A-G = 41,808; C-T = 41,766) and transversion 40,668 (A-C = 10,033; A-T = 14,360; C-G = 6,478; G-T = 9,797). In males, a total of 124,081 SNPs were also identified with the frequency of 1/58,366 nt, including transition 83,380 (A-G = 41,752; C-T = 41,628) and transversion 40,701 (A-C = 10,004; A-T = 14,264; C-G = 6,559; G-T = 9,874). Most of the repeats were repetitions of di-nucleotide or tri-nucleotide motifs. A total of 11,231 SSRs were identified with up to six nucleotide repeat lengths (Fig. 4).
Fig. 4.
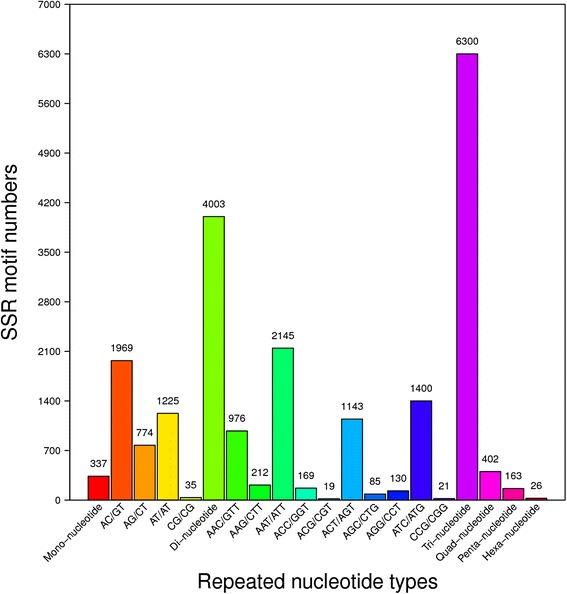
Repeated nucleotide type in Schistosoma turkestanicum transcriptome
Unigene expression differences between S. turkestanicum females and males
A total of 13,603 distinct genes were found differentially expressed between female and male worms, of which 8,021 genes were up-regulated and 5,582 genes were down-regulated in females versus males, respectively. In addition, 3,150 unigenes were identified to be expressed specifically in females and 1,014 unigenes were identified to be expressed specifically in males. The majority of sex-biased genes are in the metabolic pathways (663; 13 %) compared with other pathways. The significantly divergent pathways of differentially expressed genes mainly include pathways in cancer (234; 4.6 %), focal adhesion (220; 4.3 %), regulation of actin cytoskeleton (212; 4.2 %) and insulin signaling pathway (154; 3 %). Interestingly, several pathways associated with gonadal development and sex maintenance were found, such as the Wnt signaling pathway (103; 2 %), progesterone-mediated oocyte maturation (77; 1.5 %), GnRH signaling pathway (62; 1.2 %), TGF-beta signaling pathway (55; 1.1 %) and steroid hormone biosynthesis (12; 0.2 %) (Table 2 and Fig. 5).
Table 2.
