

Abstract
Background
Vagus nerve stimulation (VNS) paired with forelimb training drives robust, specific reorganization of movement representations in the motor cortex. The mechanisms that underlie VNS-dependent enhancement of map plasticity are largely unknown. The cholinergic nucleus basalis (NB) is a critical substrate in cortical plasticity, and several studies suggest that VNS activates cholinergic circuitry.
Objective
We examined whether the NB is required for VNS-dependent enhancement of map plasticity in the motor cortex.
Methods
Rats were trained to perform a lever pressing task and then received injections of the immunotoxin 192-IgG-saporin to selectively lesion cholinergic neurons of the NB. After lesion, rats underwent five days of motor training during which VNS was paired with successful trials. At the conclusion of behavioral training, intracortical microstimulation was used to document movement representations in motor cortex.
Results
VNS paired with forelimb training resulted in a substantial increase in the representation of proximal forelimb in rats with an intact NB compared to untrained controls. NB lesions prevent this VNS-dependent increase in proximal forelimb area and result in representations similar to untrained controls. Motor performance was similar between groups, suggesting that differences in forelimb function cannot account for the difference in proximal forelimb representation.
Conclusions
Together, these findings indicate that the NB is required for VNS-dependent enhancement of plasticity in the motor cortex and may provide insight into the mechanisms that underlie the benefits of VNS therapy.
Keywords: cortical plasticity, cortical reorganization, motor cortex, motor training, vagus nerve stimulation, vagal nerve stimulation, acetylcholine, nucleus basalis, immunotoxin
INTRODUCTION
Neuromodulatory interventions have been extensively investigated as potential therapies to reverse maladaptive plasticity or boost limited plasticity to treat neurological disease. Recently, vagus nerve stimulation (VNS) has emerged as one such potential adjunctive intervention to enhance neuroplasticity [1]. Repeated presentation of auditory stimuli paired with short bursts of VNS drives long-lasting plasticity in auditory cortex [2-4]. Moreover, VNS paired with forelimb training drives robust, specific reorganization in motor cortex [5]. Based on this enhancement of plasticity, VNS has garnered attention as a method to support recovery in the context of neurological disease.
Recently, several studies have demonstrated that VNS paired with specific rehabilitative training regimens can provide therapeutic benefits in a variety of neurological disorders. VNS paired with specific tones reverses the neural and behavioral correlates of tinnitus in a rat model, and a pilot study indicates that VNS tone therapy promotes recovery in chronic tinnitus patients [2, 3, 6, 7]. Additionally, several studies have indicated that VNS paired with motor rehabilitation improves recovery in several mechanistically distinct models of brain injury. VNS paired with rehabilitative training enhances recovery of forelimb function after cortical ischemic stroke, subcortical intracerebral hemorrhage, and traumatic brain injury [8-12]. Based on these findings, physical rehabilitation with task-concurrent VNS is now under investigation in chronic stroke patients [13, 14]. A distinct implementation using long-duration VNS is already in use in over 60,000 patients for control of intractable epilepsy and treatment-resistant depression [15-18].
Despite the demonstrated and potential efficacy in a variety of neurological diseases, the mechanisms underlying VNS-dependent enhancement of neuroplasticity and recovery are largely unknown. Previous studies have implicated the noradrenergic locus coeruleus (LC) and cholinergic nucleus basalis (NB) in the effects of VNS in the central nervous system [1, 19]. Electrical stimulation of the vagus nerve drives activity in both the LC and cholinergic basal forebrain [20-22]. VNS drives release of norepinephrine throughout the brain [23-26]. Lesions of the LC prevent the seizure-attenuating and antidepressant effects of VNS, indicating the importance of noradrenergic signaling in the effects of VNS [27, 28]. Acute antagonism of muscarinic acetylcholine receptors prevents VNS-dependent desynchronization of cortical EEG, suggesting that VNS exerts an effect on cortical processing by engaging cholinergic transmission [29]. Moreover, tones paired with either VNS or direct NB stimulation drive similar spectral and temporal features of plasticity in the auditory cortex [2, 3]. Both the LC and NB are key substrates in neural plasticity [30], and activation of these systems may underlie VNS-dependent enhancement of plasticity.
