

Abstract
Background
In addition to its role in cell adhesion and gene expression in the canonical Wingless/integrated Wnt signaling pathway, β-catenin also regulates genes that underlie the transmission of nerve impulses. Mutations of CTNNB1 (β-catenin) have recently been described in patients with a wide range of neurodevelopmental disorders (intellectual disability, microcephaly and other syndromic features). We for the first time associate CTNNB1 mutation with hyperekplexia identifying it as an additional candidate for consideration in patients with startle syndrome.
Case presentation
We describe an 11 year old male Polish patient with a de novo nonsense mutation in CTNNB1 who in addition to the major features of CTNNB1-related syndrome including intellectual disability and microcephaly, exhibited hyperekplexia and apraxia of upward gaze. The patient became symptomatic at the age of 20 months exhibiting delayed speech and psychomotor development. Social and emotional development was normal but mild hyperactivity was noted. Episodic falls when startled by noise or touch were observed from the age of 8.5 years, progressively increasing but never with loss of consciousness. Targeted gene panel next generation sequencing (NGS) and patient-parents trio analysis revealed a heterozygous de novo nonsense mutation in exon 3 of CTNNB1 identifying a novel association of β-catenin with hyperekplexia.
Conclusion
We report for the first time a clear association of mutation in CTNNB1 with an atypical syndromic heperekplexia expanding the phenotype of CTNNB1-related syndrome. Consequently CTNNB1 should be added to the growing list of genes to be considered as a cause of startle disease or syndromic hyperekplexia.
Keywords: β-catenin, Hyperekplexia, Microcephaly, Intellectual disability
Background
A novel dominant intellectual disability (ID) syndrome caused by β-catenin gene (CTNNB1) haploinsufficiency was recently identified [1]. Mutations in CTNNB1 (chr3: 41,194,837-41,260,096) are responsible for a wide spectrum of neurodevelopmental disorders. The phenotypes of all 21 patients with CTNNB1 mutations reported to date in four different studies are broad and consistently include several major features: ID and motor delay with different degrees of severity, speech impairment, behavioral anomalies, spastic paraplegia, microcephaly and overlapping facial features [1–4]. In addition, variable clinical findings may involve brain MRI abnormalities (corpus callosum thinning and enlarged ventricles) and spinal anomalies (syringomyelia or tethered cord) [2]. We report a case of an 11-year-old boy with a de novo nonsense mutation in CTNNB1 who presented with mild ID, ataxia, spastic paraplegia, mild microcephaly and dysmorphic features. Notably our patient exhibited hyperekplexia and apraxia of upward gaze, features which to date have not been described in patients with mutations of CTNNB1.
Case Presentation
The study was approved by an ethics committee of Poznan University of Medical Sciences, Poland. Parents provided written informed consent according to the Declaration of Helsinki for publication of clinical information. The index case, a male patient was born at term as the first child to healthy, young, non-consanguineous Caucasian parents. Family history was negative for the presence of neurological disorders. He weighed 2900 g, with a good Apgar score. After a normal neonatal period, delayed psychomotor and speech development was evident (at 20 months of age he was unable to walk unassisted and was able to speak only a few words). His social and emotional development was within normal range but hyperactivity was observed. No focal lesions were visualized in 1.5 T MRI; notably in cortex, thalami, midbrain and medulla, except for a 12–13 mm in diameter arachnoid cyst in the posterior fossa and an enlarged Sylvian fissure. An EEG recording showed mild slowing of background activity. The parents had noticed episodic falls since the age 8.5 years. The episodes were provoked by noise, touch or rapid movement in the neighbourhood. Sudden loss in muscle tonus of lower limbs was observed, together with an increase in axial muscle tonus. It led to a rapid flexion in knees and hips provoking a fall. The boy has never had any head injury while falling, due probably to the extension position of his trunk. Episodes were short, lasting a second or two, without loss of consciousness. The boy was able to lift up by himself very quickly directly afterwards and was able to recall the entire episode. This sequence was highly repetitive. A progressive increase in the frequency of falls was noticed. Neurological examination between episodes indicated a left hemiparesis with positive Babinski sign and mild dysarthria. EEG during episodes showed diffused fast background activity, typical for hyperekplexia. In the interictal EEG no epileptiform discharges were observed. Levetiracetam was introduced with good clinical response however the drug was withdrawn because of behavioral disturbances (aggression, tendency to cry). Brivaricetam was introduced instead and the effect was similar to Levetiracetam with fewer side effects. During the last examination at the age of 11 years problems with speech were shown to have increased and coordination problems appeared. In addition to episodes of hyperekplexia, he presents with mild ID, ataxia, truncal hypotonia, bilateral spastic paraplegia, dysarthria and apraxia of upward gaze. He needs support to walk. Ophthalmic examination showed astigmatism and hyperopia with a normal fundus of the eye. Periodical horizontal nystagmus is present but vision evoked potentials are normal. The boy has height and weight within normal ranges, he is mildly dysmorphic (asymmetric face, right frontal hair upsweep, hypotelorism, deep set eyes, strabismus, diastema, small teeth, short philtrum, thin upper lip vermillion, prominent columella, broad nasal tip, small alae nasi, hypoplastic upper crus of the inner helix), has long slender fingers and toes, mild scoliosis, gynecomastia, inverted nipples and microcephaly. He is generally a friendly child, rather anxious, avoiding eye contact, with episodes of frustration. Verbal aggression, crying and sleep disturbances have been also observed (Table 1).
