

Abstract
Background
Type VII protein secretion (T7SS) is a specialised system for excreting extracellular proteins across bacterial cell membranes and has been associated with virulence in Staphylococcus aureus. The genetic diversity of the ess locus, which encodes the T7SS, and the functions of proteins encoded within it are poorly understood.
Results
We used whole genome sequence data from 153 isolates representative of the diversity of the species to investigate the genetic variability of T7SS across S. aureus. The ess loci were found to comprise of four distinct modules based on gene content and relative conservation. Modules 1 and 4, comprising of the 5’ and 3’ modules of the ess locus, contained the most conserved clusters of genes across the species. Module 1 contained genes encoding the secreted protein EsxA, and the EsaAB and EssAB components of the T7SS machinery, and Module 4 contained two functionally uncharacterized conserved membrane proteins. Across the species four variants of Module 2 were identified containing the essC gene, each of which was associated with a specific group of downstream genes. The most diverse module of the ess locus was Module 3 comprising a highly variable arrangement of hypothetical proteins. RNA-Seq was performed on representatives of the four Module 2 variants and demonstrated strain-specific differences in the levels of transcription in the conserved Module 1 components and transcriptional linkage Module 2, and provided evidence of the expression of genes the variable regions of the ess loci.
Conclusions
The ess locus of S. aureus exhibits modularity and organisational variation across the species and transcriptional variation. In silico analysis of ess loci encoded hypothetical proteins identified potential novel secreted substrates for the T7SS. The considerable variety in operon arrangement between otherwise closely related isolates provides strong evidence for recombination at this locus. Comparison of these recombination regions with each other, and with the genomes of other Staphylococcal species, failed to identify evidence of intra- and inter-species recombination, however the analysis identified a novel T7SS in another pathogenic staphylococci, Staphylococcus lugdunensis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2426-7) contains supplementary material, which is available to authorized users.
Keywords: Staphylococcus aureus, Secretion, Type VII
Background
The secretion of virulence factors is an essential process for bacterial pathogenesis, and bacteria have evolved numerous systems through which proteins can be secreted into the environment or injected into host cells [1]. The Type I through Type VI secretion systems are found in Gram negative bacteria and mediate the transport of protein substrates across the two membranes of the cell envelope, in either a one-step or two-step mechanism. The Type VII secretion system (T7SS, variously known as the ESX-1 or ESAT-6 secretion system), by contrast, has not been functionally described in Gram negative bacteria, but is found in the Gram positive Actinobacteria and Firmicutes [2–4]. It was initially identified in pathogenic mycobacteria, with Mycobacterium tuberculosis secreting two T-cell antigens (termed ESAT-6/EsxA and CFP-10/EsxB) via the T7SS pathway. There is significant evidence that this system is an important virulence factor in mycobacteria. Genes encoding ESAT-6 and CFP-10 form part of the region of difference 1 (RD1), a cluster of genes that is deleted from the genome of the M. bovis BCG vaccine strains [5]. This deletion has been linked to the reduced virulence of the BCG strain [6], and evidence in murine models has demonstrated the importance of the ESX-1 system in enabling bacterial translocation from the phagolysosome into the cytosol, a key step in mycobacterial virulence [7].
The mycobacterial T7SS comprises a number of membrane proteins that form a large 1.5 MDa complex [7]. Central to the complex is EccC, a transmembrane protein which has three globular domains of the SpoIIIE-FtsK-like ATPase family [2]. Structural analysis of EccC has shown that the most C-terminal ATPase domain interacts with the signal sequence of the secretion substrate EsxB, which promotes oligomerisation of EccC and activates its ATPase activity [8]. EsxB and the related substrate protein EsxA are founding members of the WXG100 protein superfamily that are characterised as small helical hairpin proteins with a centrally positioned Trp-Xaa-Gly (WXG) motif [9]. Other substrates of the mycobacterial T7SS are the Pro-Glu (PE) and Pro-Pro-Glu (PPE) proteins that are larger than EsxA/EsxB but show a similar helical hairpin arrangement. It is likely that WXG100 substrates are exported as folded dimers [10].
Homologs of the mycobacterial T7SS components EsxA, EsxB and EccC are also encoded by some firmicutes and secretion of EsxA and/or EsxB have been demonstrated in Bacillus subtilis [10, 11], Bacillus anthracis [12] and Staphylococcus aureus [13, 14]. However several of the essential mycobacterial T7SS components are not found among the firmicute T7SS, with only the EccC-like ATPase and one or both of EsxA and EsxB being common across the phyla [3]. This has led to the Firmicutes’ systems being designated Type VIIb to distinguish them from the well-characterised mycobacterial secretion system [2].
S. aureus is a human commensal bacterium and an opportunistic pathogen that can cause a broad range of clinical manifestations in humans, including the majority of skin and soft tissue infections and a substantial proportion of invasive infections such as endocarditis and osteomyelitis [15–17]. The genes encoding the T7SS are found at the ess (ESAT-6-like secretion system) locus and are highly up-regulated during long-term persistence in the cystic fibrosis airway, consistent with a role in persistent infection [18]. Collectively, studies with S. aureus strains Newman, USA300 and RN6390 have shown that the ess locus codes for two secreted WXG100 family proteins (EsxA and EsxB) and two secreted proteins lacking the WXG100 motif (EsxC and EsxD). Three ess-encoded transmembrane proteins, EssA, EssB and EssC, are reportedly essential for protein secretion, but a potential function has been assigned to only one of these, the EccC-like ATPase EssC. The ubiquitin-like protein EsaB has been proposed to regulate Ess activity at either the post-transcriptional or post-translational level. Finally, the membrane protein EssD also encoded at this locus, has a non-essential role in protein secretion [13, 19–21].
S. aureus is a clonal bacterial species and is dominated by a number of successful lineages [22]. Between members of different S. aureus lineages there is variation in the extensive arsenal of immune evasion and virulence factors that modulate the host cell interaction under complex genetic regulation [23]. The ess locus has previously been described in a small number of S. aureus strains from multi-locus sequence type (MLST) clonal complex (CC) 8 (strains Newman [13, 14, 19, 21], USA300 [14, 19, 20], RN6390 [14], COL [14], SA113 [14]), CC5 (Mu50 [19, 24], N315 [3]) and CC1 (MW2 [19]). Comparative genomic analysis identified the ess locus as one of the relative few core variable regions in the S. aureus genome. However, a study of CC30 strains derived from the airways of a cystic fibrosis patient chronically infected with S. aureus found no transcription of a number of T7SS genes, including esxB, esxC and essD; subsequent sequencing of these strains found that these genes were missing from the isolates’ genomes [18]. Sequence similarity searches of published S. aureus genomes identified that in 12 strains these genes were missing, illustrating genetic diversity of the ess operon across the species.
