

Abstract
Background
Chlamydomonas reinhardtii is the model organism that serves as a reference for studies in algal genomics and physiology. It is of special interest in the study of the evolution of regulatory pathways from algae to higher plants. Additionally, it has recently gained attention as a potential source for bio-fuel and bio-hydrogen production. The genome of Chlamydomonas is available, facilitating the analysis of its transcriptome by RNA-seq data. This has produced a massive amount of data that remains fragmented making necessary the application of integrative approaches based on molecular systems biology.
Results
We constructed a gene co-expression network based on RNA-seq data and developed a web-based tool, ChlamyNET, for the exploration of the Chlamydomonas transcriptome. ChlamyNET exhibits a scale-free and small world topology. Applying clustering techniques, we identified nine gene clusters that capture the structure of the transcriptome under the analyzed conditions. One of the most central clusters was shown to be involved in carbon/nitrogen metabolism and signalling, whereas one of the most peripheral clusters was involved in DNA replication and cell cycle regulation. The transcription factors and regulators in the Chlamydomonas genome have been identified in ChlamyNET. The biological processes potentially regulated by them as well as their putative transcription factor binding sites were determined. The putative light regulated transcription factors and regulators in the Chlamydomonas genome were analyzed in order to provide a case study on the use of ChlamyNET. Finally, we used an independent data set to cross-validate the predictive power of ChlamyNET.
Conclusions
The topological properties of ChlamyNET suggest that the Chlamydomonas transcriptome posseses important characteristics related to error tolerance, vulnerability and information propagation. The central part of ChlamyNET constitutes the core of the transcriptome where most authoritative hub genes are located interconnecting key biological processes such as light response with carbon and nitrogen metabolism. Our study reveals that key elements in the regulation of carbon and nitrogen metabolism, light response and cell cycle identified in higher plants were already established in Chlamydomonas. These conserved elements are not only limited to transcription factors, regulators and their targets, but also include the cis-regulatory elements recognized by them.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2564-y) contains supplementary material, which is available to authorized users.
Keywords: Chlamydomonas reinhardtii, green algae, gene co-expression networks, molecular systems biology, transcriptomics, RNA-seq, light-regulated transcription factors and transcriptional regulators
Background
The unicellular green alga Chlamydomonas reinhardtii (Chlamydomonas) is an important model organism for genomic and physiological studies in photosynthetic organisms. Due to its evolutionary position, it diverged from land-plants over a billion years ago, Chlamydomonas is considered a living representative of the photosynthetic organisms that gave rise to the green lineage [1]. Specifically, it has been used as a model organism to study the establishment, conservation and divergence of key biological processes in photosynthetic organisms such as the photoperiod response [2–4]. Recently, Chlamydomonas has attracted substantial interest for biotechnological applications in the context of bio-fuel and bio-hydrogen production [5–7]. The main advantage of using Chlamydomonas over higher plants is that it does not compete for agricultural land use. Additionally, Chlamydomonas posseses powerful genetic tools, metabolic versatility and a haploid genome. However, an important disadvantage is the lack of sufficient functional and regulatory characterization of the molecular mechanisms underpinning these processes with biotechnological interest [8].
In order to overcome this limitation its genome was sequenced and it is currently in an advanced curated state [1, 9]. The availability of its genome has facilitated the use of Next Generation Sequencing techniques, specially RNA-seq, in order to study its complete transcriptome. This has produced a massive amount of data from a variety of genotypes grown under relevant physiological conditions [10–16]. However, these studies remain fragmented without producing global insights into the organization and regulation of the Chlamydomonas transcriptome. The first steps towards the use of molecular systems biology methodologies to characterize the Chlamydomonas transcriptome has been taken [17–19]. Nevertheless, one of the most widely used tools for the integration and study of massive amounts of transcriptomic data, gene co-expression networks, have not yet been developed for Chlamydomonas, while gene co-expression networks have been used successfully in many other photosynthetic organisms [20–22].
Gene co-expression networks integrate fragmented transcriptomic data obtained in different studies in order to characterize patterns of coordinated gene expression at the whole transcriptome level. In gene co-expression networks nodes represent genes, being nodes connected by an edge if the corresponding genes are significantly co-expressed across appropriately chosen genotypes and physiological conditions [23]. Fundamental network concepts such as node degree, neighbourhood and clustering coefficient have important applications to unravel the organization and functioning of the represented transcriptome [24, 25]. The degree of a node, that is, the number of nodes connected to it, represents the number of genes co-expressed with the corresponding gene. Therefore, genes represented by nodes with high degrees are expected to be relevant in the transcriptome since their expression is coordinated with many others. The neighborhood of a node consists of genes co-expressed with the corresponding gene. This set of genes can be used as target genes candidates when the given gene is a transcription factor or potential regulator. The transcription factor binding sites that are responsible for the coordinated expression of genes can be identified by analyzing the significance of specific motifs in the promoters of co-expressed genes [26]. Additionally, Gene Ontology (GO) term enrichment over gene neighbourhoods can be applied to determine the potential biological processes that are carried out by the orchestrated expression of any given genes. In most gene co-expression networks the probability that a node is connected with k other nodes, P (k), follows a negative exponential distribution, P (k) ~ k-λ. This is the defining property of scale-free networks [27]. In scale-free networks most nodes are connected with few nodes, whereas there exists a small number of highly connected nodes called hubs that dominate the network dynamics [28]. Genes that correspond to hub nodes play a key role in the correct functioning of biological processes and, therefore, their mutation can lead to severely affected phenotypes and even lethality [29]. The clustering coefficient of a node meassures the tendency of nodes to group together around the given node, and when applied to gene co-expression networks, this concept indicates the tendency of genes to form highly co-expressed gene clusters. Scale-free networks with an average high clustering coefficient are called small world networks [28]. In this class of networks the existence of a clustering structure around hub nodes produces short paths that connect any pair of nodes. It has been often observed that biological co-expression networks are scale-free and small world networks [20, 25].
In this study we have developed ChlamyNET, a gene co-expression network and an associated web-based software tool that integrates the massive amount of RNA-seq data available for the Chlamydomonas transcriptome, see Additional file 1: Table S1. We have used this tool to study the organization and regulation of the algal transcriptome. ChlamyNET aims at becoming an enabling technology for researchers in the Chlamydomonas transcriptome, and in a wider perspective of alga transcriptomics, being the first tool of this kind existing at this date. Researchers can explore the neighbourhood of their genes of interest in ChlamyNET looking for potential targets or regulators. Additionally, our web tool can be used to determine GO terms related to biological processes, functions and components that are significantly present in the annotation of the neighbouring genes. Finally, we have used an independent experimental data set to cross-validate the predictive power of ChlamyNET.
Results and discussion
Network construction and topology
The high resolution provided by RNA-seq data and the diverse physiological conditions and genotypes analyzed allowed us to capture the co-expression relationships between genes in the Chlamydomonas transcriptome. In order to reduce the noise in our analysis, we only considered genes that showed significant changes in at least one comparison between a condition and its corresponding control. Data processing and selection of differentially expressed genes were performed as described in the methods section. Out of the 16624 predicted genes in the Chlamydomonas genome 13699 were differentially expressed in at least one of the conditions analyzed in this study. This represents 82.40 % of the algal genome, which shows that the analyzed conditions and phenotypes are diverse enough to capture the behaviour of most of the Chlamydomonas transcriptome.
As described in the methods section, we used a range of absolute correlation thresholds to determine the co-expression level between the selected genes [30]. A compromise between the generation of a scale-free network and a high density network was established. We observed that for increasing correlation thresholds, the density of the network decreased, whereas the fit to the scale-free property increased until the cut-off value was too restrictive and the network started to deteriorate (Fig. 1a). Indeed, the scale-free model fit exhibits a maximum at a correlation value of 0.90 with an R2 equal to 0.86. According to this, we chose an absolute Pearson correlation threshold of 0.90 to consider that two genes are significantly co-expressed. The gene co-expression network generated for this threshold was called ChlamyNET.
Fig. 1.
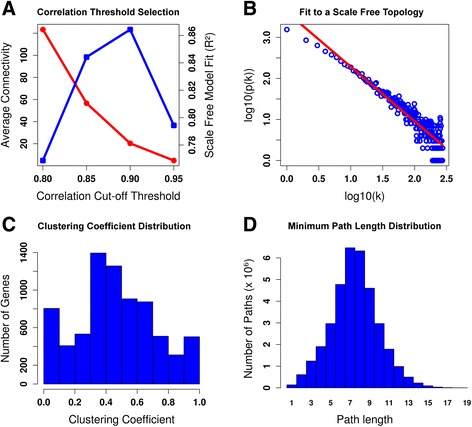
Network Construction and Topology Analysis. a Correlation Threshold Selection. The blue line shows that at the absolute value of correlation 0.90 the scale-free model fit exhibits a maximum. The red line shows that for increasing correlation thresholds the average connectivity of the network decreases, nonetheless for 0.90 it still presents a high value. According to this, the correlation threshold used to generate ChlamyNET was fixed to 0.90. b The degree distribution of a scale-free network follows an exponential negative distribution. The scale-free topology fit of ChlamyNET was computed using linear regression over the logarithmic transform of its degree distribution. c The clustering coefficient of a node or gene represents the degree of co-expression or correlation between its neighbours. Genes with a high clustering coefficient posses a high degree of co-expression or coordination among its co-expressed genes. ChlamyNET exhibits the high average clustering coefficient of 0.66. d ChlamyNET constitutes a small world network, that is a scale-free network with a high clustering coefficient. This is reflected in the fact that the average minimal path length between genes is 7.5
ChlamyNET consists of 9171 genes or nodes exhibiting an overall of 139019 co-expression relationships or edges. ChlamyNET is composed of a major connected component consisting of 8443 genes (92.1 % of the entire network) and a multitude of small components consisting of a few genes. This global connectivity property of ChlamyNET is similar to previously constructed and analyzed networks from other organisms such as Saccharomyces cerevisiae [31] and Arabidopsis thaliana [32].
The scale-free property of ChlamyNET was corroborated by computing its degree distribution and checking that it follows a negative exponential distribution. Specifically, linear regression over the logarithmic transform of the degree distribution was used (Fig. 1b). Another topological property that we analyzed in ChlamyNET was the clustering coefficient, a measurement of the density of edges or co-expression relationships around genes. The distribution of the clustering coefficient in ChlamyNET was computed (Fig. 1c). The average clustering coefficient of ChlamyNET is 0.66 which is significantlly high when compared to random scale-free networks, see the methods section. This shows that ChlamyNET constitutes a non-random scale-free network with a high clustering coefficient. This type of networks are called small-world networks since the minimal path length between genes is short when compared to random scale-free networks [33]. These properties are common in gene co-expression networks [20, 32]. In the case of ChlamyNET the average minimal path length between genes or the network diameter is 7.5 (Fig. 1d). Therefore, on average any gene on ChlamyNET can be reached from another one through approximately seven gene co-expression relationships.
The topological properties of ChlamyNET (Fig. 2a), namely scale-free and small-world properties, imply that most genes in the Chlamydomonas transcriptome are co-expressed with only a few other genes. However, there exists a low number of genes that are co-expressed with a large number of other genes. These genes are called hubs and play a key role in the structure and functioning of gene co-expression networks [28]. We determined the first 1000 hubs in terms of their degree and highlighted them in ChlamyNET, observing that they are located in specific regions of the network (Fig. 2b). Nevertheless, the definition of hubs based solely on the number of genes it is co-expressed with has been found to be incomplete and the concept of authoritative hub has been introduced [34]. Following this line, in our context, an authoritative hub gene relevant to a biological process is not considered solely on the base that they have a large number of co-expressed genes. Additionally, since its co-expressed genes are involved in the same biological process they should in turn be co-expressed among themselves, establishing links in the network between them. These authoritative genes could then be responsible for bringing together genes potentially involved in a common biological process. We identified the first 1000 authoritative hubs using the HITS algorithm [34] and represented them in ChlamyNET (Fig. 2c). We observed that the most relevant authoritative hubs are located in the center of the network. Additionally, we observe that the location of regions with high clustering coefficient is not random. These regions substantially overlap with areas where hub genes are located (Fig. 2d). In order to determine whether or not these hub genes are involved in similar biological processes we performed a gene ontology (GO) term enrichment analysis (Table 1) based on orthology relationships with Arabidopsis thaliana and on the annotation of protein families and domains available in the Pfam database [35] as described in the methods section. This analysis revealed that the central part of ChlamyNET constitutes the core of the network where the most authoritative hub genes are located. These hubs interconnect key biological processes such as protein phosphorylation and response to light stimulus with carbon/nitrogen metabolism and transmembrane transport (Table 1). Protein kinases potentially involved in developmental processes such as CrMEKK (g5375), similar to the Arabidopsis MEKK gene At5g57610, and transcription factors possibly associated with circadian rythms and photoperiodic responses like CrBbox1 (Cre03.g182700), similar to the Arabidopsis COL1 gene At5g15850, are highly authoritative hub genes in the regulation of the Chlamydomonas transcriptome with more than 250 neighbours. Relevant enzymes in the carbon/nitrogen metabolism and transmembrane transport are also authoritative hub genes in the core of ChlamyNET indicating that their expression is highly regulated and coordinated with other biological processes. For example, the nitrate transporter NRT2.3 (Cre09.g396000), nitrate reductase NIT1 (Cre09.g410950) and starch phosphorylase CrPHS1 (Cre07.g336950) are also co-expressed with more than 250 genes.
Fig. 2.
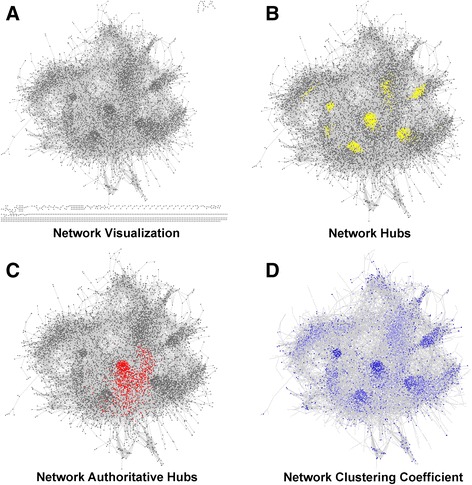
- Network Visualization, Hubs and Clustering Coefficient. a Graphical representation of ChalmyNET consisting of 9171 genes or nodes and 139019 co-expression relationships or edges. It is organized into a major connected component where most of the genes are located and a multitude of small components. b Network hubs. We have represented in yellow hub genes characterized by being co-expressed with a large number of other genes. Note that hub genes are located in specific regions of the network. c Authoritative hubs. Those hubs whose neighbours are highly connected, are mainly located at the core of the network. These authoritative hubs are represented in red. d The clustering coefficient of a gene meassures the degree of co-expression among its co-expressed genes. Genes with a high clustering coefficient are coloured in darker blue than those with a low clustering coefficient. Notice that regions of genes with a high clustering coefficient overlap with those where hubs are located
Table 1.