Key genes of the gonadal development and sex maintenance for adult females and males of Schistosoma turkestanicum
| Pathway | Pathway ID | Representative genes |
|---|---|---|
| Wnt signaling pathway | ko04310 | CL100.Contig1_All, CL100.Contig2_All, CL1218.Contig1_All, CL1218.Contig2_All, CL709.Contig2_All, CL785.Contig1_All, CL89.Contig1_All, Unigene10129_All, Unigene10266_All, Unigene10807_All, Unigene1085_All, Unigene11070_All, Unigene11334_All, Unigene1178_All, Unigene1358_All |
| Progesterone-mediated oocyte maturation | ko04914 | CL1628.Contig1_All, CL1628.Contig2_All, CL174.Contig10_All, CL174.Contig5_All, CL174.Contig6_All, CL1781.Contig1_All, CL1781.Contig3_All, CL1963.Contig1_All, CL1963.Contig2_All, CL1963.Contig3_All |
| GnRH signaling pathway | ko04912 | CL100.Contig1_All, CL100.Contig2_All, CL1536.Contig1_All, CL1536.Contig2_All, CL2320.Contig1_All, CL2423.Contig1_All, Unigene1009_All, Unigene10173_All, Unigene10497_All, Unigene10692_All, Unigene10930_All, Unigene10931_All, Unigene12392_All, Unigene12677_All |
| TGF-beta signaling pathway | ko04350 | CL1563.Contig2_All, CL1604.Contig1_All, CL1989.Contig1_All, CL1989.Contig3_All, CL272.Contig1_All, CL272.Contig2_All, Unigene10807_All, Unigene1085_All, Unigene11334_All, Unigene1178_All, Unigene1318_All, Unigene15734_All, Unigene16098_All |
| Steroid hormone biosynthesis | ko00140 | CL2632.Contig1_All, CL3396.Contig1_All, CL3396.Contig2_All, CL511.Contig1_All, CL511.Contig3_All, CL511.Contig4_All, CL688.Contig2_All, CL688.Contig3_All, Unigene11949_All, Unigene2063_All, Unigene6039_All, Unigene9729_All |
Fig. 5.
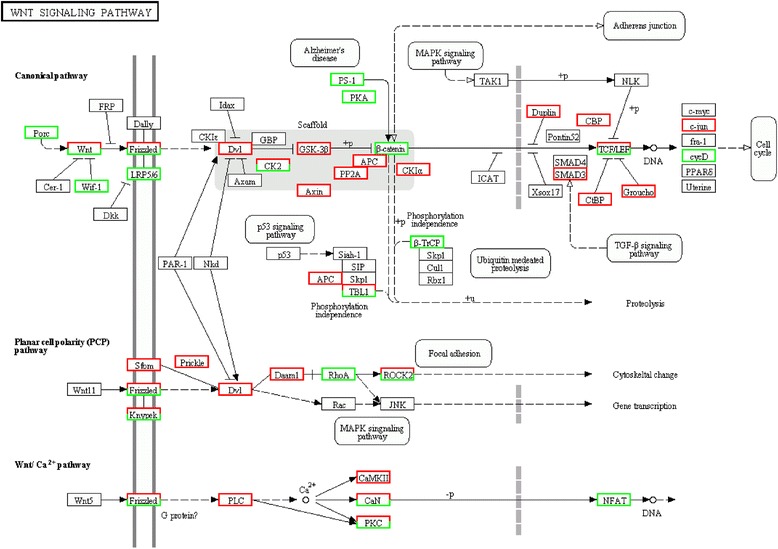
Wnt signaling pathway. Red background indicates up-regulated genes, green background indicates down-regulated genes
Discussion
Schistosomiasis caused by Schistosoma spp. remains a disease of considerable economic and medical importance in developing tropical and subtropical countries [33]. However, as a causative agent of human cercarial dermatitis and animal schistosomiasis, little is known of the transcriptome and genome of S. turkestanicum. Hence, this study decoded the trancriptomes of male and female S. turkestanicum by transcriptome sequencing.
Transcriptome sequencing is one of the most important tools for gene discovery. However, the traditional study method (large-scale EST sequencing) is time consuming and expensive. The NGS technology has become a powerful approach for high-throughput gene discovery on a genome-wide scale in organisms [9]. Due to its long read length, fast and accurate data, Solexa/Illumina have been the most widely used platforms for de novo transcriptome sequencing of many organisms, including some parasites [16, 34–37]. Recently, some studies have indicated that the relatively short reads can be effectively assembled with the great advantage of paired-end sequencing [38, 39], and the Illumina transcriptome de novo sequencing and assembly have been successfully used for distinct species [40–45]. Therefore, in the present study, a next-generation sequencing platform and powerful de novo short-read assembly method was employed to uncover a global view of the transcriptomes of female and male S. turkestanicum worms, and our results have also indicated that relatively short reads from Illumina paired-end sequencing can be effectively assembled.