In this study we describe the genetic diversity and organisational variation within the ess locus, using sequence data from a broad range of S. aureus strains that captures diversity across the species. Conducting bioinformatics analysis we have explored the diversity of this secretion system in S. aureus and postulate potential functions of additional hypothetical proteins encoded at the ess locus that are likely to contribute to the T7SS.
Results
Genes comprising the ess locus can be described in four distinct modules
Comparative genomic analysis demonstrated that the ess locus had a complex gene arrangement, which varied both between and within clonal complexes. To facilitate description of this variation we divided the region into four distinct modules, a schematic of which is shown in Fig. 1. The rationale for this was based on the observation that genes in Modules 1 and 4 were present in most isolates and were largely conserved; Module 2 comprised a gene cluster that occurred as four distinct variants; and Module 3 possessed the greatest variation, including several genes encoding hypothetical proteins that varied even between otherwise closely-related isolates.
Fig. 1.
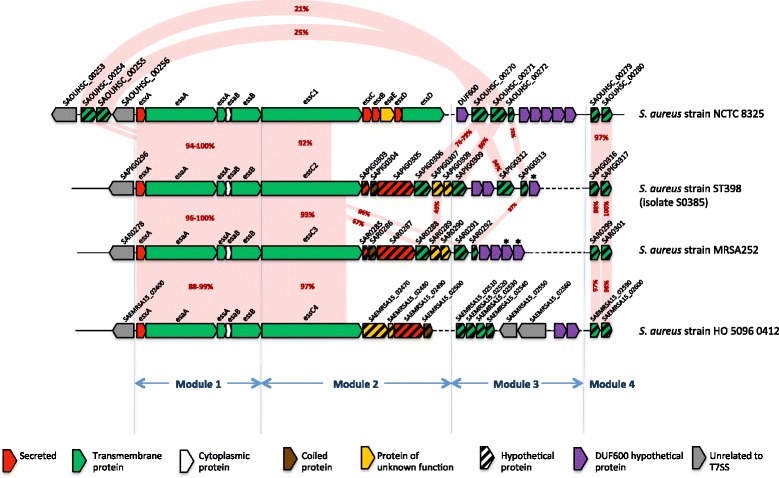
The modular structure of the T7SS loci of S. aureus. Schematic representation of the ess operon in four reference genomes strains of S. aureus, demonstrating the four modules that describe variation in the operon and the four major variants of essC and its immediate downstream gene cluster: Module 1. A conserved cluster of five genes (esxA to essB) at the 5’ end of the operon that encode one secreted protein (EsxA) and other components of the T7SS machinery; Module 2. A variable region that includes the essC gene, of which there are four variants, each associated with a distinct cluster of four to six downstream genes that are predicted to encode a number of secreted and transmembrane proteins; Module 3. A highly variable region which encodes a number of predicted transmembrane proteins and proteins possessing the domain of unknown function (DUF600); and Module 4. A 3’ conserved region, encoding two highly conserved hypothetical proteins of unknown function. Light pink shading demonstrates regions that are conserved between isolates, with the degree of amino acid identity between isolates displayed in red text. An asterisk above the gene indicates that it is present as a pseudogene
Module 1: the esxA gene and the four downstream genes are conserved in all clonal complexes
Module 1 contained the first five genes of the ess locus (esxA, esaA, essA, esaB and essB), homologs of which were present in all but one of the sequences analysed. The gene arrangement of Module 1 was as described previously [3, 13, 20], with little variation in the predicted amino acid sequence in the study isolates as presented in Fig. 1. The highly conserved nature of these genes is consistent with functional analysis showing that they encode essential components of the secretion machinery [20, 21]. A single isolate that lacked esxA, esaA, essA, esaB and essB (ASASM86) contained a genomic deletion of 9166 bp that resulted in the loss of these genes.
Module 2: the 5’ portion of essC is conserved, but there are four variants of the essC 3’ region
The essC gene encodes a membrane-bound protein with three C-terminal ATPase domains and is a key component of all T7SS. Homologs of this gene were identified in all of the S. aureus isolates investigated. The 5’ region of this protein coding sequence (CDS) (approximately 3220 bp of essC) was conserved among all isolates, however the 3’ region (approximately 1230 bp of essC) and downstream CDS fell into four distinct variants. We have termed these variants essC1-4 (Fig. 1). The essC1 variant had the highest frequency (90/153 isolates) and was found in strains belonging to CCs 1, 5, 7, 8, 9, 25, 41, 51 and 88. These included the reference strains Newman [25], USA300 [26], COL [27], Mu50 [28], N315 [28] and MW2 [29], which have previously been used to describe the biology and function of the EssC protein [3, 14, 19, 20, 24]. The next most abundant variant was essC3 (41/153 isolates), which was found in CC30 and ST239, and included the TW20 [30] and MRSA252 [31] reference isolates. By contrast, the essC4 variant was only identified in isolates belonging to CC22 (including the EMRSA-15 reference isolate HO 5096 0412) [32], and essC2 was identified in CC15 and ST398 (including the ST398 reference genome, isolate S0385) [33]. A summary of the essC variants associated with each isolate is included in Additional file 1: Tables S1 and S2. For illustrative purposes we have chosen the ess loci of NCTC 8325, S0385, MRSA252 and HO 5096 0412 reference genomes strains as representative of the four essC variants, essC1, essC2, essC3 and essC4 respectively.
It was notable that the sequence divergence associated with this module at the 5’ end occurred within the essC gene. The X-ray structure of the C-terminal 550 amino acids of EssC from Geobacillus thermodenitrificans, a thermophilic member of the firmicutes phylum has been reported [8] and encompasses two of the three ATPase domains. We mapped the variability observed in the EssC sequences onto the structure of G. thermodentrificans EssC. It is clear from Fig. 2 that the sequence variability largely encompasses the final ATPase domain. Since this C-terminal domain in the actinobacterial EccC is involved in substrate recognition [8, 34], this raises the possibility that the four EssC variants of S. aureus recognise different repertoires of secreted proteins due to the C-terminal domain variation.
Fig. 2.