Biological Process GO terms significally enriched in the 1000 most authoritative hub genes in ChlamyNET
| GO term | Representative Genes | Potential Arabidopsis Ortholog |
Number of neighbours |
|---|---|---|---|
| protein phosphorylation GO:0006468 (p-value 2.6 x 10-11) | Cre02.g108700 - Serine/Threonine Protein Kinase g2226 - VH1-Interacting Kinase Cre12.g537400 - ataurora Cre17.g742400 - Protein tyrosine kinase |
At5g08160 At1g14000 At2g45490 At1g18160 |
245 253 266 93 |
| transmembrane transport GO:0055085 (p-value 3 x 10-7) | Cre09.g396000 - Nitrate Transporter Cre10.g453400 - Mechanosensitive Channel of Small Conductance-like Cre01.g012700 - Gated Outwardly Rectifying K+ Channel |
At1g12940 At5g12080 At5g37500 |
250 233 231 |
| response to light stimulus GO:0009416 (p-value 2 x 10-5) | Cre03.g182700 - Bbox Protein Cre02.g118000 - Photolyase Cre12.g510200 - bZIP Protein g6302 - Constans-like Cre06.g295200 - Cryptochrome | At5g15850 At1g12370 At5g11260 At5g15840 At4g08920 |
259 182 133 58 57 |
| carbohydrate metabolic process GO:0005975 (p-value 8 x 10-5) | Cre07.g336950 - Alpha-glucan phosphorylase Cre08.g362450 - Alpha Amylase g3160 - Isoamylase Cre04.g215150 - Soluble Starch Synthase | At3g46970 At1g69830 At2g39930 At5g24300 |
257 39 56 24 |
| nitrogen compound metabolic process GO:0006807 (p-value 5.2 x 10-5) | Cre09.g410950 - Nitrate reductase Cre09.g410750 - Nitrite Reductase Cre03.g207250 - Glutamine synthetase | At1g37130 At2g15620 At5g35630 |
251 251 114 |
Network clustering analysis and functional annotation
The specific location of hub genes in regions exhibiting a high clustering coefficient may reflect an underlying structure in ChlamyNET relevant to physiological functions related to the Chlamydomonas transcriptome. In ChlamyNET we can observe distinct areas composed of genes with high degree and clustering coefficient that are in turn loosely connected through other genes with low degrees and clustering coefficients (compare Figs. 2b and d). This indicates the existence of relatively isolated groups of genes whose expression are highly coordinated and, hence, are potentially involved in the same biological processes. In order to corroborrate the existence of this underlying structure we applied clustering techniques over ChlamyNET using, as described in the methods section, the Pearson correlation coefficient between gene expression profiles as gene similarity measure. We compared the performance of the two most widely used clustering algorithms hierarchical clustering (HCLUST) and partition around medoids (PAM) for different number of clusters ranging from 4 to 20 clusters using the silhouette, a criterion that combines the minimization of inter-cluster similarity with the maximization of the intra-cluster similarity [36]. Our analysis concluded that the underlying structure of ChlamyNET is best described using nine clusters identified with the PAM algorithm (Fig. 3a) since this combination of clustering algorithm and number of clusters produced the highest silhouette value of 0.28 (Fig. 3b). This identified different clusters which we highlighted with different colour codes. Each gene cluster exhibits distinct expression profiles, see Additional file 2: Figure S1.
Fig. 3.
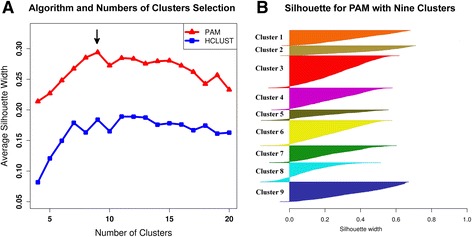
Selection of the Clustering Algorithm and Number of Clusters using as Criterion the Clustering Silhouette. a Algorithm and number of clusters selection. The absolute value of Pearson correlation coefficient between gene expression profiles was used as gene similarity measure to perform our clustering analysis. The performance of the clustering algorithms hierarchical clustering (HCLUST in red triangles) and partition around medoids (PAM in blue squares) were compared for different number of clusters ranging from 4 to 20 using the clustering silhouette. The highest silhouette value was reached for the PAM algorithm with nine cluster (marked with an arrow). b Silhouette for PAM with nine clusters. The silhouette of a clustering measures both the inter and intra cluster similarities. The best clustering silhouette obtained with the PAM algorithm for nine clusters is shown. Each horizontal line represents a gene in a given cluster. A high positive value indicates a gene with a high intra cluster similarity and a low inter cluster similarity. Whereas a negative value indicates a gene with a low intra cluster similarity and a high inter cluster similarity. Genes belonging to the same cluster are represented with the same colour. For each cluster from one to nine, the number of genes and its average silhouette are specified
Since the genes in each cluster are co-expressed throughout the diverse physiological conditions integrated in this study they are likely involved in the same biological processes. In order to study the biological processes in which each gene cluster is involved we performed a Gene Ontology (GO) term enrichment over these clusters [37]. In order to overcome the limitations imposed by the sparse annotation of the Chlamydomonas transcriptome we combined GO terms obtained using orthology information with Arabidopsis, togheter with GO terms associated to protein families defined using conserved protein domains available from the Pfam database [35]. Since the used annotation is not based on experimental studies, the results obtained here should be taken as predictions that would need further experimental validation. This is precisely one of the main goals of GO term enrichment, namely the computational prediction of gene function to be subsequently corroborated using wet laboratory experimental work [38]. The results of our clustering and functional analysis are depicted in Fig. 4 and summarized in Table 2. The nine different gene clusters are identified with different colours and numbers following the code in Fig. 3b. In order to place our clustering analysis into a physiological, biochemical and metabolic context we used the tools and databases available from the Plant Metabolic Network [39]. Specifically, we used the extensive metabolic pathway information provided by ChlamyCyc [40] to identify the metabolic pathways contained in each cluster. In the following subsections, we present in detail four gene clusters and their functional annotation.
Fig. 4.
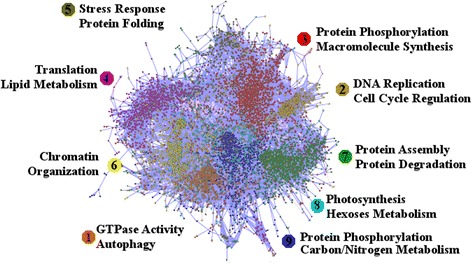
ChlamyNET Clustering and Cluster Functional Annotation. In ChlamyNET each node specifies a gene and an edge between genes represents that the corresponding gene expression profiles exhibit an absolute Pearson correlation coefficient value greater than 0.90. Therefore, edges represent co-expression relationships. Blue edges stand for positive correlation whereas pink edges stand for negative values. The nine different gene clusters are identified by numbers and different colours corresponding to the code in Fig. 3. Clusters are also annotated with the biological processes where the corresponding genes are potentially involved in
Table 2.
Biological Process GO terms significally enriched in the clusters of the gene co-expression network ChlamyNET and the Metabolic and Signalling Pathways contained in each cluster
| Cluster | Functional Annotation | Representative Genes | Metabolic/Signalling Pathways |
|---|---|---|---|
| Cluster 2 (Brown) 535 genes Silhoutte 0.44 | DNA replication (GO:0006260) | Cre01.g015250 - POLD1 Cre16.g651000 - RFA1 | Pyrimidine deoxyribonucleotides de novo biosynthesis pathway Cre16.g667850 - DUT Cre17.g715900 - THY Cre03.g190800 - TMPK |
| Chromosome organization (GO:00051276) | Cre02.g086650 - SMC2 Cre12.g4 934 00 - SMC4 | ||
| Regulation of Cell Cycle (GO:0010564) | Cre10.g466200 - CYCAB1 Cre03.g207900 - CYCA1 | ||
| Cluster 9 (Blue) 1058 genes Silhouette 0.40 | protein phosphorylation (GO:0006468) | Cre17.g742400 - PTK17 Cre12.g537400 - CrAUR3 | Starch Biosynthetic Pathway Cre04.g215150 - SSS Sucrose Biosynthetic Pathway Cre06.g283400 - SPP Nitrogen Assimilation Pathway Cre09.g410750 - NII1 |
| carbohydrate metabolic process (GO:0005975) | Cre08.g384750 - AMY Cre10.g444700 - SBE3 | ||
| transmembrane transport (GO:0055085) | Cre09.g396000 - NRT2.3 Cre13.g564650 - MRS5 | ||
| Cluster 1 (Orange) 824 genes Silhouette 0.38 | vesicle-mediated transport (GO:0016192) | Cre17.g728150 - Yky6 Cre16.g676650 - AP1G1 | TAG Biosynthetic Pathway Cre02.g106400 - PDAT Phospholipid Biosynthetic Pathway Cre01.g035500 - PI3K Coenzyme A Biosynthetic Pathway Cre01.g048050 - COAB |
| GTPase activity (GO:0043087) | Cre12.g532600 - CGL44 Cre07.g315350 - RABGAP | ||
| Autophagy (GO:0006914) | Cre09.g391500 - APG9 | ||
| Cluster 3 (Red) 1723 genes Silhouette 0.28 | protein phosphorylation (GO:0006468) | Cre02.g145500 - PTK24 Cre12.g498650 - ALK3 | TAG Biosynthetic Pathway g9572 - DGAT1 Hydrogen production Cre09.g396600 - HYDA2 MAP kinase cascade Cre10.g461150 - CrMAPKKK |
| ribosome biogenesis (GO:0042254) | Cre12.g532550 - RPL13a Cre09.g400650 - RPS6 | ||
| macromolecule biosynthesis (GO:0009059) | Cre03.g207250 - GLN4 | ||
| Cluster 4 (Purple) 1174 genes Silhouette 0.26 | translation (GO:0006412) | Cre03.g199900 - EIF4E Cre02.g117900 - RH | tRNA Charging Pathway g2951 - TrpS Amino Acid Biosynthesis Cre03.g161400 - WSN2 Pentose Phosphate Non-oxydative Cre12.g511900 - RPE1 TAG Biosynthetic Pathway Cre03.g205050 - DGAT2 |
| RNA processing (GO:0006396) | Cre16.g653050 - SpoU Cre10.g421600 - ThrRS g4 679 - RNase P | ||
| lipid metabolism (GO:0006629) | Cre09.g397250 - FAD5 Cre06.g295250 - PAP | ||
| Cluster 7 (Green) 909 genes Silhouette 0.25 | protein complex assembly (GO:0006461) | g9912 - CSN5 Cre16.g663500 - CrRPN10 | Aerobic Respiration Pathway Cre15.g638500 - CYC1 COP9 Signalling g11578 - CSN6 |
| response to misfolded protein (GO:0051788) | Cre06.g280850 - PSMB4 Cre12.g501200 - SKP1 | ||
| Cluster 6 (Yellow) 1351 genes Silhouette 0.24 | chromatin organization (GO:0006325) | g11636 - HDA Cre13.g590750 - HTB37 | Chromatin Remodelling Cre13.g591200 - HTB38 Cre13.g562400 - ABI3 |
| posttranscriptional regulation (GO:0010608) | g7250 - DCL | ||
| Cluster 5 (Dark Green) 567 genes Silhouette 0.21 | response to heat (GO:0009408) | Cre14.g617400 - HSP22F Cre08.g372100 - HSP70A | Stress Response Cre02.g098800 - ERP29 g9861 - TOR |
| protein folding (GO:0006457) | g9881 - FKBP Cre01.g047700 - CYP40 | ||
| Cluster 8 (Turquoise) 1030 genes Silhouette 0.10 | photosynthesis (GO:0015979) | Cre09.g412100 - PSAF Cre10.g44 04 50 - PSB28 | Calvin Cycle Cre12.g554800 - PRK1 TCA Cycle Cre02.g143250 - IDH2 |
| hexose metabolic process (GO:0019318) | Cre17.g725550 - GLD1 Cre02.g093450 - FBA2 |
Cluster 2, brown - DNA replication, chromosome organization and regulation of cell cycle
The most cohesive gene cluster is also the smallest one. The brown cluster is located in the periphery of ChlamyNET. It presents the highest silhouette value (0.44) in the network and contains 535 genes (Fig. 3b). Our GO term enrichment analysis reveals that this cluster is involved in cell cycle processes. Specifically, it is enriched in genes required for DNA replication (GO:0006260) such as DNA polymerase POLD1 (Cre01.g015250), replication factor RFA1 (Cre16.g651000) and origin recognition complex ORC2 (Cre03.g199400); genes associated with chromosome organization (GO:0051276) like structural maintenance of chromosomes SMC4 (Cre12.g493400) and SMC2 (Cre02.g086650) and genes involved in the regulation of cell cycle process (GO:0010564) such as the cyclin A/B CYCAB1 (Cre10.g466200) and the A-type cyclin CYCA1 (Cre03.g207900).
The metabolic pathways located in this cluster produce DNA and RNA precursors such as the pyrimidine deoxyribonucleotides de novo biosynthesis pathway. For example, the formation of the DNA-specific end product dTTP starts with the hydrolyzation of dUTP to produce dUMP by the dUTP pyrophosphatase DUT (Cre16.g667850), followed by the reductive methylation of dUMP catalyzed by thymidylate synthase THY (Cre17.g715900) which yields dTMP. Finally, the thymidylate kinase TMPK (Cre03.g190800) catalyzes the first phosphorylation of dTMP leading to dTTP. These three enzymes are members of this cluster (Additional file 3: Figure S2 and Table 2).
Cluster 9, blue - protein phosphorylation, carbohydrate metabolic process and transmembrane transport
The blue cluster located in the center of ChlamyNET is enriched with hub genes according to a p-value < 2.2°10−16 obtained using Fisher's exact test. It is the second most cohesive cluster with a silhouette value of 0.40 and 1058 genes (Fig. 3b). The most significantly over-represented GO terms in this cluster are protein phosphorylation (GO:0006468) with genes such as the protein tyrosine kinases PTK17 (Cre17.g742400) and ataurora CrAUR3 (Cre12.g537400), carbohydrate metabolic process (GO:0005975) including genes like the alpha amylase AMY (Cre08.g384750), and transmembrane transport (GO:0055085) including genes coding for magnesium and cobalt transport protein MRS5 (Cre13.g564650). An analysis of the metabolic context of this cluster reveals that core pathways in carbon and nitrogen metabolism are contained in it. Starch is the major reservoir of energy and carbon in photosynthetic organisms. The starch biosynthetic pathway constituted by the enzymes glucose-6-phosphate isomerase PGI (Cre03.g175400), phosphoglucomutase PGM (g2899), ADP glucose pyrophosphorylase APL (Cre16.g683450), starch synthase SSS (Cre04.g215150) and 1,4- -starch branching enzyme SBE3 (Cre10.g444700) is entirely contained in this cluster. In Chlamydomonas, starch is degraded to hexoses during the dark period. The derived hexoses are then used in the sucrose synthesis pathway. Key enzymes in this pathway such as glyceraldehyde 3-phosphate dehydrogenase GAP1 (Cre12.g485150) and sucrose phosphate phosphatase SPP (Cre06.g283400) are members of this cluster. The oxidative branch of the pentose phosphate pathway produces NADPH in the reactions catalized by glucose-6-phosphate dehydrogenase GLD2 (Cre08.g378150) and 6-phosphogluconate dehydrogenase GND1 (Cre12.g526800), enzymes coded by genes included in this cluster. NADPH is an important source of the reducing power required by many enzymes in central metabolic pathways. The anapleurotic pathway that fixes CO2 into oxaloacetate through the enzymes carbonic anhydrase CAH8 (Cre09.g405750) and phosphoenolpyruvate carboxylases PPC (g16646 and g11831) is also part of this cluster (Additional file 4: Figure S3). This pathway replenishes depleted Tricaboxylic Acid (TCA) cycle compounds that have been used for nitrogen assimilation or other tasks [41]. Inorganic and organic nitrogen assimilation pathways are included in this cluster (Additional file 4: Figure S3), including the nitrate transporter NRT2.3 (Cre09.g396000), nitrite transporter NAR1.4 (Cre07.g335600), nitrate reductase NIT1 (Cre09.g410950) and nitrite reductase NII1 (Cre09.g410750) yielding ammonia as a final product. In fact, these reductases need a molybdenum cofactor and the biosynthetic pathway for molybdenum cofactor constituted by the enzymes molybdopterin synthase adenylyltransferase CNX (g10007), cyclic pyranopterin monophosphate synthase CNX2 (Cre13.g602900), molybdopterin synthase sulfurylase MoaE (Cre07.g322250) and molybdopterin molybdotransferase MoeA (Cre10.g451400) is entirely included in this cluster (Additional file 4: Figure S3). Therefore, not only the enzymes, but also the pathways leading to the synthesis of the cofactors needed for nitrate assimilation are tightly co-expressed in ChlamyNET.