Table 1.
Clinical findings in patients with mutation in the N-terminal of β-catenin. * Previously published patients [2], n.e. not examined, n.r. not recorded
| Our Patient | Patient 5* | Patient 10* | Patient 13* | |
|---|---|---|---|---|
| Gender | M | F | F | M |
| Gestational weeks | 39 | 40 | 40 | 39 |
| Apgar scores | 10 | 9/10/10 | n.r. | 10/10 |
| Birth weight [g] | 2900 | 3450 | 3300 | 3160 |
| Birth length [cm] | 53 | 51 | 50.8 | 49 |
| Age at last examination [y] | 11 | 5 7/12 | 4 | 5 6/12 |
| Height [cm] | 150 | 108 | 104.5 | 112 |
| Weight [kg] | 46 | 15 | 14.15 | 17.2 |
| BMI [kg/m2] | 20.4 | 12.9 | 13.0 | 13.7 |
| OFC [cm] | 51 | 47.3 | 46 | 48.5 |
| Microcephaly | + | + | + | + |
| Croniofacial dysmorphism | ||||
| broad nasal tip | + | + | + | (+) |
| small alae nasi | + | + | + | - |
| long and/or flat philtrum | −/+ | +/+ | +/+ | −/+ |
| thin upper lip vermillion | + | + | + | + |
| Developmental and neurological abnormalities | ||||
| Truncal hypotonia | + | + | + | + |
| Peripheral hypertonia/spasticity | + | + atactic gait |
+ | + legs > arms |
| Motor delay Crowling (months) Free walking |
moderate | severe | moderate | moderate |
| did not crawl | 24 | 12 | 25 | |
| 3 years | not yet, walking frame | 4 years, falls a lot | not yet, walking frame | |
| Speech impairment | moderate, unclear speech | severe, few words (30–34), sign language | speech apraxia, ~50 words | short sentences at 6 years |
| Basic speech comprehension | + | + | + | + good |
| Intellectual disability | + | + | + | + (no formal test available) |
| Regression | + speech | ? | - | - |
| Behavioral characteristics | temper tantrums/crying, friendly personality, aggression, frustration, anxiety, sleep disturbances, stereotypic movements | sleep disturbances, stereotypic movements, autoagression | normal | normal, friendly, sensitive to loud noises |
| Seizures | - | - | - | - |
| EEG | during episodes diffused fast background activity | normal | normal | n.e. |
| Brain MRI | arachnoid cyst, enlarged Sylvius sulcus | normal | basically normal | normal |
| Hearing loss | - | - | - | - |
| Vision | hyperopia, astigmatism, strabismus | hyperopia, intermittent strabismus | normal | strabismus |
| Others | ||||
| Internal abnormalities | - | - | - | - |
| Frequent infections | - | - | - | + (infancy) |
| Miscellaneous | ginecomastia, inverted nipples | - | feeding difficulties | - |
| Hands | slender and long fingers | thin fingers, short distal phalanges | normal | clinodactyly V, hand and feet |
| Feet | sandal gap, long toes | flat feet | normal | pes equines |
The patient was followed by neuropediatrics and based upon symptoms including ataxia with progressive pyramidal syndrome, combined with supranuclear vertical gaze palsy, Niemann-Pick type C disease was assumed initially. However, sequencing of the entire coding and flanking regions of NPC1 and NPC2 failed to identify any pathogenic mutations. We further analyzed the index case using a NGS gene panel (targeted resequencing) that included 758 OMIM genes (including GLRA1, GLRB and SLCA) associated with neurological disorders (genomebiology.com/content/supplementary/s13059-015-0693-2-s4.xls). Genes were amplified and a library constructed using an AmpliSeq HiFi mix, proprietary primers and library kit (Thermo Fisher, Carlsband, CA, USA) followed by sequencing on the Ion Proton platform following the manufacturer’s protocol (Thermo Fisher, Carlsband, CA, USA). Sequences were mapped to hg19 and