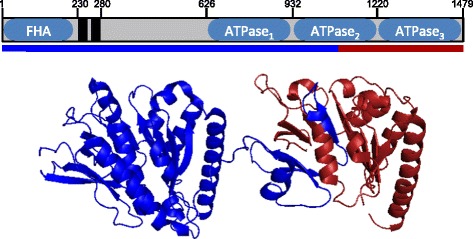
Domain architecture of EssC protein. Sequence variable region of EssC shown in red on the X-ray structure of the C-terminal ATPase domains of G. thermodentrificans EssC. The blue and red line underneath the linear representation of EssC marks the extent of the conserved and variable regions identified in the comparison between the essC1 and essC2 illustrated in Fig. 1
The five genes immediately downstream of essC1 have been previously demonstrated to be co-transcribed with the genes encoding the core secretion components [14]. Three of the five genes, esxC, esxB and esxD encode secreted T7SS substrates [13, 19, 20] whilst essD encodes a transmembrane protein and esaE a predicted soluble protein of unknown function [13, 19–21]. Although every isolate with the essC1 variant in this study had the same arrangement of these five genes, by contrast none of the isolates with the other three essC variants (essC2, essC3 and essC4) possessed any of these genes. Instead, each essC variant (Fig. 1) was associated with a unique combination of downstream genes that constituted Module 2. Since it is known for strains with essC1 sequences that Module 2 genes encode secreted substrates and accessory proteins, these findings strongly suggest that the EssC sequence variants are associated with a distinct repertoire of secreted proteins and accessory factors.
Prediction of function for Module 2 genes
Each of the essC module variants contained at least one gene encoding a protein with a WXG domain that are proposed to be secreted by T7SS (Fig. 1; esxB, SAR0287, SAEMRSA15_2490 and SAPIG0305). Consequently, each variant Ess system has at least two WXG family proteins: an EsxA protein (found in Module 1) that is highly sequence-conserved regardless of EssC subtype and a Module 2 protein, conserved with the same EssC subtype but very poorly conserved across strains with different EssC subtypes. Additional proteins sharing sequence similarity with the WXG100 family of secreted proteins were identified in the essC2 and essC3 variants (SAPIG0303 and SAR0285 respectively; Fig. 1). Although these two proteins lack the WXG motif associated with other secreted proteins in the T7SS, Interpro identified them as potential T7SS effectors in the SACOL2603 family of proteins, which are similar in length and share some sequence similarities with the WXG100 protein family.
Although functions for remaining CDSs in Module 2 could not be predicted, the conservation of protein motifs implies conserved function in this cluster. For example, SAR0286 (essC3 variant), SAEMRSA15_02500 (essC4 variant) and SAPIG0304 (essC2 variant) shared motifs associated with the CATH superfamily 1.20.5.170 [35]. This domain is characterised by single alpha-helices involved in coiled-coils or other helix-helix interfaces, is present in proteins from a wide range of organisms and is implicated in diverse functions. All six hypothetical proteins in the essC2 variant shared a motif or some peptide sequence with a hypothetical protein in the essC3 variant cluster.
Module 3: a complex arrangement of predicted genes
Immediately downstream from Module 2 was a complex arrangement of CDSs that included a variable number of predicted transmembrane proteins, interspersed with a diverse arrangement of genes coding for hypothetical proteins of approximately 166 amino acids in length with a domain of unknown function named DUF600. These genes form Module 3, as illustrated in Fig. 1. The number of unique transmembrane proteins encoded in this region appeared to be small and was a combination of homologs of the following genes: SAOUHSC_00270; SAOUHSC_00271; SAOUHSC_00272; SAOUHSC_00255 (which is sometimes found in this region downstream of essC, but in some isolates is located upstream of esxA where it is always found in association with SAOUHSC_00254); and a cluster of four genes which are always found together in the following combination: SAEMRSA15_2510, SAEMRSA15_2520, SAEMRSA15_2530 and SAEMRSA15_2540.
The combination of these genes found in each genome varied between isolates. All strains had at least one homolog of these genes; no isolates possessed homologs of them all. Our analysis suggested that there may a relationship between the essC variant and the combination of genes in this region. For example, all essC3 variants were associated with homologs of the genes SAOUHSC_00272 and SAOUHSC_00255. Some, but not all, essC3 variants were associated with the SAEMRSA15_2510 to SAEMRSA15_2540 cluster. None of the isolates with the essC3 variant were associated with the SAOUHSC_00271 gene. Different isolates within the same clonal complex appeared to possess the same combination of hypothetical transmembrane proteins encoded in Module 3. The exception was CC8, which had a wide variety of combinations. Some of the hypothetical transmembrane proteins were encoded in multiple copies in the same isolate. For example in the strain Newman, NWMN_0230 encoded a protein with almost identical amino acid sequence to NWMN_0237 (92 % amino acid identity; data not shown) [25].
The number of DUF600 genes present in representative of the different clonal complexes: the minimum number identified was two genes in CC22 isolates and the maximum number was 13 in CC8 isolates. The number of DUF600 genes also varied between isolates within the same clonal complex. In the essC1 variant gene clusters, there was variation in the 3’ region of essD, the gene immediately upstream of the first DUF600 gene. Each essD variant was associated with a different downstream DUF600 protein variant, which broadly clustered into clonal complexes.
The relationship between this cluster of transmembrane and DUF600 proteins encoded at the ess locus is unclear. Several features support an association with the T7SS, including: i) these genes were always on the same coding strand, following immediately from Modules 1 and 2 described above; ii) there was little intergenic space between coding regions, suggesting that these genes may be under the same transcriptional control as other elements of the T7SS; iii) some of the genes encoded in this region shared sequence identity with genes that flanked known secretion component genes within the ess locus. For example, SAOUHSC_00270 shared some sequence motifs with SAOUHSC_00254 (21 % identity, 39 % similarity at the amino acid level) and SAOUHSC_00271 (25 % identity, 52 % similarity at the amino acid level) with SAOUHSC_00255, genes that neighboured SAOUHSC_00256 – the gene immediately upstream of esxA. SAR0288, part of the cluster of genes immediately downstream of essC3 also shared 67 % amino acid sequence identity with SAOUHSC_00254.
Module 4: two conserved hypothetical transmembrane proteins
Downstream of the highly variable Module 3 is a region that contains two genes (SAOUHSC_00279 and SAOUHSC_00280 represented as Module 4 in Fig. 1) that were conserved in all genomes investigated. Analysis of these two proteins revealed that they contained predicted N-terminal secretion signal sequences, and protein domains also found in proteins encoded elsewhere in the ess locus: SAOUHSC_00279 contained a cystatin-like fold (DUF4467, Pfam accession PF14729) which was also present in SAEMRSA_2520, while SAOUHSC_00280 harboured the DUF4064 domain (DUF4064, PF13273) which was also present in SAEMRSA_2530.