Cluster 1, orange - intracellular transport, regulation of GTPase activity, autophagy and proteolysis
The orange cluster consists of 824 genes and is located in the periphery of ChlamyNET (Fig. 4). This cluster presents a high silhouette value of 0.38 (Fig. 3b). The GO term enrichment analysis indicates that genes within this cluster are significantly involved in processes related to intracellular transport to the endoplasmic reticulum and Golgi apparatus such as vesicle-mediated transport (GO:0016192). For instance, we can find genes coding for the endosomal R-SNARE protein Yky6 (Cre17.g728150) and gamma1-Adaptin AP1G1 (Cre16.g676650). Genes in this cluster are also significantly related to the regulation of GTPase activity (GO:0043087) such as those coding for the rab GTPase activator protein CGL44 (Cre12.g532600) and Rab/TBC domain protein (Cre07.g315350). Autophagy (GO:0006914) and proteolyis (GO:0006508) are significant GO terms in this cluster with genes coding for the Autophagy related gene 9 ATG9 (Cre09.g391500) and ubiquitin-conjugating enzyme E2 UBC9 (Cre16.g693700). Therefore, the formation of this gene cluster suggests a connection between Rab GTPase activity and autophagy. Moreover, the positive regulation of Rab GTPase activity over autophagy has been shown in Arabidopis [42].
The metabolic analysis of this cluster suggests that it is involved in triacylglycerol (TAG) biosynthesis, the major lipid reserve in plants. Many unicellular microalgae accumulate large amounts of TAG under unfavorable conditions, such as the ones leading to autophagy [43]. TAG is produced from diacylglycerol (DAG) and different acyl donors. On the one hand, DAG can be synthesized from a 1,2-diacyl-sn-glycerol 3-phosphate and the enzyme phosphatidate phosphatase PAH (Cre12.g506600), a member of this cluster. On the other hand, phospholipids (major constituents of cellular membranes) are one of the possible donors for DAG to produce TAG. In this case, the enzyme phospholipid:DAG acyltransferase PDAT (Cre02.g106400) present in this cluster catalizes this reaction (Additional file 5: Figure S4). The 3-phosphoinositide biosynthesis pathway is also included in this cluster. Phosphoinositides are involved in phospholipid biosyntehsis as well as membrane trafficking, biological processes over-represented in this cluster. The key enzymes in this pathway are phosphatidylinositol-3-kinase PI3K (Cre01.g035500), phosphatidylinositol 4-kinase PIK1 (Cre05.g245550), phosphatidylinositol-4-phosphate 5-kinase PIP5K3 (g9964) and inositol 5-phosphatase SAC1 (Cre12.g537500) which are also located in this cluster (Additional file 5: Figure S4). Other important lipid metabolic reactions are the activation and deactivation of lipids achieved by the ligation or removal of acyl-CoA. These reactions are catalized by the enzymes long-chain-fatty-acid-CoA ligase LACS (Cre03.g182050) and acyl-CoA thioesterase ACOT (Cre01.g037350) respectively, both members of this cluster. In these reactions the common acyl carrier Coenzyme A is required, and so, key enzymes in its biosynthesis such as ketopantoate hydroxymethyltransferase PAN2 (Cre12.g508550), phosphopantothenate-cysteine ligase COAB (Cre01.g048050) and phosphopantothenoylcysteine decarboxylase COAC (Cre10.g423450) are also co-expressed in this cluster (Additional file 5: Figure S4).
Cluster 3, red - protein phosphorylation, translation, ribosome biogenesis and macromolecule biosynthetic process
The red cluster expands from the periphery of ChlamyNET to its core (Fig. 4). Somehow this cluster serves as an interface between the blue cluster (hub genes involved in protein phosphorylation, carbohydrate metabolic process and transmembrane transport) and the brown cluster (cell cycle processes). This cluster is the largest one including 1723 genes and presenting a moderate silhouette value of 0.28 (Fig. 3b). According to the GO term enrichment analysis, genes in this cluster are significantly involved in diverse biological processes. The three most significant processes are protein phosphorylation (GO:0006468) including genes such as the mitogen activated protein kinase PTK24 (Cre02.g145500) and aurora-like kinase ALK3 (Cre12.g498650); translation (GO:0006412) and ribosome biogenesis (GO:0042254) with genes coding for ribosomal proteins L13 RPL13a (Cre12.g532550) and S6e RPS6 (Cre09.g400650). The next significant biological process is macromolecule biosynthetic process (GO:0009059) with genes such as the glutamine synthetase GLN4 (Cre03.g207250).
The analysis of the metabolic pathways included in this cluster identified the synthesis of triacylglycerol using exclusively as acyl donors galactolipids produced by glycolipid desaturation. The diacylglycerol O-acyltransferase DGAT1 (g9572) and monogalactosyldiacylglycerol synthase FAD6 (Cre13.g590500) are thus included in this cluster (Additional file 3: Figure S2). Although no other metabolic pathway is fully represented in cluster 3, isolated key enzymes for carbon xation, hydrogen production and oxidation such as rubisco RBCS2 (Cre02.g120150) and iron hydrogenase HYDA2 (Cre09.g396600) are co-expressed within this cluster. In fact, our study suggests that this cluster is involved in signalling and transcription control rather than in metabolism. Several serine/threonine protein kinases are included in this cluster. The genes CrAUR1 (Cre16.g669800) and ALK3 (Cre12.g498650) exhibit a high sequence similarity with the and Aurora kinases in Arabidopsis AUR1 (At4g32830) and AUR3 (At2G45490) respectively. It has been described that the diversification of plants and aurora kinases predates the origin of land plants [44]. Here we show that this diversification may be already present in Chlamydomonas. These kinases have been shown to play a key role in cell cycle related signal transduction pathways in Arabidopsis. Several other genes similar to cyclin-dependent protein kinases are located in this cluster such as CDKI1 (Cre12.g494500) and CrMAPKKK (Cre10.g461150). Cyclin-dependent protein kinases play crucial roles in the progression of the cell cycle in eukaryotes. CDKI1 (Cre12.g494500) exhibits a high sequence similarity with the Arabidopsis gene CAK4 (At1g66750), which is known to be involved in the activation of cell proliferation [45]. While CrMAPKKK (Cre10.g461150) is highly similar to the Arabidopis gene MEKK1 (At4g08500). Additionally, other genes in this cluster such as g16721, present a high similarity with the Arabidopsis Mitogen Activated Protein (MAP) kinase MAPKKK6 (At3g07980). The co-expression of these genes suggests that MAP kinase cascades are regulated not only at the posttranslational level but also at the transcriptional level in Chlamydomonas.
As it will be described in detail in the next section, this cluster is also significantly enriched in transcription factors. Several GATA transcription factors such as g7394, Cre05.g242600 and Cre08.g378800; bZIP transcriptions factors like Cre10.g438850 and Cre12.g489000 and the single DOF and CO-like transcription factors in Chlamydomonas CrDOF (Cre12.g521150) [46] and CrCO (g6302) [2] are members of this cluster.
A detailed description of the rest of clusters and their functional annotation is available for further exploration at the web page http://viridiplantae.ibvf.csic.es/ChlamyNet/. These results aim at providing researchers in the functional annotation of the Chlamydomonas transcriptome with a solid ground to design specific and targeted experimental studies to validate or refute the predictions produced in this clustering analysis.
Transcription factors and transcriptional regulators in ChlamyNET
In the previous section we performed a functional annotation of the different gene clusters identified according to GO term enrichment and metabolic pathways analysis. In this section, we further investigate the regulatory aspects of the Chlamydomonas transcriptome using ChlamyNET.
One of the most important processes involved in cellular response to internal and external stimuli is transcription or gene expression. This is a highly regulated process carried out by transcription factors and transcriptional regulators. Transcription factors (TFs) bind to specific cis-elements in the promoters of genes to activate or repress their transcription directly. On the other hand, transcriptional regulators (TRs) are involved in gene expression control but do not bind directly to gene promoters. Transcriptional regulators modulate gene expression by interacting with transcription factors, remodeling chromatin or other indirect mechanisms [47]. A genome-wide identification and classification of transcription factors and transcriptional regulators in Chlamydomonas has been previously performed [48]. This classification is available on the web portals and databases PlantTFDB (http://planttfdb.cbi.pku.edu.cn/index.php?sp=Cre) [49] and PlnTFDB (http://plntfdb.bio.uni-potsdam.de/v3.0/index.php?sp id = CRE4) [50]. Using this classification, we identified 118 TFs and 109 TRs in ChlamyNET, which constitute 2.48 % of the total number of genes in the network (Fig. 5). These TFs and TRs are classified, according to their protein domains, into 28 and 17 gene families respectively. The TFs seem not to be randomly distributed over the clusters of ChlamyNET (Fig. 5a), whereas the TRs distribution seems to be more uniformly distributed over the net (Fig. 5b). In order to asses the statistical significance of the distribution of TFs and TRs over the clusters in ChlamyNET, we performed an enrichment analysis based on Fisher's exact test. Indeed, no cluster was significantly enriched in TRs whereas the blue and red clusters were significantly enriched in TFs with p-values of 2.62°10−3 and 2.37°10−3 respectively. This complements the evidence produced by our analysis of the location of authoritative hub genes in ChlamyNET and about the key role played by the blue and red clusters in the regulation of the Chlamydomonas transcriptome under the conditions analyzed in this study.
Fig. 5.
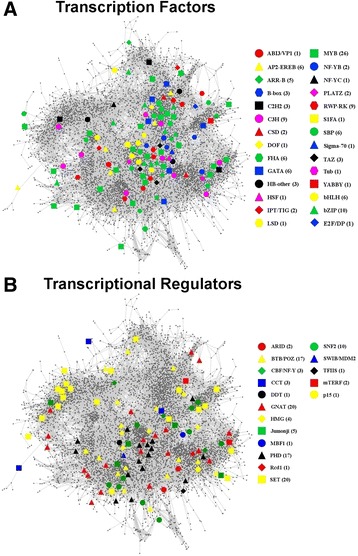
Location of Transcription Factors and Transcriptional Regulators in ChlamyNET. a Transcription Factors in ChlamyNET. We identified 118 different TFs classified into 28 different families represented using symbols with different colours and shapes. The distribution of the TFs over the clusters of ChlamyNET is not uniform. Clusters 9 (blue) and 3 (red) are enriched in TFs according to p-values of 2.62°10 −3 and 2.37°10 −3 obtained using Fisher's exact test. b Transcription Regulators in ChlamyNET. We identified 109 different TRs classified into 17 different families represented using symbols with different colours and shapes. The distribution of the TRs over ChlamyNET is uniform. Our analysis based on the Fisher's exact test did not identify any cluster significantly enriched in Trs
In our analysis, instead of classifying genes according to their sequence as previously described [48], we have studied their co-expression patterns in order to determine groups of TFs and TRs that could exert their function over target genes in a coordinated way. Similar to the previous section we applied the two most widely used clustering algorithms, hierachical clustering and partition around medoids, taking as distance among genes the correlation between their expression profiles. According to the silhouette criterion, the best description of the co-expression patterns among TFs and TRs is obtained with thirteen different groups identified with the partition around medoids algorithm (Fig. 6).
Fig. 6.
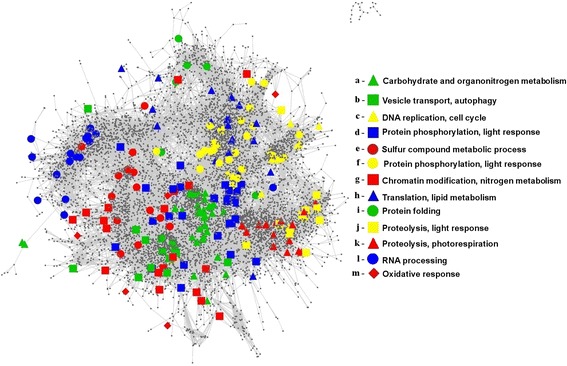
Transcription Factors and Transcriptional Regulators Clustering and Functional Annotation. According to the similarity between their gene expression profiles the TFs and TRs in ChlamyNET can be classified into 13 different groups identified by different symbols, colours and letters. The analysis of the GO terms overrepresented in the neighbourhood of each group suggest the biological processes that they might be regulating
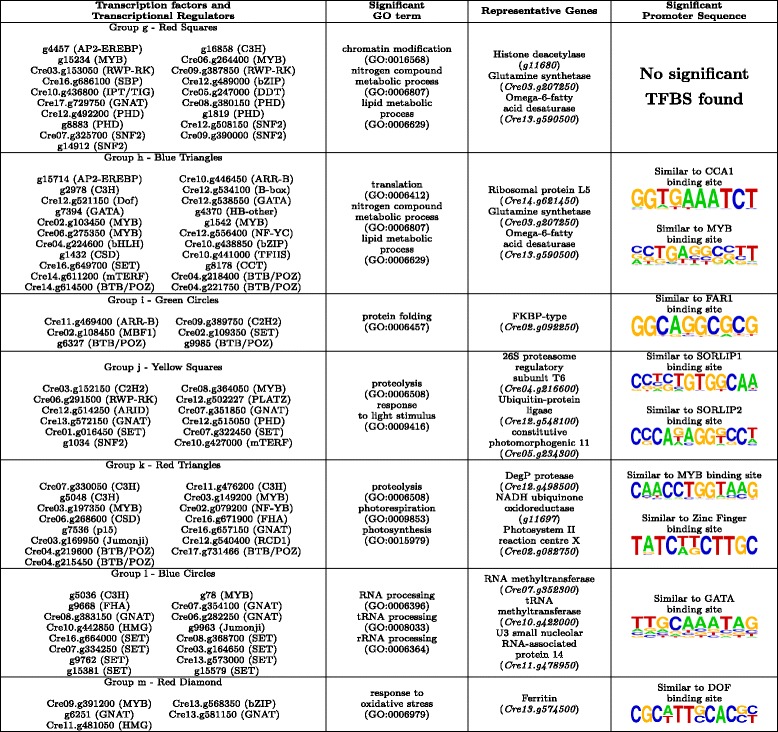
In order to determine the biological processes regulated by each group we applied GO term enrichment analysis over the genes directly linked to the corresponding TFs and TRs. Additionally, we performed a transcriptional factor binding site (TFBS) enrichment analysis over the promoters of these genes as described in the Methods section. In Tables 3 and 4 we show a summary of the results about the GO terms and TFBS significantly enriched in the genes directly linked to the TFs and TRs in each group.
Table 3.
Biological processes and transcription binding sites significantly over-represented in the neighbourhood of the TFs and TRs groups in ChlamyNET
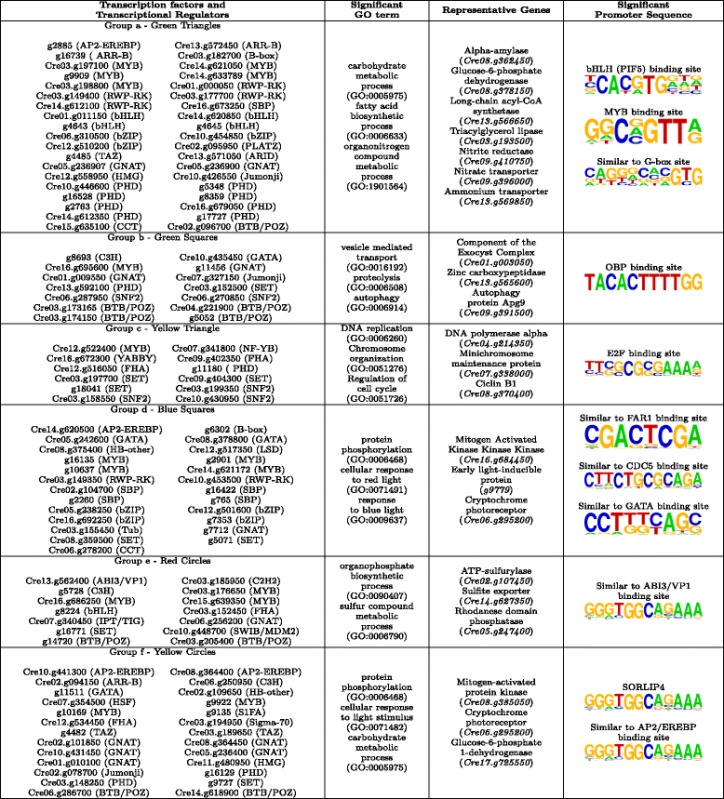
Table 4.