In the present study, a great number of unigenes could match unique known proteins in public databases, which is similar with other trematodes [14–20]. A large number of unigenes were assigned to a wide range of GO categories and COG classifications, indicating that our data represented a wide diversity of transcripts. Based on the KEGG pathway, the top five KEGG pathways were metabolic pathways, pathways in cancer, regulation of actin cytoskeleton, focal adhesion and spliceosome pathway. These results indicated the active metabolic processes in S. turkestanicum development. Therefore, these results also strongly suggested that most of the genes involved in the different metabolic processes were identified through high-throughput Illumina transcriptome sequencing. Interestingly, the ‘Immune system or Immune diseases’ sub-category, including 22 pathways, was associated with 1,802 (1,802/11,816; 15.3 %) KEGG annotated unigenes, indicating that a much higher proportion of putative KO proteins of S. turkestanicum is involved in pathways associated with the immune system or immune diseases.
In the present study, a total of 13,603 distinct genes were found differentially expressed between S. turkestanicum females and males, which is significantly higher than that of S. japonicum [46], S. mansoni [18] and S. haematobium [19]. Interestingly, several pathways associated with gonadal development and sex maintenance were found, such as Wnt signaling pathway (103; 2 %). The Wnt signaling pathway is a conserved signaling network, which takes part in embryonic development, cell differentiation and proliferation, and the process of growth regulation. Wnt is a secreted glycoprotein, and many different Wnt gene subtypes have been found in a variety of animals. Wnt4 is regarded as a sex determination gene, which plays a key role in the morphological development of female mammals [47], and can regulate the formation of the Müllerian duct and the generation of ovarian steroids [48]. At birth, sexual development in males with a mutation in Wnt4 appears to be normal; however, Wnt4-mutant females are masculinized and the Müllerian duct is absent while the Wolffian duct continues to develop. Wnt4 may also be required for the maintenance of the female germ line [49]. Many genes in the Wnt signaling pathway were found in our transcriptomic database, such as casein kinase 1, epsilon, E1A/CREB-binding protein, serine/threonine-protein phosphatase 2A regulatory subunit A, WNT inhibitory factor 1 and F-box and WD-40 domain protein 1/11. Further research is required to elucidate the roles of these genes in the reproductive process of S. turkestanicum.
Conclusions
The present study has produced the transcriptome data for adult female and male S. turkestanicum worms, and significantly enlarges the currently known gene repertoire of S. turkestanicum. These data will serve as a unique resource for future studies of S. turkestanicum gene functions, and will aid the ongoing whole genome sequencing efforts on this blood fluke. The characterization of these transcriptomic data has implications for the better understanding of the biology of S. turkestanicum, and will facilitate the development of intervention agents for this and other pathogenic flukes of human and animal health significance.
Acknowledgements
This work was supported in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), the National Key Basic Research Program (973 Program) of China (Grant No. 2015CB150300), the Special Fund for Agro-scientific Research in the Public Interest (Grant No. 201303037), and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006). BGI-Shenzhen is thanked for technical assistance. Professor Robin Gasser of the Department of Veterinary Science, The University of Melbourne, Australia is thanked for comments and suggestions on the draft manuscript.
Abbreviations
- COG
cluster of orthologous groups
- CDS
coding sequences
- Contigs
contiguous sequences
- DALYs
disability adjusted life years
- FDR
false discovery rate
- FPKM
fragments per kb per million reads
- GO
gene ontology
- ITS
internal transcribed spacer
- KEGG
kyoto encyclopedia of genes and genomes
- MISA
MIcroSAtellite
- NGS
next-generation sequencing
- NT
non-redundant nucleotide
- NR
non-redundant protein
- nt
nucleotides
- QC
quality Control
- RNA-seq
RNA sequencing
- SSRs
simple sequence repeats
- SNPs
single nucleotide polymorphisms
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ, GHL and CRW conceived and designed the study, and critically revised the manuscript. GHL performed the experiments, analysed the data and drafted the manuscript. MJX, CRW, QCC and JFG helped in study design, study implementation and manuscript revision. All authors read and approved the final manuscript.