Acetylcholine and norepinephrine act both independently and synergistically to facilitate plasticity [31, 32], and it is not clear how the effects of VNS are mediated by these neuromodulatory systems. It is possible that NB activation is necessary for the plasticity enhancing effects of VNS. Alternatively, the noradrenergic or other neuromodulatory systems may substitute in the absence of NB activation. Here, we evaluate whether the NB is necessary for the plasticity-enhancing effects of VNS paired with motor training. A clear definition of the neuromodulatory systems engaged by VNS is needed to identify factors, such as drugs or disease states, that affect neuromodulatory transmission and may interfere with the benefits of VNS therapy.
METHODS
Subjects
Twenty-six adult female Sprague-Dawley rats weighing on average 284 grams were used in this experiment. Rats were housed in a 12:12 hour reversed light cycle to increase daytime activity levels. Rats were food deprived during behavioral training, with body weights maintained above 85% to increase motivation for food pellet rewards. All handling, housing, behavioral training, and surgical procedures were approved by the University of Texas Institutional Animal Care and Use Committee.
Behavioral Procedure
Rats were trained on the bradykinesia assessment task, a quantitative, automated lever pressing task [33]. The behavioral chamber consisted of an acrylic cage with a slot located in the front right for access to a lever positioned 2.5 cm outside of the chamber (Fig. 1A). The lever was affixed to a potentiometer, which records the angle of the lever depression relative to horizontal. The lever was allowed to move 13° below horizontal, and lever depression exceeding 75 percent of the total range was considered a press. A spring provided 28 grams of resistance, returning the lever to its level resting angle. A controller board (Vulintus, Richardson, TX) sampled the potentiometer position at 100 Hz and relayed the information to custom MATLAB software that controlled the task and collected data.
Figure 1. Experimental design.
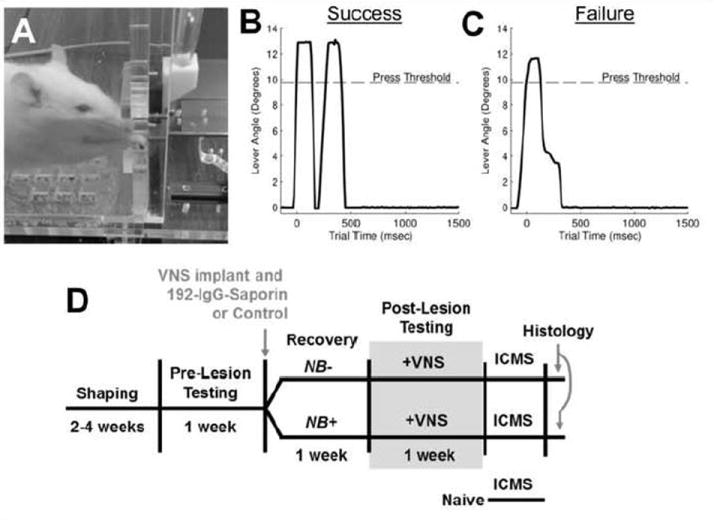
(A) Image of a rat performing the lever pressing task. (B, C) Representative data of lever pressing performance depicting a successful and unsuccessful trial. (D) Timeline of the experimental design.