Variable transcriptional landscape of the ess loci
RNA-Seq was used to examine the transcriptional profiles of the ess locus in the four S. aureus strains representative of the essC variants (NCTC 8325, S0835, MRSA252 and HO 5096 0412 as representatives of essC1, essC2, essC3 and essC4 variants, respectively) and, more specifically, to look for evidence of the expression of the hypothetical components of the ess loci identified in our analyses. Examination of the mapped strand-specific cDNA sequence data confirmed the gene prediction models of the ess regions, and revealed variation in the abundance of transcript across the ess region (Fig. 3). The highest level of transcript in the references strains was associated with the gene encoding the secreted effector protein EsxA of Module 1, a protein that has been found to play an important role in the pathogenesis of S. aureus, and also suggested as a potential target for vaccination [34]. This high level expression of esxA relative to other ess genes has been noted previously [14]. The rest of the genes in this module contained evidence of transcription, albeit at a lower level to that observed for esxA; in comparison to esaA, the next gene in the ess cluster, the transcript levels of esxA were between 60- and 70-fold greater in the four strains examined. The marked differences in transcript levels suggest separate promoters driving the expression of esxA and esaA. In their analysis of the RN6390 (a derivative of NCTC 8325) Kneuper et al. mapped the transcriptional start site upstream of esxA, and also identified a promoter in the esxA-esaA intergenic region [14]. RNA-Seq data supports this transcriptional organisation, with esxA and with the other components of the Module 1 (esaA, essA, esaB, essB) present on separate transcripts; esxA is present on a moncistronic transcript, and esaA, essA, esaB, essB on a polycistronic transcript that also contains components of Module 2, including essC and downstream CDSs.
Fig. 3.
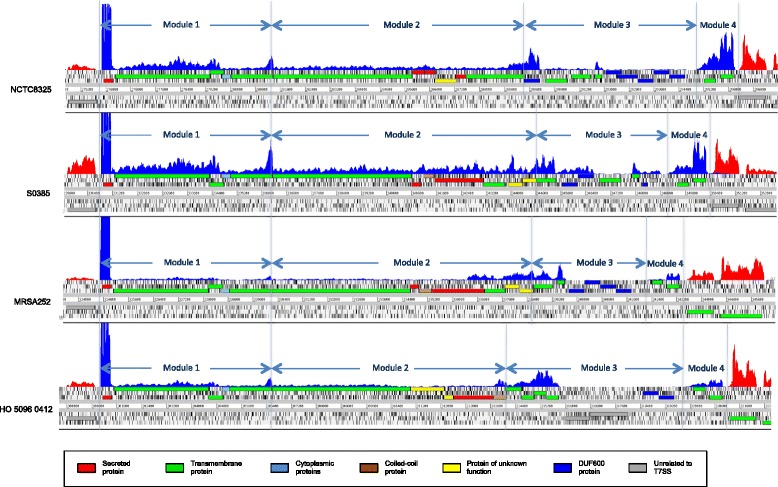
RNA-Seq profiles of the ess loci of four S. aureus reference isolates. Sequence reads from each of the four S. aureus reference isolates aligned to the respective ess loci viewed in Artemis. The mapped sequence read depth is displayed, with graphs representing the result of sequencing strand-specific cDNA mapping. The color of the graphed plots indicated the strand, with blue forward, and red reverse. CDSs from annotation are displayed in a six-frame translation, with the vertical black line indicating stop codons in frames. The coordinates in the figures indicate the position in the reference chromosome
Comparison of the relative expression conserved components of the ess clusters revealed strain-specific differences in the levels of transcription (Fig. 4). The lowest expression was observed in MRSA252, followed by HO 5096 0412, with NCTC 8325 and then S0835 exhibiting the highest levels. For all the reference strains examined there was evidence of transcription of all of the Modules 2 and 4 genes (Additional file 1: Table S5). By contrast, not all of the genes found in the Module 3 regions exhibited evidence of transcription under the assay conditions used (Fig. 3), suggesting complex transcriptional controls in this region of the ess locus. Comparison of the gene organisation of the various Module 3 regions identified multiple intergenic regions; in contrast to the operonic organisation of Module 1 and 2 regions. Of the variable genes in this region of the ess loci those containing the DUF600 domain were the most variably expressed (Fig. 3; Additional file 1: Table S5). Notably each of the reference strains contained multiple copies of the DUF600 domain containing hypothetical proteins, raising the possibility that there may be functional redundancy associated with these proteins and, as a corollary of this, differential regulation.
Fig. 4.
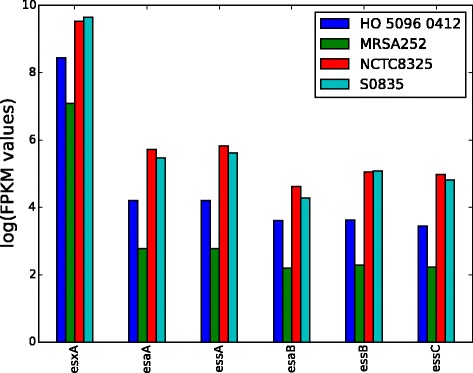
Relative expression of Module 1 components in S. aureus reference strains. RNA-Seq analysis of NCTC 8325, S0835, MRSA252 and HO 5096 0412, representatives of essC1, essC2, essC3 and essC4 variants respectively, was carried out to measure and compare the transcription of the Module 1 genes: esxA, esaA, essA, esaB, and essB. For each strain the mean fragments per kilobase of transcript per million mapped reads (FPKM) were calculated and normalized, using the expression value of the housekeeping gene rpoD. The normalized logFPKM values for each of the components of the Module 1 are plotted along with the values for essC, the first gene of Module 2 which is transcriptional linked to Modules 1 genes, esaA, essA, esaB, and essB
Evidence of recombination throughout the ess locus in S. aureus
Mapping the essC variants onto a S. aureus population framework generated from a phylogeny of MLST genes demonstrated that, although there is clustering of essC variants, there was also phylogenetic incongruence. This was suggestive of recombination at this locus across the species in a wide range of isolates (Fig. 5).
Fig. 5.