Biological processes and transcription binding sites signifcantly over-represented in the neighbourhood of the TFs and TRs groups in ChlamyNET
Herein we present in detail the results of our analysis over three groups of TFs and TRs of special interest. We discuss the conservation of their function and binding sites when compared to their putative orthologs in higher plants. The results for the remaining groups of TFs and TRs identified in our analysis are available at the web page http://viridiplantae.ibvf.csic.es/ChlamyNet/.
Core metabolic regulation, group a
This constitutes a numerous group of TFs and TRs including 38 members. They are identified in Fig. 6 using green triangles. The TFs and TRs in this group are included in the cluster 9 (blue) at the center of the network where most authoritative hub genes and carbon/nitrogen core metabolic pathways are located. These TFs and TRs seem to be of key importance in the regulation of the Chlamydomonas transcriptome under the conditions of our study since they are co-expressed on average with 87.97 other genes. Some highly authoritative hub genes in ChlamyNET are members of this group such as the B-box TF CrBbox1 (Cre03.g182700), the bHLH TFs g4643 and Cre01.g011150, the SBP TF Cre16.g673250, the RWP-RK TF NIT2 (Cre03.g177700) and the MYB TF Cre03.g197100. These TFs present a normalized authoratitave hub score higher than 0.8. GO term enrichment analysis over the genes directly linked to the TFs and TRs in this group suggests that they are mainly involved in core metabolism regulation and light response. Several GO terms related with metabolic processes are significantly enriched such as carbohydrate metabolic process, fatty acid biosynthetic process and nitrogen compound metabolic process. Representative genes in this group are the alpha-amylase AMA2 (Cre08.g362450), the long-chain acyl-CoA synthetase LACS2 (Cre13.g566650) and the nitrite reductase NII1 (Cre09.g410750), respectively.
Four bHLH transcription factors, Cre01.g011150, Cre14.g620850, g4643 and g4645, out of the 12 recognized members of this family in Chlamydomonas, are members of this group. Only bHLH Cre14.g620850 has similarity with genes present in higher plants. Specifically, its putative Arabidopsis ortholog is PAR1 (At1g69010) that has been shown to be involved in light response [51]. The rest show similarity with other bHLH genes present only in chlorophyceae. A bHLH binding site was found to be significantly present over the genes co-expressed with the TFs and TRs of this group (Table 3). This suggests that the binding site of bHLH TFs is conserved accross the green lineage. Several genes involved in carbohydrate and nitrogen metabolism contain this binding site in their promoters, for instance the glucose-6-phosphate dehydrogenase GLD2 (Cre08.g378150) and the ammonium transporter AMT4 (Cre13.g569850).
Three bZIP TFs out of the 19 identified in the Chlamydomonas genome, Cre10.g454850, Cre12.g510200 and Cre06.g310500, are members of this group. Genes CrHY5 (Cre12.g510200) and CrHYH (Cre06.g310500) present a high similarity with the Arabidopsis genes HY5 (At5g11260) and HYH (At3g17609) respectively. These TFs are known to bind to G-box sequences to regulate light response and metabolism in Arabidopsis [52, 53]. GO term and TFBS enrichment analysis suggest that this mechanism is already present in Chlamydomonas, since a sequence highly similar to the G-box has been found to be significantly present in the genes co-expressed with these two Chlamydomonas genes (Table 3).
Cre13.g572450 and g16739 that code for two ARR-B TFs and CrBbox1 (Cre03.g182700), that codes for a B-box TF, are present in this group. These genes exhibit high similarities with the Arabidopsis genes RR14 (At2g01760), TOC1 (At5g61380) and COL1 (At5g15850), respectively. They have in common a CCT domain at the carboxyl end that directly binds to DNA [54] that was found to be present in the CrCO (g6302) gene [2]. These genes are known to be involved in light response and circadian rythms in Arabidopsis [55, 56]. These functions seem to have been established already in Chlamydomonas constituting a link between circadian rythms and metabolism.
Five MYB TFs are present in this group. Some of them such as Cre14.g633789 and Cre03.g198800 are putative orthologs of the Arabidopsis genes At3g27785 and At5g61620 that have been associated with metabolic regulation [57]. MYB TF factor binding sites have been found significantly enriched in the promoters of genes co-expressed with this group of TFs and TRs. Such as, the triacylglycerol lipase CrTLL1 (Cre03.g193500) and starch phosphorylase CrPHS1 (Cre07.g336950) that present sequences highly similar to MYB binding sites in their promoters (Table 3).
Finally, several genes coding for TFs from the RWP-RK family are members of this group. One of these TFs, NIT2 (Cre03.g177700), has already been shown to be involved in nitrogen and carbohydrate metabolism regulation [58, 59], whereas the other remain to be studied. Promisingly, the RWP-RK TFs RWP14 (Cre01.g000050), RWP11 (Cre03.g149400) and RWP3 (Cre14.g612100) located in this group are putative orthologs of the Arabdidopsis genes RKD5 (At4g35590) and RKD3 (At5g66990) that have been shown to be involved in nitrogen and light response [60, 61].
Not surprisingly, TFs in this group seem to constitute an intrincate gene regulatory system with mutual regulations among them. For example, bHLH binding sites can be identified in the promoters of the B-box TF CrBbox1 (Cre03.g182700), the bZIP TF CrHY5 (Cre12.g510200), the bHLH TF Cre01.g011150 and the MYB TFs Cre03.g198800 and Cre14.g621050. In turn, G-boxes have been found in the promoters of the bHLH genes Cre01.g011150 and Cre14.g620850 and the bZIP gene Cre10.g454850. Additionally, these TFs seem to exert their regulation in a coordinated manner over the same set of genes since both bHLH and MYB binding sites have been identified in the promoters of genes such as the nitrate transporter NRT2.3 (Cre09.g396000) and the nitrate reductase NIT1 (Cre09.g410950). Such complex interactions are also common in Arabidopsis.
Autophagy regulation, group b
The TFs and TRs in this group are located in the cluster 1 (orange) identified with green squares in Fig. 6. A GO term analysis of the genes directly linked to them reveals a potential regulation over processes involved in vesicle mediated transport, catabolic process, proteolysis and autophagy. In this group we can find the C3H zinc finger TF g8693 presenting a high sequence similarity with the INOSITOL-REQUIRING ENZYME-1b gene (At5g24360) from Arabidopsis. This gene is involved in the regulation of the degradation of the endoplasmic reticulum by autophagy [62]. Directly linked to this gene we can find genes involved in autophagy such as autophagy 9 ATG9 (Cre09.g391500) and proteolysis such as signal peptide peptidase-like 2 (g18126). The GATA transcription factor Cre10.g435450 is also a member of this group and its putative ortholog in Arabidopsis, BME3 (At3g54810), has been shown to be involved in response to salt stress [63]. The MYB transcription factor Cre16.g695600 is also a member of this group whose Arabidopsis putative ortholog At5g06110, is a heat shock protein involved in stress response [64]. Two genes from the chromatin remodeling family SNF2, Cre06.g287950 and Cre06.g270850, are putative orthologs of ATRX (At1g08600) and CHR8 (At2g18760), involved in DNA damage response and recombination [65]. The induction of autophagy as a response to diverse stresses has been shown in Chlamydomonas [66]. Cre03.g173165, Cre03.g174150 and g5052 are transcriptional regulators from the BTB/POZ family that are putative orthologs of ARIA (At5g19330) involved in cellular macromolecule catabolic process [67].
In fact, the OBP binding site was found to be significantly present in the promoters of the genes directly linked to the TFs and TRs in this group (Table 3). This binding site has been shown to be present in promoters of genes induced by oxidative stress in Arabidopsis [68]. This is in agreement with the reported autophagy induction by oxidative stress in Chlamydomonas [66]. Genes related to autophagy such as ATG8 (Cre16.g689650) and ATG9 (Cre09.g391500) present the OBP binding site in their promoters. Genes involved in vesicle traficking such as Component of the Exocyst Complex SEC8 (Cre01.g003050) and Subunit f the ESCRT-I complex VPS28 (Cre16.g678100) also present the OBP binding site in their promoters (Table 3).
Cell cycle regulation, group c
The TFs and TRs of this group are included in the cluster 2 (brown) identified in the previous section as involved in DNA replication, chromosome organization and regulation of cell cycle. These TFs and TRs are highlighted using yellow triangles in Fig. 6. A GO term enrichment analysis over the genes directly linked to these TFs and TRs confirmed their potential regulation over these processes. In this group we can find a MYB3R TF Cre12.g522400 whose putative orthologs, based on their sequence similarity, are At5g11510 and At4g32730 in Arabidopsis and NtmybA1 and NtmybA2 in Nicotiana tabacum. These genes are involved in the G2/M transition during the cell cycle [69–71]. The single member of the YABBY family in Chlamydomonas that presents two high mobility group boxes, Cre16.g672300, belongs to this group. Its putative ortholog gene At4g11080 in Arabidopsis interacts with mitotic and meiotic chromosomes [72]. Another gene, ORC1 (g11180) belonging to the PHD TF family is also a member of this group. Its putative Arabidopsis ortholog At4g14700 (Origin recognition complex) has been shown to be in the core cell cycle machinery involved in the G1/S transition [73, 74]. Several TRs potentially involved in chromatin remodeling are present in this group such as Cre03.g197700 that code for SET domain containing protein that exhibits a high sequence similarity with At1g05830 a trithorax protein in Arabidopsis [75]. The rest of TFs in this group, Cre09.g402350 and Cre12.g516050, are putative orthologs of Arabidopsis genes that have been shown to be co-transcribed with other core cell cycle regulators and TFs in Arabidopsis [76].
The E2F motif [77] was found to be the only known motif significantly enriched in the promoters of the genes directly linked to the TFs and TRs in this group (Table 3). The potential orthologs of the genes that contain in their promoters the E2F motif sequence are involved in the G1/S transition such as subunits of the origin of replication complex ORC1 (g11180) and ORC4 (Cre17.g726500), pre-initiation complex subunit CDC6 (Cre06.g292850), DNA replication initiation factor CDT1 (Cre03.g163300), minichromosome maintenance protein MCM2 (Cre07.g338000) and DNA polymerase alpha POLA1 (Cre04.g214350) (Table 3). The presence of the E2F motif in genes regulating the S phase has been shown previously in Arabidopsis [78] and Nicotiana [77]. The gene Cre07.g323000, putative ortholog of the Arabidopsis E2F transcription factor, is not included in this group of TFs and TRs. Nevertheless, it is located in its vecinity, suggesting that it may function as an interface between regulation of cell cycle and other processes as it is the case for its Arabidopsis ortholog [74]. The two most significant de-novo motifs found in our study presents a high similarity with the octamer and hexamer motifs. The combination of these two motifs has been shown to confer S phase-specific transcriptional activation in plants [79]. Genes containing these motifs include B-type cyclin CYCB1 (Cre08.g370400) and cell division cycle protein CDC45 (Cre06.g270250). This suggests a remarkable conservation of cell cycle regulation in the plant kingdom not only limited to the TFs, TRs and their targets involved in this process but also in the cis-regulatory elements, TFBS, present in their promoters.
Light-regulated transcription factors and transcriptional regulators in ChlamyNET, a tutorial for ChlamyNET usage
In order to ensure the reproducibility of the results presented in this work and to facilitate further and independent studies over the Chlamydomonas transcriptome we have developed a web-based software tool also called ChlamyNET. This tool is based on WiGis, a platform for the visualization of large-scale, highly interactive graphs in a user's web browser [80]. The software tool ChlamyNET is available from the web page http://viridiplantae.ibvf.csic.es/ChlamyNet/. In this section we discuss a case study concerning the Chlamydomonas potentially light-regulated TFs and TRs that can be used as a tutorial for the use of ChlamyNET.
Light constitutes the most important source of energy for green algae. Therefore, light is a key environmental signal that affects profoundly Chlamydomonas growth and physiology. The study of the light-regulated TFs and TRs co-expression patterns, the biological processes controlled by them and the TFBS where they potentially exert their function can contribute to a better understanding of the response to this key environmental signal in Chlamydomonas. Previously, potential orthologs of the light-regulated TFs and TRs in Arabidopsis [81] have been identified in the Chlamydomonas genome [48]. Most of these TFs and TRs can be found in ChlamyNET. Suprisingly, these genes are not randomly distributed over ChlamyNET (Fig. 7). They are mainly located in clusters 9 (blue), 3 (red) and 7 (green) suggesting that they are specifically involved in carbon/nitrogen metabolism, signalling by phosphorylation and protein degradation. In order to identify genes in the network, the Search panel on the left of ChlamyNET can be used. Once the genes are found, they can be selected and information related to their name, putative Arabidopsis ortholog, topological indexes, protein domains (Pfam annotation) and neighbours is depicted on the Selected Gene Details panel. This information for light-regulated TFs and TRs in ChlamyNET is collected into Table 5.
Fig. 7.
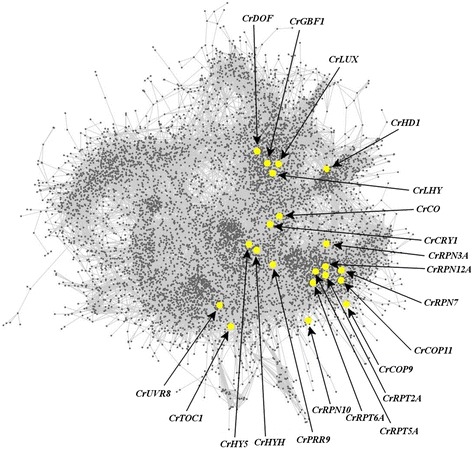
Potentially Light-regulated Transcription Factors and Transcriptional Regulators in ChlamyNET. Twenty-one TFs and TRs exhibiting a high similarity with light regulated TFs and TRs in Arabidopsis were identified in ChlamyNET. These genes are not uniformly distributed over ChlamyNET. Clusters 9 (blue), 3 (red) and 7 (green) were significantly enriched in these potentially ligh-regulated TFs and TRs, so that they are expected to be involved mainly in carbon/nitrogen metabolism, signalling by phosphorylation and protein degradation. The central location of several light-regulated TFs and TRs such as CrHY5 (Cre12.510200) and CrCRY1 (Cre06.g295200) suggests that they are highly authoritative hub genes. Indeed CrHY5 and CrCRY1 have 133 and 57 neighbouring genes respectively
Table 5.