Contributor Information
Guo-Hua Liu, Email: liuguohua5202008@163.com.
Min-Jun Xu, Email: mjxu2010@163.com.
Qiao-Cheng Chang, Email: changqiaocheng2001@163.com.
Jun-Feng Gao, Email: gaojunfeng_2005@163.com.
Chun-Ren Wang, Email: chunrenwang@126.com.
Xing-Quan Zhu, Email: xingquanzhu1@hotmail.com.
References
- 1.Xu J, Yu Q, Tchuenté LT, Bergquist R, Sacko M, Utzinger J, et al. Enhancing collaboration between China and African countries for schistosomiasis control. Lancet Infect Dis. 2016;16:376–83. doi: 10.1016/S1473-3099(15)00360-6. [DOI] [PubMed] [Google Scholar]
- 2.King CH, Dickman K, Tisch DJ. Reassessment of the cost of chronic helmintic infection: a meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet. 2005;365:1561–9. doi: 10.1016/S0140-6736(05)66457-4. [DOI] [PubMed] [Google Scholar]
- 3.Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–25. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 4.Makaula P, Sadalaki JR, Muula AS, Kayuni S, Jemu S, Bloch P. Schistosomiasis in Malawi: a systematic review. Parasit Vectors. 2014;7:570. doi: 10.1186/s13071-014-0570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doenhoff MJ, Cioli D, Utzinger J. Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr Opin Infect Dis. 2008;21:659–67. doi: 10.1097/QCO.0b013e328318978f. [DOI] [PubMed] [Google Scholar]
- 6.Wang CR, Chen J, Zhao JP, Chen AH, Zhai YQ, Li L, et al. Orientobilharzia species: neglected parasitic zoonotic agents. Acta Trop. 2009;109:171–155. doi: 10.1016/j.actatropica.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Lawton SP, Majoros G. A foreign invader or a reclusive native? DNA bar coding reveals a distinct European lineage of the zoonotic parasite Schistosoma turkestanicum (syn. Orientobilharzia turkestanicum (Dutt and Srivastava, 1955)) Infect Genet Evol. 2013;14:186–93. doi: 10.1016/j.meegid.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Knoppers BM, Zawati MH, Sénécal K. Return of genetic testing results in the era of whole-genome sequencing. Nat Rev Genet. 2015;16:553–9. doi: 10.1038/nrg3960. [DOI] [PubMed] [Google Scholar]
- 9.Mangiola S, Young ND, Korhonen P, Mondal A, Scheerlinck JP, Sternberg PW, et al. Getting the most out of parasitic helminth transcriptomes using HelmDB: implications for biology and biotechnology. Biotechnol Adv. 2013;31:1109–19. doi: 10.1016/j.biotechadv.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Cárdenas-Conejo Y, Carballo-Uicab V, Lieberman M, Aguilar-Espinosa M, Comai L, Rivera-Madrid R. De novo transcriptome sequencing in Bixa orellana to identify genes involved in methylerythritol phosphate, carotenoid and bixin biosynthesis. BMC Genomics. 2015;16:877. doi: 10.1186/s12864-015-2065-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Wang S, Qin T, Li N, Niu Y, Li D, et al. Whole transcriptome analysis of Penicillium digitatum strains treatmented with prochloraz reveals their drug-resistant mechanisms. BMC Genomics. 2015;16:855. doi: 10.1186/s12864-015-2043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petrella V, Aceto S, Musacchia F, Colonna V, Robinson M, Benes V, et al. De novo assembly and sex-specific transcriptome profiling in the sand fly Phlebotomus perniciosus (Diptera, Phlebotominae), a major Old World vector of Leishmania infantum. BMC Genomics. 2015;16:847. doi: 10.1186/s12864-015-2088-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagel JM, Morris JS, Lee EJ, Desgagné-Penix I, Bross CD, Chang L, et al. Transcriptome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol. 2015;15:227. doi: 10.1186/s12870-015-0596-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantacessi C, Mulvenna J, Young ND, Kasny M, Horak P, Aziz A, et al. A deep exploration of the transcriptome and "excretory/secretory" proteome of adult Fascioloides magna. Mol Cell Proteomics. 