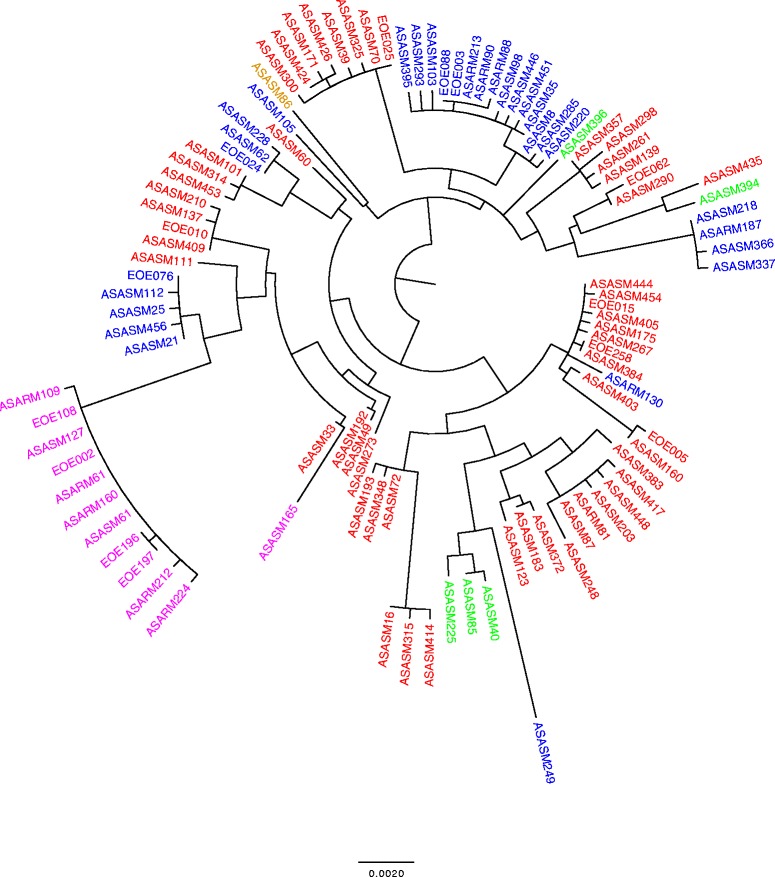
The distribution of essC variants across the S. aureus population. Maximum likelihood reconstruction of the phylogeny of 103 UKCRC S. aureus isolates based on MLST allele phylogeny, annotated by essC variant. Variant essC1 is highlighted in red; essC2 in green; essC3 in blue and essC4 in purple. The single variant where essC has been deleted is colored in ochre
We applied further recombination detection methods using the Phi test within the PhiPack package [36] on individual genes within the ess locus. This method reported statistically significant P values for a number of genes in this locus, including those with a variable C-terminal region (essC (P = 0.0), essD (P = 0.0)). Although some conserved genes did not show evidence of recombination (e.g. esxA) other genes that are encoded in the conserved part of the ess locus did reach statistical significance using this method (esaB (P = 0.017), essB (P < 0.0001)). These indicate that the variation between the same gene in different isolates was more likely to have occurred by recombination than convergent selection or the random accumulation of homoplastic SNPs. To illustrate this, Fig. 6 shows the homoplastic SNPs in the essD genes of isolates with the essC1 subtype, an apparent hotspot of recombination events within the ess operon.
Fig. 6.
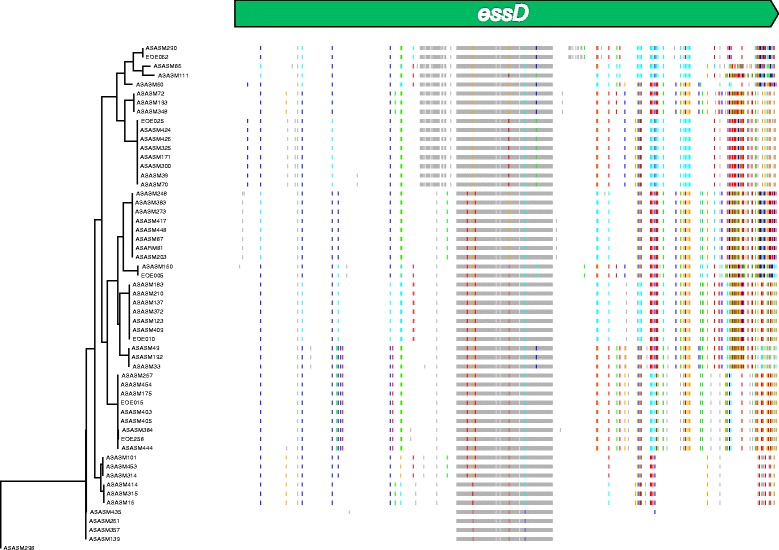
Evidence of recombination in the essD gene. On the left is a maximum likelihood phylogeny constructed with essD gene sequences from isolates containing the essC1 subtype. To the right, each vertical colored line represents a SNP position in the essD sequence, top. Colored vertical lines along the tracks represent bases that differ from the ancestral sequence: grey = non-homoplastic change; colored lines represent homoplastic bases: red = A, blue = T, green = C, orange = G. The pattern of lines provide a barcode of essD similarity between strains and identifies regions containing homoplasies that are incongruent with the phylogeny and are associated with recombination
Components of the ess operon in other staphylococci
Comparative genomic analysis of S. aureus with publicly available reference genomes for other species in the Staphylococcus genus was conducted to investigate the wider distribution and genetic arrangement of this locus. As shown in Fig. 7, the ess locus was present in Staphylococcus lugdunensis, with homologs of the conserved portion of the locus from esxA to essB (SLGD_01975 to SLGD_01971) and a gene similar to the essC4 variant (SLGD_01970). There was also a cluster of genes similar to those downstream of essC4, including homologs of SAEMRSA15_02470 to SAEMRSA_02500, SAEMRSA_02520 and SAEMRSA_02540 (SLGD_01964 to SLGD_01969). There were two genes with homology to those coding for DUF600 proteins (SLGD_01962 and SLGD_01963), but no obvious homologs of the two conserved genes found in Module 4 of the ess operon (SAOUHSC_00279 and SAOUHSC_00280).
Fig. 7.
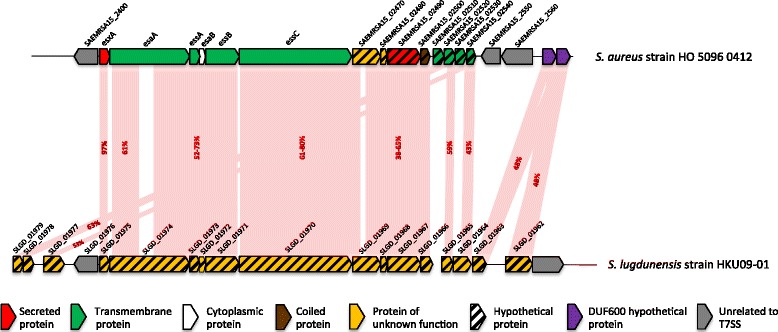
T7SS locus identified in S. lugdunensis. Comparison of the ess operon of S. aureus (strain HO 5096 0412) and S. lugdunensis (strain HKU09-01). Light pink shading demonstrates regions that are conserved between strains, with the degree of amino acid identity between strains marked in red
Of the other species examined within the Staphylococcus genus, none possessed the entire ess operon. However, Staphylococcus pasteuri contained one gene with sequence identity to the DUF600 gene (STP1_1226) and homologs of the cluster of four hypothetical transmembrane-protein coding genes, SAEMRSA_02510 to SAEMRSA_02540 (STP1_1228 to STP1_1231). Staphylococcus warneri also possessed homologous genes to this cluster (A284_00845 to A284_00860). None of the genes identify in these staphylococci exhibited high DNA identity to S. aureus homologs suggestive of interspecies recombination. A complete list of the species and isolates examined can be found in Additional file 1: Table S3.