Potentially Light Regulated TFs and TRs in ChlamyNET. Their potential Arabidopsis ortholog and topological indexes are indicated as well
| Chlamydomonas gene | Putative Arabidopsis Ortholog | Number of neighbours | Normalized hub score | Clustering coefficient |
|---|---|---|---|---|
| Cre06.g295200 CPH1 / CrCRYl | At4g08920 CRYPTOCHROME 1 | 57 | 8.12 x 10-5 | 0.39 |
| Cre01.g043150 CrGBF1 | At4g36730 G-BOX BINDING FACTOR 1 | 182 | 7.36 x 10-7 | 0.31 |
| Cre12.g510200 CrHY5 |
At5g11260 ELONGATED HYPOCOTYL 5 | 133 | 0.32 | 0.47 |
| Cre06.g310500 CrHYH |
At3g17609 HY5-HOMOLOG | 39 | 0.12 | 0.71 |
| Cre12.g521150 CrDOF |
At5g39660 CYCLING DOF FACTOR 2 | 27 | 3.79 x 10-8 | 0.34 |
| g6302 CrCO |
At5g15840 CONSTANS | 58 | 2.29 x 10-4 | 0.40 |
| Cre02.g094150 CrPRR9 |
At2g46790 PSEUDO-RESPONSE REGULATOR 9 | 1 | 7.72 x 10-7 | 0 |
| Cre06.g275350 CrLHY |
At1g01060 LATE ELONGATED HYPOCOTYL | 78 | 5.86 x 10-7 | 0.38 |
| g1542 CrLUX |
At3g46640 LUX | 38 | 2.23 x 10-7 | 0.40 |
| Cre06.g277350 CrHD1 |
At4g38130 HISTONE DEACETYLASE 1 | 1 | 5.12 x 10-18 | 0 |
| g16739 CrTOC1 |
At5g61380 TIMING OF CAB EXPRESSION 1 | 3 | 1.49 x 10-5 | 0.33 |
| Cre14.g617350 CrUVR8 |
At5g63860 UVB-RESISTANCE 8 | 1 | 4.09 x 10-14 | 0 |
| Cre05.g234300 CrCOP11 |
At3g61140 CONSTITUTIVE PHOTOMORPHOGENIC 11 | 36 | 7.81 x 10-14 | 0.37 |
| Cre14.g608850 CrCOP9 |
At4g14110 CONSTITUTIVE PHOTOMORPHOGENIC 9 | 11 | 1.26 x 10-12 | 0.27 |
| Cre17.g708300 CrRPN12A |
At1g64520 REGULATORY PARTICLE NON-ATPASE 12A | 57 | 7.44 x 10-14 | 0.39 |
| Cre10.g439150 CrRPT5A |
At3g05530 REGULATORY PARTICLE TRIPLE-A ATPASE 5A | 51 | 7.49 x 10-14 | 0.45 |
| Cre13.g581450 CrRPN7 |
At4g24820 REGULATORY PARTICLE NON-ATPASE 7 | 59 | 1.52 x 10-13 | 0.28 |
| Cre06.g275650 CrRPN3A |
At1g20200 REGULATORY PARTICLE NON-ATPASE 3A | 14 | 2.80 x 10-15 | 0.41 |
| Cre16.g663500 CrRPN10 |
At4g38630 REGULATORY PARTICLE NON-ATPASE 10 | 4 | 3.48 x 10-14 | 0 |
| Cre04.g216600 CrRPT6A |
At5g19990 REGULATORY PARTICLE TRIPLE-A ATPASE 6A | 19 | 1.74 x 10-12 | 0.25 |
| Cre07.g329700 CrRPT2A |
At4g29040 REGULATORY PARTICLE TRIPLE-A ATPASE 2A | 3 | 3.44 x 10-16 | 0.33 |
According to this information several light-regulated TFs are highly authoritative hub genes in ChlamyNET such as CrGBF1 (Cre01.043150) and CrHY5 (Cre12.510200) that are co-expressed with more than 130 genes. These genes are involved in photomorphogenesis in Arabidopsis, yet their function in Chlamydomonas is unknown. Others light-regulated TFs and TRs that constitute hub genes that are co-expressed with more than 50 genes, are CrCRY1 (Cre06.g295200), CrCO (g6302), CrLHY (Cre06.g275350) and the different subunits of the 26S proteasome CrRPN12A (Cre17.g708300), CrRPT5A (Cre10.g439150) and CrRPN7 (Cre13.g581450). CrCRY1, also known as CPH1, codes for a putative ortholog of CRY1 in Arabidopsis and it is a well known photoreceptor that responds to light stimulus [82]. On the other hand, CrCO expression is affected by photoperiod and regulates carbon metabolism and cell cycle progression [2]. Silencing and over-expression of these genes have been shown to massively disrupt Chlamydomonas cell growth and proliferation supporting their function as hubs in the network [2]. The potential role of CrLHY in circadian rythms and the proteolytic function of CrRPN12A, CrRPT5A and CrRPN7 are yet to be tested experimentally. The potentially light-regulated genes CrHYH (Cre06.g310500), CrDOF (Cre12.g521150), CrLUX (g1542) and CrCOP11 (Cre05.g234300) whose putative Arabidopsis orthologs are involved in photomorphogenesis, photoperiod response, circardian rythms and protein degradation respectively are co-expressed with around 30 other genes. Recently, CrDOF expression has been shown to be influenced by circadian rythms and the photoperiod whereas it directly regulates the expression of CrCO [46]. The rest of potentially light-regulated TFs and TRs identified in ChlamyNET are co-expressed with fewer than 20 other genes and are not considered hubs in the network. Most of these genes exhibit a high clustering coeficient in ChlamyNET suggesting a high level of coordination among their co-expressed genes.
In order to study co-expression patterns among a set of selected genes we can generate heatmaps using ChlamyNET. In this type of graphs we represent the correlation among expression profiles of a selected set of genes. Heatmaps can be generated in ChlamyNET using the Analysis section located in the Search panel once a set of genes have been selected. Additionally, genes at distance one, two or three from the selected genes can be included in the heatmap. As an example, a heatmap depicting the co-expression patterns among the potentially light-regulated TFs and TRs previously selected is presented in Fig. 8. We can observe three different groups. Genes in the same group exhibit a high positive correlation (red/yellow colours) whereas genes from different groups present a very low negative correlation (blue/purple colours). In group 1 we can distinguish two subgroups. On the one hand, in subgroup 1a it can be observed that CrCRY1, CrPRR9, CrCO, CrHY5 and CrHYH are highly co-expressed. This could indicate co-regulation of these genes by the same factors or regulation among themselves. On the other hand, in subgroup 1b we find that CrLUX, CrLHY, CrGBF1 and CrDOF have a very high co-expression value. These two subgroups are also co-expressed and hence form a single group. Group 2 is composed of three genes, CrUVR8, CrTOC1 and CrHD1. This group of genes presents a negative correlation with genes in subgroup 1b which makes them constitute a separate group. For instance, CrLHY and CrTOC1 are negatively co-expressed as expected if they are true orthologs of the Arabidopsis circadian genes LHY/CCA1 and TOC1. These genes form a negative feedback loop that constitutes the core of the circadian clock in Arabdiopsis. Group 3 exhibits a very low negative correlation with group 1 and a moderate negative correlation with group 2. The genes in this group are different members of the 26S proteasome and signalosome subunits such as CrRPN7 and CrCOP9. Their putative Arabidopsis orthologs have been described to degrade proteins involved in light response [61]. This suggests an antagonist expression pattern between light-regulated TFs and different subunits of the 26S proteasome and signalosome that is already established in Chlamydomonas. This would need further research and experimental validation, but can be an important clue for a preliminary investigation.
Fig. 8.
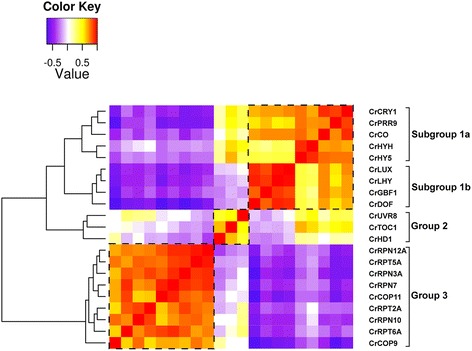
Heatmap Representing the Co-expression Patterns among the Potentially Light-regulated TFs and TRs in ChlamyNET. High positive correlation between the corresponding gene profiles is represented by red/yellow colours, low negative correlation is represented by blue/purple colours. Three different groups are apparent. The first group can be divided into two subgroups. We can observe negative correlations between genes in the subgroup 1b and genes in the second group such as between CrLHY and CrTOC1 which indicates that these two genes may be true orthologs of the circadian clock Arabidopsis genes LHY/CCA1 and TOC1. Very low negative correlations are observed between genes in Group 1 and genes in Group 3. Genes coding for different 26S proteasome and signalosome subunits such as CrRPN7 and CrCOP9 can be found in. Group 3. Their putative Arabidopsis orthologs have been described to degrade proteins involved in light response that exhibit a high sequence similarity with those coded by genes in Group 1
The biological processes potentially controlled by the light-regulated TFs and TRs in ChlamyNET can be deduced by applying GO terms enrichment over their co-expressed genes. This can be performed by using the Analysis section located in the Search panel once the neighbouring genes have been selected. As described in the Methods section we can combine the significative GO term identified, based on orthology, with those determined based on conserved protein domains (Additional file 6: Table S2). According to this methodology, the potentially light-regulated TFs and TRs are co-expressed with genes involved in ion transport, for example the nitrate transporter NRT2.2 (Cre09.g410800) and Mo-molybdopterin cofactor biosynthesis such as MoeA (Cre10.g451400), which produces essential cofactors for the nitrate reductase, a key enzyme in the nitrate metabolism. Additionally, carbohydrate metabolism appears as a significative GO term, including genes involved in starch and glucose degradation such as the starch phosphorylase CrPHS2 (Cre12.g552200), the alpha-amylase AMY (Cre08.g384750) and the glucose-6-phosphate dehydrogenase GLD2 (Cre08.g378150). Finally, protein phosphorylation is another relevant significative GO term with genes potentially involved in cell cycle control such as the mitogen-activated protein kinase kinase kinases CrMKKK1 (Cre07.g347000) and CrMKKK2 (Cre02.g108650). Therefore, this analysis suggests a potential regulation of carbon/nitrogen metabolism and cell cycle through protein phosphorylation by these potentially light-regulated TFs and TRs that needs to be experimentally validated.
Finally, selecting the switch Promoter Sequence Enrichment in the Search panel, a transcriptional factor binding site (TFBS) enrichment analysis over the promoters of selected genes can be performed . In this case, several significative light-regulated TFBS in Arabidopsis such as SORLIP2, SORLIP3 and SORLREP5 [83] were identified. For example, the CrGBF1, CrHYH and CrLUX genes present the sequence SORLIP3 in their promoters. This is in agreement with their high co-expression values (Fig. 8). The presence of these TFBS in the promoters of the genes studied here suggests a high conservation of light regulated TFBS across the green lineage.
A more detailed presentation of this case study is available from the web page of ChlamyNET, http://viridiplantae.ibvf.csic.es/ChlamyNet/.
Experimental validation
ChlamyNET aims at becoming an enabling technology for researchers on the Chlamydomonas transcriptome. For example, ChlamyNET can be used to predict changes in gene expression. When a specific gene is mutated or overexpressed, ChlamyNET predicts that the expression of genes located in the neighbourhood of the mutated or overexpressed gene will be affected, whereas genes in distant regions will not substantially change their expression profile. Specifically, another application of ChlamyNET is predicting targets of specific TFs. TFs and their targets tend to be strongly co-expressed since TFs directly regulate the expression of their target genes. Therefore, the targets of a TF should be contained in its neighbourhood, possibly directly linked to it. Additionally, when the sequences recognised by a TF are well characterized, the identification of these sequences in the promoters of genes co-expressed with it provides more convincing evidence for these genes being direct targets of the corresponding TF. In this way, the analysis of gene neighbourhoods and the significance of TFBS in gene promoters can be studied using ChlamyNET constituting a powerful tool for gene expression analysis.
In this section, we present an experimental validation of the predictive power of ChlamyNET based on one of the most widely used methodologies, cross-validation. In this methodology, once a predictor has been constructed, an independent data set is used to evaluate its performance. As it is common in the study of Chlamydomonas, the data used to construct ChlamyNET was obtained from algae grown on continuous light (LL) conditions without a dark period. In spite of this, probably due to the fact that the different experiments were carried out with different light intensities, we were able to identify patterns of co-expression between TFs and TRs that potentially respond to light stimuli as discussed in the previous section. Additionally, none of the genotypes used in the construction of ChlamyNET consisted of an overexpressor of a gene involved in photoperiod response. Therefore, in order to use a totally independent data set from those used to construct ChlamyNET we generated RNA-seq data from algae grown in long day (LD) conditions (16 hours of light and 8 hours of dark) and in short day (SD) conditions (8 hours of light and 16 hours of dark). Two different genotypes were analyzed. As wild type we used the Chlamydomonas strain CW15 and for a strain affecting the response to photoperiod we chose algae that overespressed the CrDOF gene under a nitrate inducible promoter. As discussed in the previous section the gene CrDOF (Cre12.g521150) is a potentially light-regulated transcription factor that has recently been shown to response to photoperiod and circadian rythms [46]. This last Chlamydomonas strain was called CrDOFin. For more details on algae material, growth conditions and RNA-seq see the Methods section. Using the RNA-seq data analysis protocol described in the Methods section we determined the gene expression level fold-change when comparing CrDOFin to CW15 (Fig. 9). According to the predictions provided by ChlamyNET the 216 genes in the neighbourhood at distance two from the CrDOF gene, yellow dots in Fig. 9a, are expected to increase their expression. Indeed, 69.44 % of these CrDOF neighbouring genes increased their expression level in the CrDOFin strain when compared to the wild type strain CW15 in LD conditions. Actually, the CrDOF neighbouring genes showed an average fold-change increase of 2.7 which is significantly higher than the fold-change in the rest of ChlamyNET with a p-value of 5.63°10−3. Additionally, we identified in ChlamyNET those genes that increased their expression level a fold-change of four in the genotype CrDOFin when compared to the wild type CW15 in LD and SD conditions (Fig. 9b and c). The neighbourhood of the gene CrDOF at distance two was shown to be significantly enriched with a p-value of 0.029 in genes that increase their expression level a fold-change of 4 in LD conditions. This further supports the predictive power of ChlamyNET in LD conditons (Fig. 9b). Nevertheless, the highly activated genes in SD conditions appeared scattered all over ChlamyNET without concentrating in the CrDOF neighbourhood at distance two (Fig. 9c). This shows a limitation in the predictive power of ChlamyNET. It should be recalled that all data collected for the construction of ChlamyNET correspond to LL conditions. The data from LD conditions is somehow similar to these data and therefore, ChlamyNET was a good predictor for this condition. On the other hand, SD conditions, in which the dark period is longer than the light period, represent very different conditions from those used for the data with which ChlamyNET was generated. Therefore, ChlamyNET performs poorly as a predictor for this condition.
Fig. 9.
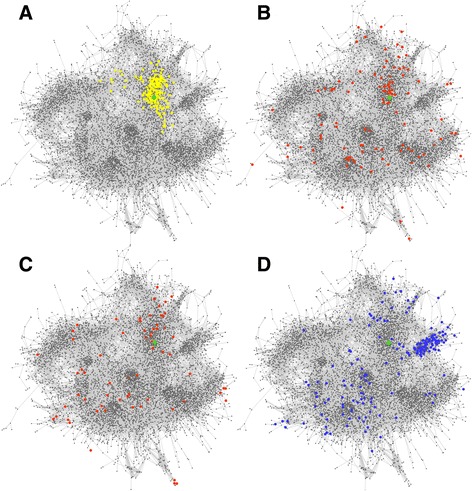
Experimental Cross-validation of the Predictive Power of ChlamyNET using RNA-seq Data from Algae Overexpressing the CrDOF gene. a The CrDOF gene (identified as a green diamond in ChlamyNET) has a neighbourhood at distance two consisting of 216 genes represented in yellow. These genes showed an average fold-change increase of 2.7 which is significantly higher than the fold-change in the rest of ChlamyNET with a p-value of 5.63° 10 −3. b Genes increasing their expression level in LD conditions at least by a four fold-change in the CrDOF genotype when compared to the wild type CW15 are represented in red. Note that the neighbourhood of the CrDOF gene, represented in green, is enriched in these genes according to a p-value of 0.029 obtained using Fisher's exact test. c Genes increasing their expression level in SD conditions at least by a four fold-change in the CrDOF genotype when compared to the wild type CW15 are represented in red. These genes tend to group around the CrDOF gene, represented in green. d Inhibited genes in LD conditions in the CrDOFin genotype when compared to the wild type CW15 with at least by a four fold-change are represented in blue. Note that cluster 2 (brown) involved in DNA replication and cell cycle processes is significantly enriched in these genes
We also identified in ChlamyNET those genes that showed a 4-fold decrease in expression level in CrDOFin compared to the wild type CW15 in LD conditions (Fig. 9d). These genes were not located in the neighbourhood of CrDOF, suggesting that it acts as a direct activator and, possibly, as an indirect repressor in LD conditions. Instead, cluster 2 (brown) was significantly enriched with these highly inhibited genes with a p-value of 2.2°10−16. This provides evidence about CrDOF being involved in cell-cycle regulation, which indeed was experimentally validated [46].