During behavioral testing, a timer was initiated on the first press of the lever. If the lever was depressed a second time within 500 msec, the trial was recorded as a success and a reward pellet (45 mg dustless precision pellet, BioServ, Frenchtown, NJ) was delivered (Fig. 1B). A tone provided an auditory cue for successful tasks. If the lever was not pressed again or the second press occurred more than 500 msec later, the trial was recorded as a failure and no reward or VNS was given (Fig. 1C). Training was conducted in stages, as previously described [33]. Behavioral training and testing was performed in two thirty minute sessions per day, five days a week, with at least two hours between daily training sessions. Rats continued behavioral training until they performed at least 100 successful trials during each training session. Once proficient, rats underwent 192-IgG-saporin or control injections and stimulating cuff implants. One week after lesion, rats returned for behavioral testing. After habituating to the stimulating cable, rats underwent 5 days of training with VNS paired with successful trials.
Cortical Cholinergic Depletion
Cholinergic lesions were performed similar to previous reports [34-36]. Rats were anesthetized with ketamine hydrochloride (80 mg/kg, i.p.) and xylazine (10 mg/k, i.p.), and given supplemental doses as needed to maintain anesthesia levels. After placing the rat in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) burr holes were drilled over the nucleus basalis bilaterally. Rats received injections of either conjugated 192-IgG-saporin (Advanced Targeting Systems, San Diego, CA) to selectively lesion cholinergic neurons in the basal forebrain, or control injections of an untargeted antibody and saporin, which does not enter cells and induce cell death. Toxin or control peptide (0.375 mg/mL in saline) was injected through a pulled glass needle at 0.1 μL/min using a Nanoliter 2010 injector (World Precision Instruments, Sarasota, FL). Injections were made at the following sites (site 1&2: 0.3 μL, AP: -1.4, ML: +/- 2.5, DV: -8.0; sites 3&4: 0.2 μL, AP: -2.6, ML: +/- 4, DV: -7.0). A schematic of injection locations can be found in the supplementary data. The needle remained in place for 4-5 minutes after each injection to allow for diffusion and prevent backflow. Burr holes were sealed with bone wax.
Vagus Nerve Cuff Implant
Vagus nerve cuff implantations were performed as described previously [8-11]. Immediately following 192-IgG-saporin or control injections, four bone screws were manually drilled into the skull at points near the lamboid suture and over the cerebellum. A two-channel connector was attached to the cranial screws with acrylic. An incision and blunt dissection of the neck muscles exposed the left vagus nerve. The vagus nerve was isolated with blunt dissection and placed in a bipolar stimulating cuff electrode with platinum-iridium leads (~5 kΩ impedance). Cuff leads were tunneled subcutaneously and attached to the skull mounted connector and encapsulated with acrylic. Neck and scalp incisions were sutured and treated with topical antibiotic ointment. Rats were provided with amoxicillin (5 mg) and carprofen (1 mg) tablets for 3 days following the surgeries and allowed to recover for one week before returning to behavioral training.
Vagus Nerve Stimulation Procedure
Upon returning to behavioral testing following surgery, rats were given 1-5 days to become habituated to the stimulating cable connected to the headcap while performing the task. Once rats consistently performed 200 successful trials per day while connected to the stimulator, VNS pairing commenced. VNS was delivered on successful trials in all rats, as previously described [9, 11]. VNS was delivered as a 500-ms train of 15 pulses at 30 Hz. Each biphasic pulse was 0.8 mA in amplitude and 100 μs in phase duration. These parameters are identical to previous studies [5, 8, 9, 11, 12]. Rats received VNS paired with behavioral training for 5 days before ICMS.