Discussion
In this study we have described, for the first time, the organisational variation in the ess locus using S. aureus isolates from a broad range of clonal complexes from two distinct genome collections. We identified genes conserved across all isolates and characterised variants in the ess operon, including a number of further candidate substrate proteins that may be secreted by the S. aureus T7SS.
Despite the variation elsewhere in the ess locus, it was striking that the cluster of genes in Module 1, from esxA to essB, was conserved among almost all isolates, suggesting a strong survival benefit or functional constraint on this cluster. In-frame deletions of each of these genes have been shown to abolish secretion of T7SS substrate proteins, supporting the assertion that these genes encode core components of the T7SS [13, 14, 19].
A key finding in this study was the identification of four variants of the essC gene. This membrane-bound protein is essential for secretion of all effector proteins in the T7SS [13, 20]. Variation within this gene occurred in the region encoding the C-terminus of EssC, covering the final ATPase domain. We have shown that each essC variant was associated with a unique cluster of downstream genes, suggesting that the EssC C-terminus may play a role in interacting with the proteins encoded immediately downstream from essC. One of the proteins encoded downstream of each EssC variant is from the WXG superfamily, and in the commonly studied S. aureus Newman, USA300 and RN6390 strains it is EsxB. Studies have shown that esxB is expressed at a very similar level to essC [14], and structural analysis with the related EccC protein of Actinobacteria has shown that EsxB binds to the C-terminal ATPase domain of the EssC homolog and promotes its multimerisation [8]. We postulate that the WXG protein encoded downstream of each essC variant interacts with the C-terminal domain of its cognate EssC protein to activate secretion. It is also possible that this sequence-variable EssC domain may recognise variant-specific substrate proteins or accessory factors. While the genetic basis of this variation and its functional consequence remains unclear, the arrangement of genes and their incongruence with MLST suggest that recombination has played a role in the evolution of this locus. Comparative genomic analysis of S. aureus has revealed that recombination in the core genome is relatively rare [37]. It is therefore notable that the T7SS undergoes such a degree of recombination and suggests that the recombination-driven generation of variation may be an important system for the biology of the organism.
The function of many of the proteins in Module 2, downstream from essC, remains unclear. The best characterised cluster was downstream from essC1, which has been studied in a small number of CC8 isolates and includes genes encoding the secreted proteins EsxB, EsxC and EsxD and the transmembrane protein EssD. These proteins appear to have a functional relationship, with reports that strains lacking each of these proteins affect the stability or expression of the others, while EsxB and EsxD have been shown to form heterodimers [20]. Further work is required to determine whether a similar relationship is present between proteins encoded in the other ess variants.
Although there is a limit to information that can be derived from the bioinformatics analysis of four variants of Module 2 identified in this study, the observation that different variants encoded proteins that shared sequence motifs, particularly the modules associated with essC3 and essC2, implied some redundancy in their function. However, there were also marked differences between variants. For example, the essC2, essC3 and essC4 variants all contained a hypothetical protein with the WXG motif (SAPIG0305, SAR0287, SAEMRSA15_02490 respectively) that was much larger (440–556 aa) than the previously identified T7SS substrates associated with essC1 (for example EsxB and EsxD are 104 and 105 amino acids in length respectively). There was also no transmembrane protein of a comparable size to EssD encoded in the other variants.
The association between EssD and the DUF600 hypothetical proteins has been observed previously, with the C-terminus of some EssD sequences in S. aureus (as well as the EssD-like protein in B. subtilis, YeeE) containing the DUF600 motif [21]. We observed that there was a relationship between the C-terminal sequence of EssD and the immediately downstream DUF600 protein. However, the molecular relationship between these two proteins is unclear. We have also identified a range of isolates that did not possess the essD gene, which raised the question of whether other genes in these variants serve an equivalent function.
Previous evidence has suggested that there is very little variation within coding regions between isolates belonging to the same lineage [38]. However, we have shown that the copy number of DUF600 proteins varied between isolates of the same clonal complex, and also there is differential expression of these proteins within ess clusters. A recent study by Baek and colleagues demonstrated that the greatest sequence variation between strains of the same S. aureus lineage (NCTC 8325) occurred between homologs of the DUF600 proteins [39]. This included variation in the copy number of DUF600 proteins encoded in each genome as well as the sequence of each copy. The reason for this variation is unknown, but the fact that it occurs between such closely related lineages suggests considerable selection pressure on these genes.
There are many hypothetical transmembrane proteins encoded within Module 3 in the ess operon, associated with the DUF600 proteins. Their function and relationship to the T7SS will require further investigation. The presence of these hypothetical transmembrane proteins without the remainder of the T7SS apparatus in the genomes of S. warneri and S. pasteuri may suggest they are not key components of this system. However it is unclear why these genes are located in the same region of the S. aureus genome, often flanked by DUF600 proteins.
The T7SS has been described in other Gram positive bacteria [2, 3, 6, 7]. We describe here homologs of a large number of genes encoding this system in S. lugdunensis, a medium-pathogenic coagulase-negative staphylococcus which shares a number of clinical features with S. aureus, including the ability to form abscesses [40]. The mechanisms of virulence of this organism are poorly understood, but it is possible that, as with S. aureus, the T7SS could have a role in abscess development and therefore in pathogenicity. Investigation into a broader range of staphylococcal isolates may provide clues as to the evolution of the ess locus.
Conclusions
Having described a range of hypothetical proteins that will require further molecular research to determine their role and function, our study provides further avenues for investigation. Recent studies have suggested that the expression of key genes of the T7SS, including esxA, are under complex regulatory control and vary between closely related isolates in the same clonal complex [14]. In this study we have demonstrated a far greater diversity of genetic organisation of the ess operon, which is likely to have implications for gene regulation in the broader range of clonal complexes. Mutants in the T7SS have impaired ability to persist in the complex interplay between host and pathogen during colonisation and abscess formation [14, 41]. However, published research has focused on a narrow range of S. aureus isolates, all of which possess the essC1 operon arrangement. It is unclear what effect other arrangements will have on the pathogenicity of S. aureus, or its abscess-forming phenotype.
Methods
Bacterial isolates and whole genome sequencing
S. aureus whole genome sequence data utilized in this study was generated by the Wellcome Trust Sanger Institute (WTSI). The S. aureus isolates were obtained from human bacteremia cases at Addenbrooke’s Hospital, Cambridge, UK between 2006 and 2012. Further stored isolates from neighbouring hospitals in the East of England, collected between 1998 and 2011, were also included. From this broad collection, 103 isolates were chosen to represent as many different clonal complex (CC) and MLST types (ST) as possible, to provide a snapshot of the genetic diversity of disease causing isolates from this large scale UK collection. To augment the collection, a further 50 published and publicly available S. aureus sequences from the European Bioinformatics Institute were included in the analysis. A full list of isolates used in this study, along with accession numbers, is presented in Additional file 1: Tables S1, S2 and S3. All samples were sequenced as multiplexed libraries using the Illumina HiSeq 2000 analyzers as previously described [23].