2012;11:1340–53. doi: 10.1074/mcp.M112.019844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y, Chen W, Wang X, Liu H, Chen Y, Guo L, et al. The carcinogenic liver fluke, Clonorchis sinensis: new assembly, reannotation and analysis of the genome and characterization of tissue transcriptomes. PLoS One. 2013;8:e54732. doi: 10.1371/journal.pone.0054732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu GH, Xu MJ, Song HQ, Wang CR, Zhu XQ. De novo assembly and characterization of the transcriptome of the pancreatic fluke Eurytrema pancreaticum (Trematoda: Dicrocoeliidae) using Illumina paired-end sequencing. Gene. 2016;576:333–8. doi: 10.1016/j.gene.2015.10.045. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Xu X, Lu X, Zhang Y, Pan W. Transcriptome bioinformatical analysis of vertebrate stages of Schistosoma japonicum reveals alternative splicing events. PLoS One. 2015;10:e0138470. doi: 10.1371/journal.pone.0138470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson L, Amaral MS, Beckedorff F, Silva LF, Dazzani B, Oliveira KC, et al. Schistosoma mansoni Egg, Adult male and female comparative gene expression analysis and identification of novel genes by RNA-Seq. PLoS Negl Trop Dis. 2015;9:e0004334. doi: 10.1371/journal.pntd.0004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young ND, Jex AR, Li B, Liu S, Yang L, Xiong Z, et al. Whole-genome sequence of Schistosoma haematobium. Nat Genet. 2012;44:221–215. doi: 10.1038/ng.1065. [DOI] [PubMed] [Google Scholar]
- 20.Pomaznoy MY, Logacheva MD, Young ND, Penin AA, Ershov NI, Katokhin AV, et al. Whole transcriptome profiling of adult and infective stages of the trematode Opisthorchis felineus. Parasitol Int. 2015;65:12–9. doi: 10.1016/j.parint.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Choudhary V, Garg S, Chourasia R, Hasnani JJ, Patel PV, Shah TM, et al. Transcriptome analysis of the adult rumen fluke Paramphistomum cervi following next generation sequencing. Gene. 2015;570:64–70. doi: 10.1016/j.gene.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Cwiklinski K, Dalton JP, Dufresne PJ, La Course J, Williams DJ, Hodgkinson J, et al. The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol. 2015;16:71. doi: 10.1186/s13059-015-0632-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dutt SC, Srivastava HD. A revision of the genus Ornithobilharzia Odhner (1912), with the creation of two genera Orientobilharzia Dutt and Srivastava (1955) and Sinobilharzia Dutt and Srivastava (1955) (Trematoda: Schistosomatidae) Indian J Helminthol. 1961;13:61–73. [Google Scholar]
- 24.Wang CR, Li L, Ni HB, Zhai YQ, Chen AH, Chen J, et al. Orientobilharzia turkestanicum is a member of Schistosoma genus based on phylogenetic analysis using ribosomal DNA sequences. Exp Parasitol. 2009;121:193–7. doi: 10.1016/j.exppara.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–2. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 27.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;138–48. [PubMed]
- 28.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 29.Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:293–7. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor Appl Genet. 2003;106:411–22. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 31.Li R, Li Y, Fang X, Yang H, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–32. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 33.Lai YS, Biedermann P, Ekpo UF, Garba A, Mathieu E, Midzi N, et al. Spatial distribution of schistosomiasis and treatment needs in sub-Saharan Africa: a systematic review and geostatistical analysis. Lancet Infect Dis. 2015;15:927–40. doi: 10.1016/S1473-3099(15)00066-3. [DOI] [PubMed] [Google Scholar]