Intracortical Microstimulation Mapping
Within 24 hours of the final VNS paired training session rats underwent intracortical microstimulation (ICMS) of the left motor cortex to derive functional maps in cortex contralateral to the trained limb using standard procedures [5, 37-39]. An additional 8 rats that did not receive motor training underwent ICMS as naïve controls. Rats were anesthetized with ketamine hydrochloride (70 mg/kg, i.p.) and xylazine (5 mg/k, i.p.) and received supplementary doses as needed. To prevent swelling, a small incision was made in the cisterna magna. A craniotomy and duratomy exposed the left motor cortex, contralateral to the trained forelimb. A tungsten electrode (~0.7 MΩ impedance) was inserted following a grid with 500 μm spacing to a depth of 1.8 mm. Sequential electrode placements were made at least 1 mm apart where possible. Stimulation consisted of a 40 ms pulse train of ten 200 μs monophasic cathodal pulses delivered at 286 Hz. Stimulation intensity was gradually increased from 20 μA to 200 μA until a movement was observed. If no movement was observed at 200 μA, responses were evaluated at 1.6 mm and 2.0 mm electrode depths to account for variability in cortical thickness. If no movement was observed at any depth at the maximal stimulation, the site was deemed nonresponsive. The borders of primary motor cortex were defined based on unresponsive sites and stopped at the posterior-lateral vibrissae area, which is known to overlap the somatosensory cortex [40].
Motor mapping procedures were conducted with two experimenters as previously described [5]. The first experimenter placed the electrode and recorded the data for each site. The second experimenter was blind to the treatment group and electrode position to avoid potential biasing. The second experimenter delivered stimulations and observed and classified movements. Movements were classified at the threshold current, but in some cases, slightly higher currents were used to disambiguate movements too small to be classified at threshold. Stimulation sites were randomly chosen and did not extend beyond established border (i.e., unresponsive) sites. Movements of the shoulder and elbow were classified as “proximal forelimb.” Movements of the wrist and digits were classified as “distal forelimb.” “Hindlimb” included any movement in the hindlimb of the rat. Neck, vibrissa, and jaw movements were classified as such. Cortical area was calculated by multiplying the number of sites eliciting a response by the area surrounding a site (0.25 mm2). Complete borders were determined when possible, but some maps do not have complete borders. All maps were used for analysis, and raw ICMS maps from all subjects can be found in the online supplement.
Histology and Quantification of ACh Depletion
Following ICMS, rats were transcardially perfused with 250 mL of 0.02% heparin/100mM phosphate-buffered (PB) solution, followed by 450 mL of 4% paraformaldehyde/100 mM PB solution. Brains were removed and postfixed in 4% paraformaldehyde/100 mM PB solution, and then cryoprotected in a 30% sucrose/0.1 M PB solution. The full extent of the motor cortex was sectioned at a 40 μm thickness. Three sections from motor cortex contralateral to the trained limb were randomly selected and stained for acetylcholinesterase (AChE) activity using standard protocol [41]. In brief, free floating sections were washed in a Tris-Maleate buffer solution containing 6 mg/ml promethazine. After a series of washes in Tris-Maleate buffer, tissue was incubated in a solution containing 10 mM sodium citrate, 30 mM cupric sulfate, 5.0 mM potassium ferricyanide, and 0.5 mg/ml acetylcholine iodide. After washes in a Tris-HCl buffer, tissue was processed for DAB to intensify the labeling. Tissue was then mounted, dehydrated, and cover slipped.
Stained tissue was imaged using a NanoZoomer (Hamamatsu, Tokyo, Japan). Analysis of cortical cholinergic innervation was performed by counting AChE positive fibers crossing of a grid overlay, as described previously [42, 43]. A region of layer V of motor cortex from each section was randomly selected for analysis by an independent experimenter blind to the condition of each rat. A 6×6 grid (250 μm × 250 μm) was manually superimposed on the area using Adobe Photoshop CS4 (Figure 2). All intersections between AChE stained fibers and a gridline were manually identified and counted by an experimenter using coded images. Four rats that received injections of 192-IgG-saporin failed to show greater than 90% depletion of cholinergic fibers and were excluded.
Figure 2. 192-IgG-saporin lesions deplete cortical cholinergic innervation.

Representative images of AChE fiber staining in layer V motor cortex of an NB+ control lesioned subject (A) and an NB- 192-IgG-saporin lesioned subject (B). The right-most panel shows a further magnification with white arrowheads marking fiber crossings. Calibrations are 1 mm for main image, and the spacing between gridlines is 50 μm for inset.