variants were called and annotated using the ion torrent pipeline (Thermo Fisher, Carlsband, CA, USA). Sequencing identified 2985 variants relative to hg19. By excluding previously reported variants (present in RefSeq, OMIM, Genbank, dbSNP, 1000 genomes, in-house database) and only focusing on non-synonymous, splicing, frameshift, indel and nonsense variants we decreased the number to 10 heterozygous changes each in a different gene (Fig. 1A). All changes were validated by Sanger sequencing of the index case and parents. Nine changes were inherited with the only other variant being confirmed as a heterozygous de novo nonsense mutation in exon 3 of CTNNB1 (NM_001098209): c.C232T; p.Q78X (Fig. 1B).
Fig. 1.
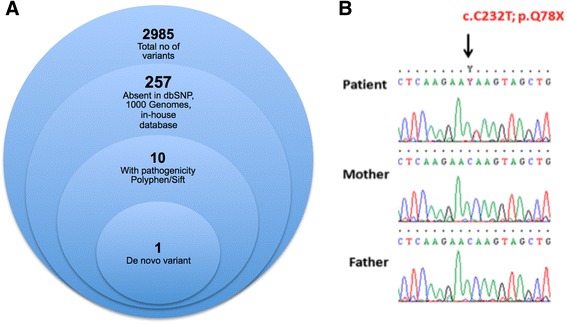
Filtering strategy used for identification of a causative mutation using trio analysis and NGS-gene panel (a). DNA electrophoregram with the c.C232T; p.Q78X mutation in exon 3 of CTNNB1 (b)
Conclusions
In this study we present an 11 year old boy in which we detected a de novo nonsense mutation in β-catenin (CTNNB1). Mutations of CTNNB1 were recently reported to be associated with a rare ID syndrome with consistent clinical features including significant motor delay with hypotonia of the trunk, progressive distal hypertonia/spasticity of the legs, speech impairment, behavioral anomalies, frequently microcephaly and overlapping dysmorphic facial features [1–4]. Our patient’s phenotype was consistent with, albeit generally milder than major signs of this entity. However, the boy also suffers from hyperekplexia and apraxia of upward gaze. These features have not been previously described in patients with a mutation of CTNNB1.
Hyperekplexia, or human startle disease, (OMIM#149400) is a rare, nonepileptic, paroxysmal neurogenetic disorder characterized by hypertonia and exaggerated persistent startle reflex to unexpected auditory, visual, and somatosensorial stimuli [5, 6]. The cluster of these symptoms is associated with failure of glycinergic synaptic transmission often resulting from mutations in genes encoding presynaptic and postsynaptic proteins [7]. Hyperekplexia results from mutations in GLRA1 and GLRB encoding the human glycine receptor and also the glycine transporter SLCA5 [8, 9]. Later, mutations in the proteins (GPHN and ARHGEF9) that cluster and localize the inhibitory glycine and GABA receptors were identified [10, 11]. However, patients with defects in these synaptic clustering proteins presented a disorder that had only a degree of phenotypic overlap with hyperexplexia. The genetic basis of hyperekplexia in a large proportion of cases remains to be discovered [12]. In our case the clinical diagnosis of hyperekplexia is based on the presence of its typical features: falls as a reaction to sudden stimuli, paroxysmal character, shortness and repetitiveness of episodes (that differenciate hyperekplexia from episodic falls and dyskinesias), no epileptiform EEG discharges during episode nor loss of conciousness (that are observed in atonic seizures), rapid recovery (that is never seen in cataplexy). However, the hyperekplexia in our patient lacks some of cardinal features of hyperekplexia such as rigidity during first year of life, general increase of muscle tonus during an episode and frequent head injuries. Presence of progressive neurologic impairment is also not typical for hyperekplexia. Therefore, we propose a diagnosis of atypical hyperekplexia for this patient`s neurological condition.