In order to illustrate the effect of the overexpression of CrDOF over its neighbouring genes we chose three genes at distance two from it and represented their expression level in the four conditions, CrDOFin and CW15 grown in LD and SD. We chose three genes that represent the main biological processes affected by the potentially light-regulated TFs and TRs according to the results from the previous section. These genes are a fatty acid desaturase FAD6 (Cre13.g590500) involved in carbon metabolism, a glutamate dehydrogenase GDH2 (Cre05.g232150) involved in nitrogen metabolism and a serine/threonine kinase MAPKKK2 (Cre16.g684450) possibly involved in cell cycle regulation (Fig. 10). As expected, these genes increased their expression levels in the CrDOFin genotype when compared to CW15. Additionally, we chose three genes located in the cluster 4 (purple), far away from CrDOF, to show that the expression level of distant genes tend to remain unchanged. These genes are a glycinamide ribonucleotide synthetase CrGARS (g18106), a phosphoribosylglycinamide formyltransferase PGFT (Cre12.g550700), both involved in purine biosynthesis and a plastid TF PTAC3 (Cre12.g497350) involved in regulation of plastid genes. All these experimental tests validated the predictive power of ChlamyNET.
Fig. 10.
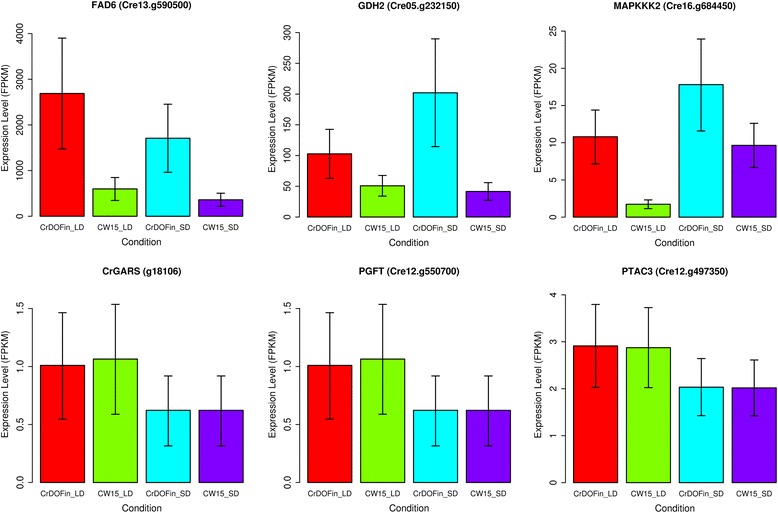
Expression Levels (FPKM) of several CrDOF Neighbouring Genes and Distant Genes in the CrDOFin and CW15 Genotypes Grown in LD and SD Conditions. Three genes in the neighbourhood at distance two from CrDOF were chosen to illustrate the correct prediction provided by ChlamyNET with respect to their increase in expression level in the CrDOFin genotype when compared to CW15. These genes are fatty acid desaturase FAD6 (Cre13.g590500) involved in carbon metabolism, glutamate dehydrogenase GDH2 (Cre05.g232150) involved in nitrogen metabolism and serine/threonine kinase MAPKKK2 (Cre16.g684450) possibly involved in cell cycle regulation. Additionally, we selected three genes from the purple cluster located far away from the CrDOF gene in ChlamyNET to show that distant genes expression is not substantially affected by CrDOF overexpression. These genes are glycinamide ribonucleotide synthetase CrGARS (g18106), phosphoribosylglycinamide formyltransferase PGFT (Cre12.g550700), both involved in purine biosynthesis and plastid transcription factor PTAC3 (Cre12.g497350) involved in regulation of plastid genes
Conclusions
This gene co-expression network and its associated web-based tool ChlamyNET constitute one of the first integrative approaches to the study of the Chlamydomonas transcriptome. They aim at providing researchers with an enabling technology that will allow them to study gene co-expression patterns, determine significant biological processes, molecular functions and cellular components for a set of genes of interest as well as to identify significant TFBS in the promoters of a given set of genes.
In this work, we have shown that ChlamyNET exhibits non-random topological properties, namely scale-free and small-world properties. This suggests that the Chlamydomonas transcriptome posseses relevant characteristics related to error tolerance, vulnerability and information propagation [28]. On theone hand, the scale-free property implies robustness against random gene mutations or error tolerance, which means that since most genes are only co-expressed with a few other genes, a random mutation is likely to affect a non important gene altering the expression of a reduced number of other genes.Nevertheless, the existence of key authoritative hub genes produces fragility or vulnerability to targeted attacks against this type of genes. The removal or mutation of an authoritative hub gene would affect a large number of other genes co-expressed with it, massively disrupting the functioning of the Chlamydomonas transcriptome. This can lead to lethality or defective growth. For example, this has been shown for the authoritative hub gene CrCO (g6302) whose over-expression and silencing are detrimental for cellular growth [2]. Additionally, the small-world property facilitates a quick spreading of information throughout ChlamyNET.
The analysis of the location of hub genes and genes with high clustering coefficient shows that both of them group together in specific regions of ChlamyNET. This indicates the existence of gene clusters whose expressions are highly coordinated, possibly to perform related biological processes. Indeed, we identified nine gene clusters that present a high intra-cluster and a low inter-cluster co-expression. Among these clusters we highlight the results obtained in two of them, clusters 1 and 2.
The most central cluster (blue cluster), where most authoritative hub genes are located, is significantly enriched in genes involved in carbon/nitrogen metabolism, signalling through protein phosphorylation and light response. This cluster is also significantly enriched in TFs, revealing a high transcriptional control over carbon/nitrogen metabolism induced by light. Several bHLH TFs are contained in this cluster and only one of them, CrbHLH1 (Cre14.g620850), presents a potential Arabidopsis ortholog, PAR1 (At1g69010), which has been shown to be involved in light response, and indeed the E-box sequence, a bHLH binding site, was found to be significantly present in the promoters of genes in this cluster. Two TFs from the bZIP family are also located in this cluster, CrHY5 (Cre12.g510200) and CrHYH (Cre06.g310500), potential orthologs of the Arabidopsis HY5 (At5g11260) and HYH (At3g17609) genes, respectively. Again, the G-box sequence, a TFBS recognized by HY5 and HYH in Arabidopsis, was found to be significantly present in the promoters of their co-expressed genes. Additionally, this cluster contains two ARR-B TFs (Cre13.g572450 and g16739) and the B-box TF CrBbox1 (Cre03.g182700) whose potential Arabidopsis orthologs RR14 (At2g01760), TOC1 (At5g61380) and COL1 (At5g15850) are involved in circadian rythms. This suggests a key role played by light in the regulation of central metabolism inChlamydomonas mediated by TFs from the bHLH and bZIP families. It also provides evidence for an input from circadian rythms exerted by genes from the ARR-B and B-box families.
One of the most peripheral clusters in ChlamyNET, cluster 2 (brown), was shown to be involved in DNA replication and transitions between the cell cycle phases. This cluster contains potential orthologs of Arabidopsis genes involved in the G1/S transition such as origin of replication complex ORC1 (g11180) and ORC4 (Cre17.g726500), pre-initiation complex subunit CDC6 (Cre06.g292850), DNA replication initiation factor CDT1 (Cre03.g163300), minichromosome maintenance protein MCM2 (Cre07.g338000) and DNA polymerase alpha POLA1 (Cre04.g214350). The E2F sequence was found to be significantly present in the promoters of these genes. Additionally, the presence of a combination of the octamer and hexamer motifs was significantly enriched in the promoters of the genes from this cluster such as the B-type cyclin CYCB1 (Cre08.g370400). The E2F sequence and the combination of octamer and hexamer motifs have been shown to confer S phase-specific transcriptional activation in higher plants.
These results suggest that key elements in the regulation of cell cycle, light response and carbon/nitrogen metabolism are already established in Chlamydomonas and conserved in higher plants such as Arabidospsis. The conserved elements are not only limited to TFs, TRs and their targets but also include the cis-regulatory elements, TFBS, present in their promoters.
The web-based software tool ChlamyNET (http://viridiplantae.ibvf.csic.es/ChlamyNet/) was developed to ensure the reproducibility of the results presented in this work and to facilitate further and independent studies over the Chlamydomonas transcriptome. We used potentially light-regulated TFs and TRs to illustrate its functionalities. Our case study suggests that these genes regulate carbon/nitrogen metabolism and cell cycle. Additionally, light regulated TFBS in Arabidopsis such as SORLIP2, SORLIP3 and SORLREP5 were identified in their gene promoters. Several other cases studies are available from the web page of ChlamyNET, http://viridiplantae.ibvf.csic.es/ChlamyNet/.
Finally, RNA-seq data from algae overexpressing the transcription factor CrDOF involved in photoperiod response were used as an independent data set to experimentally cross-validate the predictive power of ChlamyNET.
Methods
Data acquisition and processing
In this study we used RNA-seq data from the Chlamydomonas transcriptome publicly available at the Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra/) [84], a database resource at the National Center for Biotechnology Information (NCBI) that stores more than 500 TeraBases of next-generation sequencing data. We collected more than 287 GigaBytes of information produced by seven different studies [10–16] consisting of 50 samples representing eight different genotypes under diverse physiological conditions, see Additional file 1: Table S1. These data provide a general overview of the Chlamydomonas transcriptome in a physiologically relevant context. All these data were obtained using the same next-generation sequencing platform, Illumina Genome Analyzer [85], in order to facilitate the comparison between samples from different experiments.
The available Chlamydomonas sequenced genome (version 5.3) [1] was downloaded from Phytozome (http://www.phytozome.net/) [86], a web-based platform for green plant genomics, in order to be used as the reference genome in our study. Additionally, we also obtained from the same web resource the corresponding Augustus u11.6 gene annotation that was used as a reference transcriptome.
The processing of raw sequencing data when a reference genome is available can be divided into three different stages: (i) filtering out low quality reads and alignment of reads to the reference genome; (ii) assembly of transcripts; and (iii) estimation of gene expression [87]. In our study, we followed the methodology described in [88] that makes use of the free software packages Tophat [89] and Cufflinks [90]. First, we perfomed the preprocessing of the raw data consisting of the fastq files from each sample. The read sequences of low quality were filtered out according to their Phred quality scores [91, 92] and the remaining ones were aligned to the reference genome with the software package Tophat that in turn makes use of the fast and memory efficient short read aligner Bowtie [93]. Most of the analyzed samples were of good quality and produced a high alignment rate greater that 80 %. The alignments of read sequences to the reference genome produced in this step were stored in BAM (binary aligment maps) files.
In the second step, we used the alignments in the BAM files and the known transcripts from the Augustus u11.6 annotation for the assembly of the sample specific transcriptomes using the software package Cufflinks. The whole transcriptome identified in all the samples was integrated and stored in a GTF (gene transfer format) file using Cuffmerge, a utility program within the Cufflinks package. We performed this refinement of the currently available annoted Chlamydomonas transcriptome in order to avoid incomplete or incorrect annotation that could reduce accuracy [90] in our study.
Finally, the gene expression levels in the different conditions integrated in our study were estimated using Cuffdiff, a program included in the Cufflinks package. In order to avoid biases due to transcript length and the total number of reads generated in each experiment, we used as unit of measurement fragments per kilobase of transcript per million mapped fragments (FPKM) [90, 94]. Additionally, recent suggestions for normalization methods [95] that reduce the bias due to the non-uniform distribution of mapped reads within transcripts were taken into account by setting the corresponding parameters in Cuffdiff. These normalizations remove the biases in the data while preserving the variation in gene expression that occurs because of biologically relevant changes in transcription, allowing the comparison of gene expression across multiple experiments. Subsequent analysis and visualization of the results were performed using the R package cummeRbund [96].
Selection of di erentially expressed genes
The selection of differentially expressed genes was performed using the standard methodology applied to the analysis of RNA-seq data described in [97]. The logarithm of the levels of expression measured in FPKM were computed and the delta method to estimate the variance of the log odds was used. Those genes that exhibited an adjusted p-value for the multiple testing lower than 0.05 were considered to be differentially expressed.
Gene Co-expression criterion and network construction
We used the absolute value of the Pearson correlation coefficient between gene expression profiles across the different conditions to determine the level of co-expression between the selected genes [30]. For each possible correlation value cor, we represented the co-expression relationships between genes that exceed this value in the Chlamydomonas transcriptome using an undirected weighted network Gcor = (Vcor,Ecor). The nodes or vertices in Vcor correspond to the genes. An undirected edge (g1,g2) in Ecor with associated weight w > cor indicates the existence of a significant co-expression relationship between genes g1 and g2 with an absolute value of the Pearson correlation coefficient between the corresponding expression profiles equal to w.
In order to decide which correlation value is high enough to consider that two genes are significantly co-expressed we used a criterion that establishes a compromise between the generation of a scale-free network and a high network density. Most biological networks characterized so far are scale-free, which makes this property the most common metric for the rational selection of a gene correlation threshold. In order to facilitate the detection of clusters or modules of genes in the constructed network we also added the restriction of generating a network with a high density [25].
A range of correlation thresholds were considered. For each possible correlation cut-off value we determined how close the corresponding network was to fullfil the scale-free condition by computing the R2 of the linear regression for the logarithmic transform of the node degree distribution. Additionally, for each possible cut-off value we used the average node degree as a measurement of the network density.
Network visualization
Graphical representations of the network were perfomed using Cytoscape [98], a software package for network visualization and data integration. Specifically, the organic layout method was applied to visualize ChlamyNET. This algorithm consists of a variant of the force-directed layout. Nodes produce repulsive forces whereas edges induce attractive forces. Nodes are then placed such that the sum of these forces are minimised. The organic layout has the effect of exposing the clustering structure of a network. In particular, this layout tends to locate tighly connected nodes with many interactions or hub nodes together in central areas of the network.
Significance of topological properties
In order to determine the statistical significance of the scale-free property of ChlamyNET we generated 104 random networks with the same number of nodes and edges as ChlamyNET following the Erdös and Renyi random graph model [99]. None of these random networks exhibited a scale-free topology similar to ChlamyNET. This indicates that the scale-free topology of ChlamyNET is not random but rather it is the product of a self-organizing process. It has been suggested that scale-free networks emerge from a growth process by which newly added nodes preferentially attach to already existing nodes with a high number of neighbours [27]. In the case of ChlamyNET the scale-free feature can be a consequence of two mechanisms in the evolution of gene co-expression networks: (i) gene co-expression networks are not static, instead new genes may appear; and (ii) new genes are preferentially co-expressed with genes that already exhibit a large number of co-expressed genes.
We also studied the clustering coefficient in ChlamyNET, a measurement of the density of edges or co-expression relationships around genes. The clustering coefficient of a gene is calculated as the ratio of the actual number of co-expression relationships among all its neighbours and the maximal possible number of such co-expression relationships.
In order to determine that the global clustering coefficient of ChlamyNET is significantly high we generated 104 random scale-free networks with the same number of nodes and edges as ChlamyNET following the Barabasi random scale-free graph model [27]. None of these random networks exhibited a clustering coefficient higher than ChlamyNET.
Clustering techniques
In a general way, clustering techniques aim at identifying groups or clusters whose individuals exhibit high similarities, whereas individuals from different groups or clusters present low similarities. When clustering techniques are applied to co-expression networks, the similarity among genes is meassured using the correlation among the corresponding gene profiles or co-expression. Therefore, the goal of clustering techniques, when applied to gene co-expression data, consists on identifying disjoint groups or clusters of genes so that the co-expression between genes in the same cluster is high (intra-cluster similarity) whereas the co-expression between genes from different clusters is low (inter-cluster similarity) [36]. In this respect, the silhouette [36] a criterion that combines the minimization of inter-cluster similarity with the maximization of the intra-cluster similarity, is one of the most popular measurements for the assesment of a clustering analysis. In our study we used this criterion to determine which clustering algorithm and number of clusters best describes the underlying structure in ChlamyNET. We compared the performance of the two most widely used clustering algorithms, hierarchical clustering and partition around medoids (PAM) for different number of clusters ranging from 4 to 20 clusters using the R package clValid [36].