Statistics
All data are reported in the main text as mean ± SEM. All comparisons were planned in the experimental design a priori, and significant differences were determined using one-way ANOVA and t-tests where appropriate. Statistical tests for each comparison are noted in the text. Paired t-tests were used to compare performance before and after lesion, and unpaired t-tests were used to compare measures across groups. Alpha level was set at 0.05 for single comparisons, and Bonferroni-corrected to 0.017 for multiple comparisons where applicable. Error bars indicate SEM in all figures, and * denotes p < 0.05.
RESULTS
192-IgG-Saporin lesions deplete cortical cholinergic innervation
Rats were trained to proficiency on an automated lever pressing task that required use of the proximal forelimb (Fig.1) [33]. Once they reached proficiency, rats were randomly assigned to receive either an NB lesion (NB-) or control procedure (NB+). NB lesion subjects received bilateral NB injections of 192-IgG-saporin, a toxin that selectively lesions cortically-projecting cholinergic neurons while leaving other NB neurons intact [44, 45]. Depletion is complete by the seventh day post-injection [46]. Control subjects received bilateral injections of a saporin conjugated with a control antibody, which does not induce cell death. To confirm cholinergic lesion after 192-IgG-Saporin injections, AChE containing fibers were analyzed in motor cortex (Fig. 2A&B). Fiber count analysis indicates that the 192-IgG-Saporin injections caused substantial cholinergic denervation. 192-IgG-saporin resulted in an 96.6 ± 1.0% reduction in cortical AChE fiber staining compared to control injections (Fig. 2C; NB-, n = 8; NB+, n = 9).
Cholinergic depletion prevents VNS-dependent cortical plasticity
After injection, both the NB+ and NB- groups underwent five days of motor training during which successful trials were paired with a burst of VNS. On the day after the final session of VNS paired training, all subjects underwent ICMS mapping of motor cortex. Additionally, ICMS mapping was performed on a cohort of untrained subjects (Naïve, n = 8).
VNS paired with motor training in subjects with an intact NB increases proximal forelimb representation in motor cortex (Fig. 3, maps for all subjects can be found in the Online Supplement). ANOVA on proximal forelimb representation revealed a significant effect (One-way ANOVA, F[2,24] = 9.45, p = 9.39 × 10-4). VNS paired with successful trials on the lever pressing task resulted in a 159% increase in proximal forelimb representation in cholinergically intact rats compared to untrained controls (Fig. 4; NB+: 2.02 ± 0.19 mm2; Naïve: 0.78 ± 0.19 mm2; unpaired t-test, p = 2.21 × 10-4). Depletion of cortical cholinergic projections substantially reduced VNS-dependent map expansion. Rats with cholinergic lesions exhibited a 54% smaller proximal forelimb representation compared to cholinergically intact rats (NB-: 0.94 ± 0.37 mm2; unpaired t-test vs. NB+, p = 0.012) and similar to that observed in untrained controls (unpaired t-test NB- vs. Naïve; p = 0.693). These results indicate cholinergic innervation is necessary for VNS-dependent map plasticity in motor cortex.
Figure 3. Representative ICMS maps.

(A,B) Example motor cortex maps from an untrained control rat. (C,D) Example maps depicting the substantial increase in proximal forelimb representation in rats with an intact NB that received VNS paired with motor training. (E,F) Example maps from rats with an NB lesion that received VNS paired with motor training. Note the similarity to the untrained control map. Each square represents a 0.25 mm2 (0.5 × 0.5 mm) area. Electrode penetrations occurred in the middle of each square. Raw maps from all subjects can be found in the supplementary data.
Figure 4. NB lesions prevent VNS-dependent motor cortex map reorganization.