β-catenin (CTNNB1) belongs to the armadillo family of proteins responsible for regulation of gene expression in the canonical Wnt signaling pathway, in addition to their function in cell adhesion [13]. Wnt family secreted glycoproteins are highly conserved and modulate cell-cell communication in cellular processes including central nervous system development [14]. A central component of this pathway, β-catenin binds the TCF/LEF transcription factors that are involved in regulation of many genes [15]. In the thalamus, β-catenin regulates the expression of genes encoding proteins associated with excitation of neurons. In the mouse, it binds to Gabra 3 encoding the GABA receptor, and also to genes responsible for voltage and ligand-gated ion channels: Cacna1g and Kcna6. This indicates a role of β-catenin in the maintenance of neuronal excitability [13].
During axonal developmental, the specification of the axon initial segment (AIS), characterized by a high density of voltage-gated Na+ and K+ channels, is critical in initiating and modulating action potentials [16]. These ion channels are tethered by ankyrinG (intracellular protein) to the membrane of the AIS and during this process, β-catenin is progressively accumulated at the AIS and is an essential component of this neuronal domain [17]. Its enrichment plays a role in dendritic morphogenesis, axonal growth and the maturation of AIS functions, modulating neuronal excitability and voltage-gated sodium channels [13, 18]. β-catenin knockdown decreases voltage-gated sodium currents at the AIS. The sodium current reduction is most likely due to diminished ankyrinG tethering at the AIS and the loss of AIS integrity when β-catenin is absent [17]. This molecular mechanism underlying neuronal action potential generation explains β-catenin involvement in disorders related to neuronal development and excitability [17].
Our patient expresses hyperekplexia, a unique symptom that has not been seen among other reported CTNNB1 patients. Only in one other case (P14) a pronounced startle response on both auditory and visual stimuli provoking a breath holding spell was noted without a clear diagnosis of hyperekplexia [2]. Our patient has a de novo mutation in CTNNB1 (c.C232T; p.Q78X) which is localized in the N-terminal domain of β-catenin. Three other patients (P5, P10 and P13) described by Kuechler’s et al. also had mutations in the N-terminal helical domain [2]. This region contains a conserved short linear motif responsible for binding β-TrCPE3 ubiquitin ligase, but only when it is phosphorylated. Degradation of β-catenin is regulated by this N-terminal segment. When comparing the clinical findings for all four patients with mutation in the N-terminal of β-catenin, consistent phenotypic features emerge with exception of atypical hyperekplexia and impaired upward gaze unique to our case (Table 1). In some cases with CTNNB1 haploinsufficiency syndrome, there are patients with additional features not seen in others [4]. There is also, perhaps not surprisingly, inconsistency in the phenotype resulting from heterozygous loss-of-function mouse and human mutations, with phenotype in humans being much more severe [3]. This wide spectrum of clinical features in cases with CTNNB1 mutations may result from compensatory activity of other protein partners that can assume the role of β-catenin in its absence [3]. Plakoglobin for instance (also called γ-catenin) has a very similar structure and ligand binding capacity to β-catenin [19]. The data presented here provides new insight into the role of β-catenin in brain development and its association with neurodevelopmental disorders including hyperekplexia. CTNNB1 should be considered a candidate in patients with hyperekplexia in whom classical genetic mechanisms identified to date are negative.
Consent
Written informed consent was obtained from the patient’s parents for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgements
The authors would like to thank the patient and parents for their cooperation and consent in relation to this study
Abbreviations
- Wnt
Wingless/Integrated
- NGS
Next Generation Sequencing
- CTNNB1
β-catenin protein
- GABA
γ-aminobutyric acid
- EEG
Electroencephalogram
- TCF/LEF
T-cell factor-1 (Tcf-1) and lymphoid enhancing factor-1 (Lef-1)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AWW, MBS, BS and ALB performed the medical assessment. AMN, MN and DM performed genetic analysis. AWW, MBS and DM drafted the manuscript with the help of all other authors. All authors read and approved the final manuscript.