- 34.Sahoo PK, Kar B, Mohapatra A, Mohanty J. De novo whole transcriptome analysis of the fish louse, Argulus siamensis: first molecular insights into characterization of Toll downstream signalling molecules of crustaceans. Exp Parasitol. 2013;135:629–41. doi: 10.1016/j.exppara.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Kumar M, Gantasala NP, Roychowdhury T, Thakur PK, Banakar P, Shukla RN, et al. De novo transcriptome sequencing and analysis of the cereal cyst nematode, Heterodera avenae. PLoS One. 2014;9:e96311. doi: 10.1371/journal.pone.0096311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F, Li D, Wang Z, Dong A, Liu L, Wang B, et al. Transcriptomic analysis of the rice white tip nematode, Aphelenchoides besseyi (Nematoda: Aphelenchoididae) PLoS One. 2014;9:e91591. doi: 10.1371/journal.pone.0091591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun J, Wang SW, Li C, Hu W, Ren YJ, Wang JQ. Transcriptome profilings of female Schistosoma japonicum reveal significant differential expression of genes after pairing. Parasitol Res. 2014;113:881–92. doi: 10.1007/s00436-013-3719-2. [DOI] [PubMed] [Google Scholar]
- 38.Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A. 2009;106:12353–8. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Qiao G, Jiang J, Yang H, Xie L, Xie J, et al. Transcriptome sequencing and de novo analysis for Ma bamboo (Dendrocalamus latiflorus Munro) using the Illumina platform. PLoS One. 2012;7:e46766. doi: 10.1371/journal.pone.0046766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010;11:726. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong F, Li H, Sun P, Zhou Y, Mao Y. De novo assembly and characterization of the transcriptome of seagrass Zostera marina using Illumina paired-end sequencing. PLoS One. 2014;9:e112245. doi: 10.1371/journal.pone.0112245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Kuang H, Lai Z. Transcriptome analysis by Illumina high-throughout paired-end sequencing reveals the complexity of differential gene expression during in vitro plantlet growth and flowering in Amaranthus tricolor L. PLoS One. 2014;9:e100919. doi: 10.1371/journal.pone.0100919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muriira NG, Xu W, Muchugi A, Xu J, Liu A. De novo sequencing and assembly analysis of transcriptome in the Sodom apple (Calotropis gigantea) BMC Genomics. 2015;16:723. doi: 10.1186/s12864-015-1908-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sudheesh S, Sawbridge TI, Cogan NO, Kennedy P, Forster JW, Kaur S. De novo assembly and characterisation of the field pea transcriptome using RNA-Seq. BMC Genomics. 2015;16:611. doi: 10.1186/s12864-015-1815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu SJ, Song SH, Wang WQ, Song SQ. De novo assembly and characterization of germinating lettuce seed transcriptome using Illumina paired-end sequencing. Plant Physiol Biochem. 2015;96:154–62. doi: 10.1016/j.plaphy.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 46.Piao X, Cai P, Liu S, Hou N, Hao L, Yang F, et al. Global expression analysis revealed novel gender-specific gene expression features in the blood fluke parasite Schistosoma japonicum. PLoS One. 2011;6:e18267. doi: 10.1371/journal.pone.0018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jordan BK, Mohammed M, Ching ST, Délot E, Chen XN, Dewing P, et al. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet. 2001;68:1102–9. doi: 10.1086/320125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med. 2004;351:792–8. doi: 10.1056/NEJMoa040533. [DOI] [PubMed] [Google Scholar]
- 49.Vainio S, Heikkilä M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397:405–9. doi: 10.1038/17068. [DOI] [PubMed] [Google Scholar]