Ethics
Written informed consent from patients was not required as all bacterial isolates were collected, processed and stored as part of routine clinical care. The study protocol was approved by the National Research Ethics Service (reference 11/EE/0499), and by the Cambridge University Hospitals NHS Foundation Trust Research and Development Department (reference A092428).
Transcriptional analyses
Strains of S. aureus (Newman, S0385, MRSA252 and HO 5096 0412 reference genomes strains) were incubated shaking at 200 rpm for 16 h at 37 °C. The overnight culture was used to inoculate 25 ml TSB media to an OD600 of 0.03. 2 ml of OD600 1.0 cells were harvested by centrifugation at 13000 g for 2 min and processed for RNA extraction using the Ambion Ribo-PureTM Bacteria kit as per the manufacturers’ guidance. Three biological and three technical replicates were performed for each strain. The RNA quality and concentration was assessed by Agilent BionalyserTM, NanodropTM and visually on a 1 % agarose gel containing 1 % gel red (Biotium) and visualised by gel doc imaging system (Bio-Rad).
For RNA-Seq, total RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen). Actinomycin D (6 μg/ml, Sigma) was added to the reaction to avoid spurious second-strand cDNA synthesis [35]. cDNA was purified using QIAquick PCR purification kit (Qiagen) and used for single stranded cDNA library construction as previously [25, 36]. FRT-seq Illumina libraries were constructed as previously described [37]. RNA sequencing was performed using an Illumina HiSeq 2000 sequencer, and the reads processed as previously described [38].
Bioinfomatic analyses
Assemblies were created with Velvet v1.2.09 [24] using the VelvetOptimiser.pl v2.2.5 (http://www.vicbioinformatics.com/software.velvetoptimiser.shtml) script to optimize the kmer length. Automatic annotation of the Accessory genome contigs was carried out using PROKKA [25]. Detailed comparisons of individual sequences were conducted on the de novo assemblies using BlastN [26], and was facilitated by using the Artemis Comparison Tool (ACT) [27].
Individual T7SS genes were identified from the publicly available Newman strain reference genome (the most commonly studied S. aureus strain in the T7SS literature) [28]. Primers were designed for the terminal 30 nucleotides of each of these genes. In silico PCR using these primers was performed on the de novo assemblies for all isolates using a Python script, to determine the presence or absence of each gene in each isolate. In brief, the script used approximate regular expressions to identify all best matches to each primer sequence in each genome, allowing up to 3 mismatches per match. Where forward and reverse primers were in the correct orientation relative to each other and produced a product of approximately the correct length (defined as <10 kb), products were extracted as fasta files. If a gene could not be identified in an isolate using this method, tblastx comparisons [26] were generated comparing the novel isolate with the reference. This comparison was then visualised graphically using the Artemis Comparison Tool [27] to confirm the absence of the gene or identify major variants in the ess locus. In silico primers were developed for all novel genes identified and the in silico PCR process repeated until all genes in the ess locus had been identified. All primers used in this analysis are presented in Additional file 1: Table S4.
Bioinformatic searches on all genes and putative gene products were conducted using the web-resources NCBI BLASTn [29]) (available at www.ncbi.nlm.nih.gov/blast/), and Pfam [30] (available at http://pfam.xfam.org/), InterPro [31] (available at http://www.ebi.ac.uk/interpro/). Maximum likelihood phylogenetic trees were generated for whole genome sequences of the 103 isolates sequenced using RAxML [33]. Further maximum-likelihood phylogenetic trees based on single genes or clusters of genes in the ess locus were generated using PhyML 3.1 [34].
For the RNA-Seq data samples were mapped to the appropriate reference genomes, and transcripts were assembled, and expression values for the assembled genes and transcripts were computed. Reads were aligned using the Tophat aligner [40]. One of the sequence samples in the MRSA252 strain RNA-Seq experiments produced a very poor yield, therefore this was excluded from the rest of the analysis. After merging the biological with the corresponding technical aligned samples, transcriptome assembly for each sample in the strains was carried out using cufflinks [41]. To acquire a single trasncriptome for each strain, we merged the three assemblies produced by cufflinks and we quantified the abundances of each sample using cuffquant. Differential expression (cuffdiff) as well as normalization (cuffnorm) within the samples of each strain was carried out to acquire the expression values of assembled genes and transcripts.
To compare the expression levels of the genes of interest (esxA, esaA, essA, esaB, essB and essC) in the different strains and remove any extra source of bias in the data, we performed an extra normalization, using the expression value of a housekeeping gene, which is rpoD in our case. More specifically, we scaled the fragments per kilobase of transcript per million (FPKM) mapped reads values of the housekeeping gene across the four strains (housekeeping for the specific strain, hs) and we resized the FPKM values of all the genes based on the hs.
Availability of supporting data
All sequences from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under the study numbers ERP000871, ERP001009, and ERP001320; individual accession numbers are given in Additional file 1: Table S1. RNA-Seq data has been submitted to the European Nucleotide Archive with accession number ERP009279 and in Array express under accession number E-ERAD-362.
Acknowledgements
We thank the core sequencing and informatics teams at the Sanger Institute for their assistance and The Wellcome Trust for its support of the Sanger Institute Pathogen Genomics and Biology groups. SRH, JP and MTGH were supported by Wellcome Trust grant 098051. Bioinformatics and Computational Biology analyses were supported by the University of St Andrews Bioinformatics Unit that is funded by a Wellcome Trust ISSF award (grant 105621/Z/14/Z). SP is funded by the UKCRC Translational Infection Research Initiative, and the NIHR Cambridge Biomedical Research Centre. CPH is supported by the Wellcome Trust (grant number 104241/z/14/z) TP is a Royal Society/Wolfson Merit Award Holder. We thank Drs James Chalmers and Holger Kneuper for helpful discussion, and acknowledge Dr Holger Kneuper’s assistance in preparing Fig. 2.
Abbreviations
- CC
clonal complex
- CDS
protein coding sequence
- DUF
domain of unknown function
- MLST
multi-locus sequence type
- RNA-Seq
RNA sequencing
- T7SS
Type VII protein secretion
Additional file
Table S1. List of all whole genome sequenced S. aureus isolates used in this study. Table S2. List of publicly available reference strains of S. aureus used in this study. Table S3. List of publicly available genomes of reference strains of non-S. aureus staphylococci analysed in this study. Table S4. List of primer sequences used for in silico PCR analysis. Table S5. RNA-Seq transcriptional analysis of the ess clusters of four reference genomes strains of S. aureus. (DOC 405 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BW, SJP, SRH and MTGH designed the study. BW, SRH, AV and MTGH carried out research and analyzed data. CPH, NSW, TP and JP supplied isolates, metadata, and whole genome sequencing. BW, TP and MTGH wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ben Warne, Email: benwarne@cantab.net.