Contributor Information
Anna Winczewska-Wiktor, Email: awwiktor@ump.edu.pl.
Magdalena Badura-Stronka, Phone: +48607566527, Email: bstronka@gmail.com.
Anna Monies-Nowicka, Email: aniamonies@gmail.com.
Michal Maciej Nowicki, Email: mmnowickimd@gmail.com.
Barbara Steinborn, Email: bstein@ump.edu.pl.
Anna Latos-Bieleńska, Email: alatos@ump.edu.pl.
Dorota Monies, Email: dmonies@kfshrc.edu.sa.
References
- 1.de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 2.Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. 2015;134(1):97–109. doi: 10.1007/s00439-014-1498-1. [DOI] [PubMed] [Google Scholar]
- 3.Tucci V, Kleefstra T, Hardy A, Heise I, Maggi S, Willemsen MH, et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest. 2014;124(4):1468–1482. doi: 10.1172/JCI70372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A. 2014;164A(6):1571–1575. doi: 10.1002/ajmg.a.36484. [DOI] [PubMed] [Google Scholar]
- 5.Al-Futaisi AM, Al-Kindi MN, Al-Mawali AM, Koul RL, Al-Adawi S, Al-Yahyaee SA. Novel mutation of GLRA1 in Omani families with hyperekplexia and mild mental retardation. Pediatr Neurol. 2012;46(2):89–93. doi: 10.1016/j.pediatrneurol.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Bakker MJ, van Dijk JG, van den Maagdenberg AM, Tijssen MA. Startle syndromes. Lancet Neurol. 2006;5(6):513–524. doi: 10.1016/S1474-4422(06)70470-7. [DOI] [PubMed] [Google Scholar]
- 7.Thomas RH, Chung SK, Wood SE, Cushion TD, Drew CJ, Hammond CL, et al. Genotype-phenotype correlations in hyperekplexia: apnoeas, learning difficulties and speech delay. Brain. 2013;136(Pt 10):3085–3095. doi: 10.1093/brain/awt207. [DOI] [PubMed] [Google Scholar]
- 8.Chung SK, Bode A, Cushion TD, Thomas RH, Hunt C, Wood SE, et al. GLRB is the third major gene of effect in hyperekplexia. Hum Mol Genet. 2013;22(5):927–940. doi: 10.1093/hmg/dds498. [DOI] [PubMed] [Google Scholar]
- 9.Carta E, Chung SK, James VM, Robinson A, Gill JL, Remy N, et al. Mutations in the GlyT2 gene (SLC6A5) are a second major cause of startle disease. J Biol Chem. 2012;287(34):28975–28985. doi: 10.1074/jbc.M112.372094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rees MI, Harvey K, Ward H, White JH, Evans L, Duguid IC, et al. Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J Biol Chem. 2003;278(27):24688–24696. doi: 10.1074/jbc.M301070200. [DOI] [PubMed] [Google Scholar]
- 11.Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci. 2004;24(25):5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bode A, Lynch JW. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain. 2014;7:2. doi: 10.1186/1756-6606-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wisniewska MB, Nagalski A, Dabrowski M, Misztal K, Kuznicki J. Novel beta-catenin target genes identified in thalamic neurons encode modulators of neuronal excitability. BMC Genomics. 2012;13:635. doi: 10.1186/1471-2164-13-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maguschak KA, Ressler KJ. The dynamic role of beta-catenin in synaptic plasticity. Neuropharmacology. 2012;62(1):78–88. doi: 10.1016/j.neuropharm.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narasipura SD, Henderson LJ, Fu SW, Chen L, Kashanchi F, Al-Harthi L. Role of beta-catenin and TCF/LEF family members in transcriptional activity of HIV in astrocytes. J Virol. 2012;86(4):1911–1921. doi: 10.1128/JVI.06266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11(2):178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- 17.Tapia M, Del Puerto A, Puime A, Sanchez-Ponce D, Fronzaroli-Molinieres L, Pallas-Bazarra N, et al. GSK3 and beta-catenin determines functional expression of sodium channels at the axon initial segment. Cell Mol Life Sci. 2013;70(1):105–120. doi: 10.1007/s00018-012-1059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu X, Malenka RC. Multiple functions for the cadherin/catenin complex during neuronal development. Neuropharmacology. 2004;47(5):779–786. doi: 10.1016/j.neuropharm.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 19.Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148(3):567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]