Gene ontology term enrichment analysis
The Chlamydomonas transcriptome is very sparsely annotated since experimental validation of the different computationally predicted functions is still missing for most genes. In order to overcome this lack of GO term annotation, we followed two diffeerent complementary approaches. In our first approach, we assigned to each Chlamydomonas gene the GO terms associated with its potential Arabidopsis ortholog based on sequence similarity. In our second approach, we used the annotation about protein domains and tools available from the Pfam database [35] to determine the protein family to which each Chlamydomonas gene belongs to. The GO terms associated to the identified protein family were then assigned to the corresponding gene. Our methodology for the identification of the GO terms over-represented in each cluster is a combination of both approaches. We identified as overrepresented GO terms those found by both approaches or by only one of them with a very high statistical signi cance (a p-value lower than 10 −6). The R package topGO was used to perform GO term enrichment using Fisher's exact test. As gene background we selected the entire Chlamydomonas gene set as indentified in the Phytozome 9.1 database.
Transcription factor binding site enrichment analysis
Transcription Factor Binding Sites (TFBS) enrichment analysis was performed using HOMER [100] and the known TFBS sequences in plants from the databases AGRIS [101], JASPAR [102] and AthaMap [103]. The findMotifs.pl script, applying the default parameters, was used to perform a known and de-novo motif over-representation analysis.
The background used for the over-representation analysis consists of all the gene promoters annotated in the current version of the Chlamydomonas genome. These data were downloaded using the BioMart functionality associated with Phytozome.
Alga material, growth conditions and RNA sequencing
Two independent biological replicates of Chlamydomonas reinhardtii wild type CW15 [104], and the transgenic line CrDOFin [46], were grown in flasks with the induction media Sueoka NO3- [105] under LD (16 h light/8 h dark) or SD (8 h light/16 h dark) conditions at 50 E light intensity with 22 C (during light period) and 18 C (during night period) in a model SG-1400 phytotron (Radiber SA, Spain). Algal cells were grown during 4 days and then, were harvested 4 hours after the light went on, which was considered at Zeitgebertime zero (ZT0). The RNA isolation was performed by TRIZOL (Invitrogen) method following the manufacturer instruction. RNA quality was tested employing a ND-1000 Spectrophotometer (Nanodrop). Library preparation was carried out following the manufacturer's recommendations. Sequencing of RNA libraries was performed with the Illumina HiSeq 2000 sequencer, yielding approximately 40 million 50 bp long reads for each sample.
Availability of supporting data
The RNA-seq data set used to cross-validate the preditive power of ChlamyNET is available at European Nucleotide Archive identified with the accession number PRJEB6682.
The processed RNA-seq data, R scripts used in the construction and analysis of our network as well as the network itself in gml format are available from the web page http://viridiplantae.ibvf.csic.es/ChlamyNet/.
Acknowledgements
This work was supported with funding from projects CSD2007-00057, BIO2011-28847-C02-00 and BIO2014-52425-P (Spanish Ministry of Economy and Competitiveness, MINECO) and Excellence project P08-AGR-03582 (Junta de Andaluc a) partially supported by FEDER funding to Federico Valverde and Jose M. Romero. Eva Lucas-Reina was funded by a CSIC-JAE fellowship which is partly supported by structure funding from the EU (SEF). Funding from the JdC program (MINECO) and the Excellence project P08-TIC-04200 (Junta de Andaluc a) to Francisco J. Romero-Campero is also acknowledged. The authors acknowledge the support of the high-performance computational resources from the Centro Informatico Cientifico de Andalucia (CICA).
Additional files
Brief description of the data used in this analysis including genotypes, conditions and sequence information. (PDF 19 kb)
Gene Cluster Expression Profiles. The normalized expression of every gene in each cluster is represented in grey. For each cluster the red line represents the mean positive expression profile whereas the blue line represents the mean negative expression profile. Recall that the absolute value of the correlation is used to define co-expression. Note that the expression profiles for each cluster are distinct. (TIFF 7917 kb)
{kind=link}
Metabolic Pathways Contained in Clusters 2 (Brown), 3 (Red) and 7 (Green) from Figure 4. Analysis of the Chlamydomonas metabolic pathways revealed that cluster 2 (brown) in Figure 4 is enriched in metabolic processes involved in the DNA and RNA synthesis such as the pyrimidine synthesis pathways. Nevertheless, clusters 3 (red) and 7 (green) (Figure 4) were not so significantly enriched in metabolic pathways. Nevertheless, in cluster 3 (red), with a low significance, the synthesis pathway for triacylglycerol using galactolypids produced by glycolipid desaturation as acyl donors, was identified. In cluster 7 (green), the aerobic respiration pathway was fully included. (PNG 50 kb)
{kind=link}
Metabolic Pathways Contained in Clusters 9 (Blue) and 8 (Turquoise) from Figure 4. Analysis of the Chlamydomonas metabolic pathways revealed that cluster 9 (blue) and 8 (turquoise) (Figure 4) are enriched in diverse metabolic processes. Cluster 9 (blue) contains genes involved in core carbon/nitrogen metabolic pathways such as starch biosynthesis, the oxydative branch of the pentose phosphate pathway and nitrogen assimilation pathways co-expressed with the needed molybdenum cofactor synthetic pathway. Cluster 8 (turquoise), the two most important metabolic pathways in photosynthetic organisms, the Calvin cycle and the TCA cycle, have a significant number of genes represented. (PNG 98 kb)
{kind=link}
Metabolic Pathways Contained in Cluster 1 (Orange) and 4 (Purple) from Figure 4. Analysis of the Chlamydomonas metabolic pathways revealed that cluster 1 (orange) and 4 (purple) (Figure 4) are enriched in specific metabolic processes. Cluster 4 (purple) comprises metabolic pathways associated with protein synthesis such as the tRNA charging pathways, aminoacid biosynthesis and the non-oxydative branch of the pentose phosphate pathway. The TAG biosynthetic pathway that preferentially uses as acyl donors galactolipids is also a member of this cluster. Cluster 1 (orange) is enriched in genes involved in lipid metabolism, including the TAG biosynthetic pathway which uses phospholipids that are in turn produced in the phosphatidylinositol pathway as acyl donors. The pathway for the synthesis of the necessary coenzyme A, is also present in this cluster. (PNG 119 kb)
Biological Processes Potentially Controlled by the Light-regulated TFs and TRs in ChlamyNET (PDF 8 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author's contributions
FJRC performed the bioinformatics analysis, contributed to the design and development of the web-based tool ChlamyNET and wrote the paper. ELR performed the experimental work for the validation of ChlamyNET and contributed to the bioinformatics analysis. IPH contributed to the design and development of the web-based tool ChlamyNET. FV and JMR supervised the research, discussed the biological interpretation of the results, designed the experiments for the cross-validation of ChlamyNET and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Francisco J. Romero-Campero, Email: fran@us.es
Ignacio Perez-Hurtado, Email: perezh@us.es.
Eva Lucas-Reina, Email: eva.lucas@ibvf.csic.es.
Jose M. Romero, Email: jmromero@us.es
Federico Valverde, Email: federico.valverde@ibvf.csic.es.
References
- 1.Merchant S, Prochnik S, Vallon O, Harris E, Karpowicz S, Witman G, Terry A, Salamov A, Fritz-Laylin L. Marechal-Drouard Lea: The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007;318(5848):245–250. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano G, Herrera-Palau R, Romero JM, Serrano A, Coupland G, Valverde F. Chlamydomonas CONSTANS and the evolution of plant photoperiodic signalling. Curr Biol. 2009;19:359–368. doi: 10.1016/j.cub.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 3.Romero JM, Valverde F. Evolutionarily conserved photoperiod mechanisms in plants: when did plant photoperiodic signaling appear? Plant Signal Behav. 2009;4(7):642–644. doi: 10.4161/psb.4.7.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valverde F. CONSTANS and the evolutionary origin of photoperiodic timing of owering. J Exp Bot. 2011;62(8):2453–2463. doi: 10.1093/jxb/erq449. [DOI] [PubMed] [Google Scholar]
- 5.Rittmann B. Opportunities for renewable bioenergy using microorganisms. Biotechnol Bioeng. 2008;100(2):203–212. doi: 10.1002/bit.21875. [DOI] [PubMed] [Google Scholar]
- 6.Sivakumar G, Vail D, Xu J, Burner D, Lay J, Ge X, Weathers P. Bioethanol and biodiesel: Alternative liquid fuels for future generations. Eng Life Sci. 2010;10:8–18. doi: 10.1002/elsc.200900061. [DOI] [Google Scholar]
- 7.Kruse O, Hankamer B. Microalgal hydrogen production. Curr Opin Biotechnol. 2010;21(3):238–243. doi: 10.1016/j.copbio.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Pienkos P, Darzins A. The promise and challenges of microalgal-derived biofuels. Biofuels Bioproducts Biore ning-Biofpr. 2009;3(4):331–340. [Google Scholar]
- 9.Blaby I, Blaby-Haas C, Tourasse N, Hom E, Lopez D, Aksoy M, Grossman A, Umen J, Dutcher S, Porter M, King S, Witman G, Stanke M, Harris E, Goodstein D, Grimwood J, Schmutz J, Vallon O, Merchant S, Prochnik S. The Chlamydomonas genome project: a decade on. Trends Plant Sci. 2014;19(10):672–680. doi: 10.1016/j.tplants.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle NR, Page MD, Liu B, Blaby IK, Casero D, Kropat J, Cokus SJ, Hong-Hermesdorf A, Shaw J, Karpowicz SJ, Gallaher SD, Johnson S, Benning C, Pellegrini M, Grossman A, Merchant S. Three acyltransferases and nitrogen-responsive regulator are implicated in nitrogen starvation-induced triacylglycerol accumulation in Chlamydomonas. J Biol Chem. 2012;287(19):15811–158825. doi: 10.1074/jbc.M111.334052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urzica EI, Adler LN, Page MD, Linster CL, Arbing MA, Casero D, Pellegrini M, Merchant SS, Clarke SG. Impact of oxidative stress on ascorbate biosynthesis in Chlamydomonas via regulation of the VTC2 gene encoding a GDP-L-galactose phosphorylase. J Biol Chem. 2012;287(17):14234–14245. doi: 10.1074/jbc.M112.341982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer B, Ledford H, Wakao S, Huang S, Casero D, Pellegrini M, Merchant S, Koller A, RI E, Niyogi K. SINGLET OXYGEN RESISTANT 1 links reactive electrophile signaling to singlet oxygen acclimation in Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A. 2012;109(20):E1301–E1311. doi: 10.1073/pnas.1116843109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kropat J, Hong-Hermesdorf A, Casero D, Ent P, Castruita M, Pellegrini M, Merchant SS, DM. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011;66:770–80. [DOI] [PMC free article] [PubMed]
- 14.Castruita M, Casero D, Karpowicz S, Kropat J, Vieler A, Hsieh S, Yan W, Cokus S, Loo J, Benning C, Pellegrini M, Merchant S. Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell. 2011;23(4):1273–1292. doi: 10.1105/tpc.111.084400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller R, Wu G, Deshpande R, Vieler A, Gartner K, Li X, Moellering ER, Zauner S, Cornish AJ, Liu B, Bullard B, Sears BB, Kuo M, Hegg EL, Shachar-Hill Y, Shiu S, Benning C. Changes in Transcript Abundance in Chlamydomonas reinhardtii following Nitrogen Deprivation Predict Diversion of Metabolism. Plant Physiol. 2010;154:1737–1752. doi: 10.1104/pp.110.165159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Ballester D, Casero C, Cokus S, Pellegrini M, Merchant SS, Grossman A. RNA-Seq Analysis of Sulfur-Deprived Chlamydomonas Cells Reveals Aspects of Acclimation Critical for Cell Survival. Plant Cell. 2010;22:2058–2084. doi: 10.1105/tpc.109.071167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dal'Molin C, Quek L, Palfreyman R, Nielsen L. AlgaGEM - a genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genomics. 2011;12:S5. doi: 10.1186/1471-2164-12-S4-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez D, Casero D, Cokus S, Merchant S, Pellegrini M. Algal Functional Annotation Tool: a web-based analysis suite to functionally interpret large gene lists using integrated annotation and expression data. BMC Bioinf. 2011;12:282. doi: 10.1186/1471-2105-12-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng H, Chiang-Hsieh Y, Chien C, Hsu B, Liu T, Chen C, Chang W. AlgaePath: comprehensive analysis of metabolic pathways using transcript abundance data from next-generation sequencing in green algae. BMC Genomics. 2014;15:196. doi: 10.1186/1471-2164-15-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassel G, Lan H, Glaab E, Gibbs D, Gerjets T, Krasnogor N, Bonner A, Holdsworth M, Provart N. Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc Natl Acad Sci. 2011;108(23):9709–9714. doi: 10.1073/pnas.1100958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai B, Li C, Huang J. Systematic identi cation of cell-wall related genes in Populus based on analysis of functional modules in co-expression network. PLoS One. 2014;9(4):e95176. doi: 10.1371/journal.pone.0095176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang Y, Cai B, Chen F, Wang G, Wang M, Zhong Y, Z C. Construction and validation of a gene co-expression network in grapevine (Vitis vinifera. L.). Horticulture Res. 2014;1:14040. [DOI] [PMC free article] [PubMed]
- 23.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article 17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 24.Dong J, Horvath S. Understanding network concepts in modules. BMC Syst Biol. 2007;1:24. doi: 10.1186/1752-0509-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoki K, Ogata Y, Shibata D. Approaches for Extracting Practical Information from Gene Coexpression Networks in Plant Biology. Plant Cell Physiol. 2007;48(3):381–390. doi: 10.1093/pcp/pcm013. [DOI] [PubMed] [Google Scholar]
- 26.Gao Z, Zhao R, Ruan J. A genome-wide cis-regulatory element discovery method based on promoter sequences and gene co-expression networks. BMC Genomics. 2013;14:S4. doi: 10.1186/1471-2164-14-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barabasi A, Albert R. Emergence of Scaling in Random Networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Chen G. Complex networks: small-world, scale-free, and beyond. IEEE Circuits Syst Mag. 2003;3:6–20. doi: 10.1109/MCAS.2003.1228503. [DOI] [Google Scholar]
- 29.Jeong H, Mason S, Barabasi A, Oltvai Z. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 30.Song L, Langfelder P. H S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinf. 2012;13:328. [DOI] [PMC free article] [PubMed]
- 31.Batada N, Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ. Stratus Not Altocumulus: A New View of the Yeast Protein Interaction Network. PLoS Biol. 2006;10(4):e317. doi: 10.1371/journal.pbio.0040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao L, Van Hemert J, Sudhansu DD, Dickerson J. Arabidopsis gene co-expression network and its functional modules. BMC Bioinf. 2009;10:346. doi: 10.1186/1471-2105-10-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watts D, Strogatz S. Collective dynamics of 'small-world' networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 34.Kelinberg J. Authoritative Sources in a Hyperlinked Environment. J ACM. 1999;46(5):604–632. doi: 10.1145/324133.324140. [DOI] [Google Scholar]
- 35.Punta M, Coggill P, Eberhardt R, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer E, Eddy S, Bateman A, Finn R. The Pfam protein families database. Nucleid Acids Res. 2012;40(D1):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brock G, Pihur V, Datta S. Datta S: clValid: An R Package for Cluster Validation. J Stat Softw. 2008;25:4.