Catriona P. Harkins, Email: ch246@st-andrews.ac.uk
Simon R. Harris, Email: sh16@sanger.ac.uk
Alexandra Vatsiou, Email: alex.vatsiou@gmail.com.
Nicola Stanley-Wall, Email: n.r.stanleywall@dundee.ac.uk.
Julian Parkhill, Email: parkhill@sanger.ac.uk.
Sharon J. Peacock, Email: sjp97@medschl.cam.ac.uk
Tracy Palmer, Email: t.palmer@dundee.ac.uk.
Matthew T. G. Holden, Email: mtgh@st-andrews.ac.uk
References
- 1.Costa TRD, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, et al. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Micro. 2015;13:343–359. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 2.Abdallah AM, Gey van Pittius NC, Champion PAD, Cox J, Luirink J, Vandenbroucke-Grauls CMJE, et al. Type VII secretion--mycobacteria show the way. Nat Rev Micro. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 3.Pallen MJ. The ESAT-6/WXG100 superfamily -- and a new Gram-positive secretion system? Trends Microbiol. 2002;10:209–212. doi: 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 4.Schneewind O, Missiakas DM. Protein secretion and surface display in Gram-positive bacteria. Philos Trans R Soc Lond B Biol Sci. 2012;367:1123–1139. doi: 10.1098/rstb.2011.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majlessi L, Prados-Rosales R, Casadevall A, Brosch R. Release of mycobacterial antigens. Immunol Rev. 2015;264:25–45. doi: 10.1111/imr.12251. [DOI] [PubMed] [Google Scholar]
- 6.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, et al. Substrates control multimerization and activation of the multi-domain ATPase motor of type VII secretion. Cell. 2015;161:501–512. doi: 10.1016/j.cell.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poulsen C, Panjikar S, Holton SJ, Wilmanns M, Song Y-H. WXG100 protein superfamily consists of three subfamilies and exhibits an α-helical C-terminal conserved residue pattern. PLoS One. 2014;9:e89313. doi: 10.1371/journal.pone.0089313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sysoeva TA, Zepeda-Rivera MA, Huppert LA, Burton BM. Dimer recognition and secretion by the ESX secretion system in Bacillus subtilis. Proc Natl Acad Sci U S A. 2014;111:7653–7658. doi: 10.1073/pnas.1322200111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baptista C, Barreto HC, São-José C. High levels of DegU-P activate an Esat-6-like secretion system in Bacillus subtilis. PLoS One. 2013;8:e67840. doi: 10.1371/journal.pone.0067840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garufi G, Butler E, Missiakas D. ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol. 2008;190:7004–7011. doi: 10.1128/JB.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burts ML, Williams WA, DeBord K, Missiakas DM. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A. 2005;102:1169–1174. doi: 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kneuper H, Cao ZP, Twomey KB, Zoltner M, Jäger F, Cargill JS, et al. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/Type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol Microbiol. 2014;93:928–943. doi: 10.1111/mmi.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowy FD. Staphylococcus aureus Infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 16.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 17.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 18.Windmuller N, Witten A, Block D, Bunk B, Sproer C, Kahl BC, et al. Transcriptional adaptations during long-term persistence of Staphylococcus aureus in the airways of a cystic fibrosis patient. Int J Med Microbiol. 2015;305:38–46. doi: 10.1016/j.ijmm.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Burts ML, DeDent AC, Missiakas DM. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol Microbiol. 2008;69:736–746. doi: 10.1111/j.1365-2958.2008.06324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson M, Aly KA, Chen Y-H, Missiakas D. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol Microbiol. 2013;90:734–743. doi: 10.1111/mmi.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson M, Chen Y-H, Butler EK, Missiakas DM. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J Bacteriol. 2011;193:1583–1589. doi: 10.1128/JB.01096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA) Proc Natl Acad Sci. 2002;99:7687–7692. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindsay JA. Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int J Med Microbiol. 2014;304:103–109. doi: 10.1016/j.ijmm.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka Y, Kuroda M, Yasutake Y, Yao M, Tsumoto K, Watanabe N, et al. Crystal structure analysis reveals a novel forkhead-associated domain of ESAT-6 secretion system C protein in Staphylococcus aureus. Proteins. 2007;69:659–664. doi: 10.1002/prot.21302. [DOI] [PubMed] [Google Scholar]
- 25.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol. 2008;190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 27.Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 29.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K-I, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 30.Holden MTG, Feil EJ, Lindsay JA, Peacock SJ, Day NPJ, Enright MC, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A. 2004;101:9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holden MTG, Lindsay JA, Corton C, Quail MA, Cockfield JD, Pathak S, et al. Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW) J Bacteriol. 2010;192:888–892. doi: 10.1128/JB.01255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holden MTG, Hsu L-Y, Kurt K, Weinert LA, Mather AE, Harris SR, et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013;23:653–664. doi: 10.1101/gr.147710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schijffelen MJ, Boel CH, van Strijp JA, Fluit AC. Whole genome analysis of a livestock-associated methicillin-resistant Staphylococcus aureus ST398 isolate from a case of human endocarditis. BMC Genomics. 2010;11:376. doi: 10.1186/1471-2164-11-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Champion PAD, Champion MM, Manzanillo P, Cox JS. ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol Microbiol. 2009;73:950–962. doi: 10.1111/j.1365-2958.2009.06821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sillitoe I, Cuff AL, Dessailly BH, Dawson NL, Furnham N, Lee D, et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 2013;41(Database issue):D490–8. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo-Ramírez S, Corander J, Marttinen P, Aldeljawi M, Hanage WP, Westh H, et al. Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol. 2012;13:R126. doi: 10.1186/gb-2012-13-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindsay JA. Genomic variation and evolution of Staphylococcus aureus. Int J Med Microbiol. 2010;300:98–103. doi: 10.1016/j.ijmm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 39.Bæk KT, Frees D, Renzoni A, Barras C, Rodriguez N, Manzano C, et al. Genetic variation in the Staphylococcus aureus 8325 strain lineage revealed by whole-genome sequencing. PLoS One. 2013;8:e77122. doi: 10.1371/journal.pone.0077122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank KL, Del Pozo JL, Patel R. From clinical microbiology to infection pathogenesis: how daring to be different works for Staphylococcus lugdunensis. Clin Microbiol Rev. 2008;21:111–133. doi: 10.1128/CMR.00036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng AG, DeDent AC, Schneewind O, Missiakas D. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 2011;19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]