- 37.Rhee S, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nat Rev Genet. 2008;9:509–515. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- 38.Marcotte E, Date S. Exploiting big biology: integrating large-scale biological data for function inference. Brief Bioinform. 2001;2:363–374. doi: 10.1093/bib/2.4.363. [DOI] [PubMed] [Google Scholar]
- 39.Chae L, Lee I, Shin J, Rhee S. Towards understanding how molecular networks evolve in plants. Curr Opin Plant Biol. 2012;15(2):177–184. doi: 10.1016/j.pbi.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 40.May P, Christian J, Kempa S, Walther D. ChlamyCyc: an integrative systems biology database and web-portal for Chlamydomonas reinhardtii. BMC Genomics. 2009;10:209. doi: 10.1186/1471-2164-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gennidakis S, Rao S, Greenham K, Uhrig R, O'Leary B, Snedden W, Lu C, Plaxton W. Bacterial- and plant-type phosphoenolpyruvate carboxylase polypeptides interact in the hetero-oligomeric Class-2 PEPC complex of developing castor oil seeds. Plant J. 2007;52(5):839–849. doi: 10.1111/j.1365-313X.2007.03274.x. [DOI] [PubMed] [Google Scholar]
- 42.Kwon S, Cho H, Kim S, Park O. The Rab GTPase RabG3b Positively Regulates Autophagy and Immunity-Associated Hypersensitive Cell Death in Arabidopsis. Plant Physiol. 2013;161(4):1722–1736. doi: 10.1104/pp.112.208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon K, Han D, Li Y, Sommerfeld M, Hu Q. Phospholipid:Diacylglycerol Acyltransferase Is a Multifunctional Enzyme Involved in Membrane Lipid Turnover and Degradation While Synthesizing Triacylglycerol in the Unicellular Green Microalga Chlamydomonas reinhardtii. Plant Cell. 2012;24(9):3708–3724. doi: 10.1105/tpc.112.100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demidov D, VanDamme D, Geelen D, Blattner F, Houbena A. Identi cation and Dynamics of Two Classes of Aurora-Like Kinases in Arabidopsis and Other Plants. Plant Cell. 2005;17:836–848. doi: 10.1105/tpc.104.029710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Umeda M, Shimotohno A, Y M. Control of Cell Division and Transcription by Cyclin-dependent Kinase-activating Kinases in Plants. Plant Cell Physiol. 2005;46(9):1437–42. [DOI] [PubMed]
- 46.Lucas-Reina E, Romero-Campero F, Romero J, Valverde F. An evolutionarily conserved DOF-CONSTANS module controls plant photoperiodic signalling. Plant Physiol. 2015;168(2):561–574. doi: 10.1104/pp.15.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaufmann K, Pajoro A, Angenent G. Regulation of transcription in plants: mechanisms controlling developmental switches. Nat Rev Genet. 2010;11:830–842. doi: 10.1038/nrg2885. [DOI] [PubMed] [Google Scholar]
- 48.Riaño-Pachon D, Guedes-Correa L, Trejos-Espinosa R, MR B. Green Transcription Factors: A Chlamydomonas Overview. Genetics. 2008;179:31–9. [DOI] [PMC free article] [PubMed]
- 49.Jin J, Zhang H, Kong L, Gao G, Luo J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2013;42(D1):D1182–D1187. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pérez-Rodríguez P, Riaño-Pachón DM, Corrêa LG, Rensing SA, Kersten B, Mueller-Roeber B. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010;38(suppl 1):D822–D827. doi: 10.1093/nar/gkp805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cifuentes-Esquivel N, Bou-Torrent J, Galstyan A, Gallemí M, Sessa G, Salla-Martret M, Roig-Villanova I, Ruberti I, Mart nez-García J. The bHLH proteins BEE and BIM positively modulate the shade avoidance syndrome in Arabidopsis seedlings. Plant J. 2013;75(6):989–1002. doi: 10.1111/tpj.12264. [DOI] [PubMed] [Google Scholar]
- 52.Chattopadhyay S, Ang L, Puente P, Deng X, W N. Arabidopsis bZIP protein HY5 directly interacts with light-responsive promoters in mediating light control of gene expression. Plant Cell. 1998;10(5):673–83. [DOI] [PMC free article] [PubMed]
- 53.Jonassen E, Sandsmark B, Lillo C. Unique status of NIA2 in nitrate assimilation: NIA2 expression is promoted by HY5/HYH and inhibited by PIF4. Plant Signal Behav. 2009;4(11):1084–1086. doi: 10.4161/psb.4.11.9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wenkel S, Turck F, Singer K, Gissot L, Le Gourrierec J, Samach A, Coupland G. CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate owering of Arabidopsis. Plant Cell. 2006;18(11):2971–2984. doi: 10.1105/tpc.106.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gendron J, Pruneda-Paz J, Doherty C, Gross A, Kang S, Kay S. Arabidopsis circadian clock protein, TOC1, is a DNA-binding transcription factor. Proc Natl Acad Sci U S A. 2012;109(8):3167–3172. doi: 10.1073/pnas.1200355109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ledger S, Strayer C, Ashton F, Kay S, Putterill J. Analysis of the function of two circadian-regulated CONSTANS-LIKE genes. Plant J. 2012;26:15–22. doi: 10.1046/j.1365-313x.2001.01003.x. [DOI] [PubMed] [Google Scholar]
- 57.Yanhui C, Xiaoyuan Y, Kun H, Meihua L, Jigang L, Zhaofeng G, Zhiqiang L, Yunfei Z, Xiaoxiao W, Xiaoming Q, Yunping S, Li Z, Xiaohui D, Jingchu L, Xing-Wang D, Zhangliang C, Hongya G, Li-Jia Q. The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol Biol. 2006;60:107–124. doi: 10.1007/s11103-005-2910-y. [DOI] [PubMed] [Google Scholar]
- 58.Camargo A, Llamas A, Schnell R, Higuera J, Gonzalez-Ballester D, Lefebvre P, Fernandez E, Galvan A. Nitrate Signaling by the Regulatory Gene NIT2 in Chlamydomonas. Plant Cell. 2007;19(11):3491–3503. doi: 10.1105/tpc.106.045922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Remacle C, Eppe G, Coosemans N, Fernandez E, Vigeolas H. Combined intracellular nitrate and NIT2 e ects on storage carbohydrate metabolism in Chlamydomonas. J Exp Bot. 2014;65:23–33. doi: 10.1093/jxb/ert339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scheible W, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi M, Stitt M. Genome-Wide Reprogramming of Primary and Secondary Metabolism, Protein Synthesis, Cellular Growth Processes, and the Regulatory Infrastructure of Arabidopsis in Response to Nitrogen. Plant Physiol. 2004;136:2483–2499. doi: 10.1104/pp.104.047019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiao Y, Lau O, Deng X. Light-regulated transcriptional networks in higher plants. Nat Rev. 2007;8:217–230. doi: 10.1038/nrg2049. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, Burgos J, Deng Y, Srivastava R, Howell S, Bassham D. Degradation of the Endoplasmic Reticulum by Autophagy during Endoplasmic Reticulum Stress in Arabidopsis. Plant Cell. 2012;24(11):4635–4651. doi: 10.1105/tpc.112.101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sottosanto J, Saranga Y, Blumwald E. Impact of AtNHX1, a vacuolar Na+/H+ antiporter, upon gene expression during short- and long-term salt stress in Arabidopsis thaliana. BMC Plant Biol. 2007;7:18. doi: 10.1186/1471-2229-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ascencio-Ibanez J, Sozzani R, Lee T, Chu T, Wolnger R, Cella R, Hanley-Bowdoin L. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 2008;148:436–54. [DOI] [PMC free article] [PubMed]
- 65.Shaked H, Avivi-Ragolsky N, Levy A. Involvement of the Arabidopsis SWI2/SNF2 chromatin remodeling gene family in DNA damage response and recombination. Genetics. 2006;173(2):985–994. doi: 10.1534/genetics.105.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perez-Perez M, Florencio F, Crespo J. Inhibition of TOR signaling and stress activate autophagy in Chlamydomonas reinhardtii. Plant Physiol. 2010;152(4):1874–1888. doi: 10.1104/pp.109.152520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim S, Choi H, Ryu H, Park J, Kim M, Kim S. ARIA, an Arabidopsis Arm Repeat Protein Interacting with a Transcriptional Regulator of Abscisic Acid-Responsive Gene Expression, Is a Novel Abscisic Acid Signaling Component. Plant Physiol. 2004;136(3):3639–3648. doi: 10.1104/pp.104.049189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen W, Chao G, Singh K. The promoter of a H2O2-inducible, Arabidopsis glutathione S-transferase gene contains closely linked OBF- and OBP1-binding sites. Plant J. 1996;10(6):955–966. doi: 10.1046/j.1365-313X.1996.10060955.x. [DOI] [PubMed] [Google Scholar]
- 69.Ito M, Araki S, Matsunaga S, Itoh T, Nishihama R, Machida Y, Doonan J, Watanabe A. G2/M-Phase-Specific Transcription during the Plant Cell Cycle Is Mediated by c-Myb-Like Transcription Factors. Plant Cell. 2001;13:1891–1905. doi: 10.1105/tpc.13.8.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haga N, Kato K, Murase M, Araki S, Kubo M, Demura T, Suzuki K, Müller I, Vo U, Jürgens G, Ito M. R1R2R3-Myb proteins positively regulate cytokinesis through activation of KNOLLE transcription in Arabidopsis thaliana. Development. 2007;134:1101–1110. doi: 10.1242/dev.02801. [DOI] [PubMed] [Google Scholar]
- 71.Araki S, Kato K, Suzuki T, Okumura T, Machida Y, Ito M. Cosuppression of NtmybA1 and NtmybA2 causes downregulation of G2/M phase-expressed genes and negatively a ects both cell division and expansion in tobacco. Plant Signal Behav. 2013;9:e26780. doi: 10.4161/psb.26780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pedersen D, Coppens F, Ma L, Antosch M, Marktl B, Merkle T, Beemster G, Houben A, Grasser K. The plant-specific family of DNA-binding proteins containing three HMG-box domains interacts with mitotic and meiotic chromosomes. New Phytol. 2011;192(3):577–589. doi: 10.1111/j.1469-8137.2011.03828.x. [DOI] [PubMed] [Google Scholar]
- 73.Van Leene J, Hollunder J, Eeckhout D, Persiau G, Van De Slijke E, Stals H, Van Isterdael G, Verkest A, Neirynck S, Buel Y, De Bodt S, Maere S, Laukens K, Pharazyn A, Ferreira P, Eloy N, Renne C, Meyer C, Faure J, Steinbrenner J, Beynon J, Larkin J, Van de Peer Y, Hilson P, Kuiper M, De Veylder L, Van Onckelen H, Inze D, Witters E, De Jaeger G. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol Syst Biol. 2010;6:397. doi: 10.1038/msb.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gutierrez C. The Arabidopsis Cell Division Cycle. Arabidopsis Book. 2009;7:e0120. doi: 10.1199/tab.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Avramova Z. Evolution and pleiotropy of TRITHORAX function in Arabidopsis. Int J Dev Biol. 2009;53:371–381. doi: 10.1387/ijdb.082664za. [DOI] [PubMed] [Google Scholar]
- 76.Wang Y, Yang M. In Silico Identi cation of Co-transcribed Core Cell Cycle Regulators and Transcription Factors in Arabidopsis. J Integr Plant Biol. 2007;49(8):1253–1260. doi: 10.1111/j.1672-9072.2007.00547.x. [DOI] [Google Scholar]
- 77.Chaboute M, Clement B, Sekine M, Philipps G, Chaubet-Gigot N. Cell cycle regulation of the tobacco ribonucleotide reductase small subunit gene is mediated by E2F-like elements. Plant Cell. 2000;12(10):1987–2000. doi: 10.1105/tpc.12.10.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Jager S, Menges M, Bauer U, Murray J. Arabidopsis E2F1 binds a sequence present in the promoter of S-phase-regulated gene AtCDC6 and is a member of a multigene family with differential activities. Plant Mol Biol. 2001;47(4):555–568. doi: 10.1023/A:1011848528377. [DOI] [PubMed] [Google Scholar]
- 79.Taoka K, Kaya H, Nakayama T, Araki T, Meshi T, Iwabuchi M. Identification of three kinds of mutually related composite elements conferring S phase-speci c transcriptional activation. Plant J. 1999;18(6):611–623. doi: 10.1046/j.1365-313x.1999.00486.x. [DOI] [PubMed] [Google Scholar]
- 80.Gretarsson B, Bostandjiev S, Donovan J, Hllerer T. International Symposium on Graph Drawing. 2009. WiGis: A framework for Web-based interactive graph visualizations; pp. 119–134. [Google Scholar]
- 81.Jiao Y, Yang H, Ma L, Sun N, Yu H, Liu T, Gao Y, Gu H, Chen Z, Wada M, Gerstein M, Zhao M, Qu L, Deng X. A Genome-Wide Analysis of Blue-Light Regulation of Arabidopsis Transcription Factor Gene Expression during Seedling Development. Plant Physiol. 2003;133(4):1480–1493. doi: 10.1104/pp.103.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reisdorph N, Small G. The CPH1 Gene of Chlamydomonas reinhardtii Encodes Two Forms of Cryptochrome Whose Levels Are Controlled by Light-Induced Proteolysis. Plant Physiol. 2004;134:1546–1554. doi: 10.1104/pp.103.031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hudson M, Quail P. Identification of Promoter Motifs Involved in the Network of Phytochrome A-Regulated Gene Expression by Combined Analysis of Genomic Sequence and Microarray Data. Plant Physiol. 2003;133(4):1605–1616. doi: 10.1104/pp.103.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W, Kenton DL, Khovayko O, Lipman DJ, Madden TL, Maglott DR, Ostell J, Pontius JU, Pruitt KD, Schuler GD, Schriml LM, Sequeira E, Sherry ST, Sirotkin K, Starchenko G, Suzek TO, Tatusov R, Tatusova TA, L W, E Y. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–45. [DOI] [PMC free article] [PubMed]
- 85.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HRea. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–9. [DOI] [PMC free article] [PubMed]
- 86.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, S RD. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–86. [DOI] [PMC free article] [PubMed]
- 87.Garber M, Grabherr M, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 88.Trapnell C, Roberts A, Go L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg S, Rinn J, L P. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed]
- 89.Trapnell C, Pachter L, Salzberg S. TopHat: discovering splice junctios with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cea T. Transcript assembly and quanti cation by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8(3):175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 92.Ewing B, Green P. Base-calling of automated sequencer traces using phred, II. Error probabilities. Genome Res. 1998;8(3):186–194. doi: 10.1101/gr.8.3.186. [DOI] [PubMed] [Google Scholar]
- 93.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-e cient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mortazavi A, Williams B. McCue k, Schae er L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–8. [DOI] [PubMed]
- 95.Roberts A, Trapnell C, Donaghey J, Rinn J, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Go L, Trapnell C: cummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. 2011. [R package version 1.2.0].
- 97.Bullard J, Purdom E, Hansen K, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinf. 2010;11(94):1–13. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smoot M, Ono K, Ruscheinski J, Peng-Liang W, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Erdos P, Renyi A. On the evolution of random graphs. Publ Math Inst Hung Acad Sci. 1960;5:17–61. [Google Scholar]
- 100.Heinz S, Benner C, Spann N, Bertolino E, Lin Y, Laslo P, Cheng J, Murre C, Singh H, Glass C. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davuluri R, Sun H, Palaniswamy S, Matthews N, Molina C, Kurtz M, Grotewold E. AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinf. 2003;4:25. doi: 10.1186/1471-2105-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sandelin A, Alkema W, Engstrom P, Wasserman W, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding pro les. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Steffens N, Galuschka C, Schindler M, Bulow L, Hehl R. AthaMap: an online resource for in silico transcription factor binding sites in the Arabidopsis thaliana genome. Nucleic Acids Res. 2004;32:D368–D372. doi: 10.1093/nar/gkh017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Davies D, Plaskitt A. Genetical and structural analyses of cell-wall formation in Chlamydomonas reinhardtii. Gen Res. 1971;17:33–43. doi: 10.1017/S0016672300012015. [DOI] [Google Scholar]
- 105.Sueoka N, Chiang K, Kates R. Deoxyribonucleic acid replication in meiosis of Chlamydomonas reinhardtii. J Mol Biol. 1967;25:47–66. doi: 10.1016/0022-2836(67)90278-1. [DOI] [PubMed] [Google Scholar]