Total area of multiple movement representations in motor cortex. VNS paired with motor training in rats with an intact NB results in significantly greater proximal forelimb and jaw representations. NB lesions prevent VNS-dependent expansion of movement representations. Other movement representations are unchanged. * indicates p < 0.05 compared to NB+ group for each movement representation.
Jaw representation in the motor cortex was also altered by VNS paired with motor training. ANOVA on jaw representation revealed a significant effect of group (One-way ANOVA, F[2,24] = 9.84, p = 7.56 × 10-4). Post hoc comparison indicated that VNS paired with motor training drove a 234% expansion in jaw representation in the NB+ group compared to NB- group (Fig. 4; NB+: 1.47 ± 0.26 mm2; NB-: 0.44 ± 0.17 mm2; unpaired t-test, p = 4.00 × 10-3) and a 194% expansion compared to untrained controls (Naïve: 0.50 ± 0.20 mm2; unpaired t-test v. NB+, p = 7.50 × 10-3). Jaw representation was similar in the NB- and Naïve groups (unpaired t-test NB- v. Naïve, p = 0.802), suggesting that the expansion of jaw map area is dependent on acetylcholine.
Pairing VNS with training on a lever pressing task did not alter of distal forelimb representations in any group (Fig. 4; One-way ANOVA, F[2,24] = 0.45, p = 0.646). There was also no difference in vibrissa, neck, or hindlimb representation between groups (One-way ANOVA, Vibrissa: F[2,24] = 1.55, p = 0.233; Neck: F[2,24] = 0.01, p = 0.986; Hindlimb: F[2,24] = 0.24, p = 0.790). The total area of motor cortex was not significantly different between groups (One-way ANOVA, F[2,24] = 2.52, p = 0.10). Consistent with previous reports, no differences in stimulation threshold were observed (One-way ANOVA, F[2,24] = 0.36, p = 0.70).
Depletion of cortical acetylcholine does not alter behavioral performance
Behavioral changes could potentially contribute to the observed differences in cortical representations. Prior to lesion or control surgery, there was no difference between groups in hit rate (Fig. 5A; Pre; NB+: 67.7 ± 3.2%, NB-: 71.9 ± 3.7%; unpaired t-test, p = 0.39), number of trials per day (Fig. 5B; Pre; NB+: 398 ± 27 trials, NB-: 374 ± 44 trials; unpaired t-test, p = 0.645), or interpress interval (Fig. 5C; Pre; NB+: 404 ± 39 msec, NB-: 363 ± 41 msec; unpaired t-test, p = 0.49). Consistent with previous studies, NB lesions did not alter task performance group (Fig. 5; Post; Hit Rate: NB+: 64.4 ± 5.6%, paired t-test v. Pre, p = 0.53; NB-: 66.3 ± 5.6%, paired t-test v. Pre, p = 0.14; Trials per Day: NB+: 361 ± 34 trials, paired t-test v. Pre, p = 0.33; NB-: 341 ± 46 trials, paired t-test v. Pre, p = 0.48; Interpress Interval: NB+: 432 ± 54 msec, paired t-test v. Pre, p = 0.43; NB-: 409 ± 54 msec, paired t-test v. Pre, p = 0.24) [47]. No differences in performance were observed across groups after surgery (Post, NB+ v. NB-; unpaired t-test, Hit Rate: p = 0.81; Trials per Day: p = 0.72; Interpress Interval: p = 0.77). Additionally, the number of stimulations received in each group was similar (Fig. 5D; NB+: 1084 ± 65; NB-: 1009 ± 96, unpaired t-test, p = 0.52). These results indicate that differences in task performance and amount of VNS cannot account for the observed differences in cortical representations.
Figure 5. NB lesions do not change forelimb performance.

No differences in forelimb performance measures, including hit rate (A), total number of trials per day (B), or speed of lever presses (C), were observed between groups before or after NB lesion. Additionally, both groups received a similar number of VNS stimulations (D).
DISCUSSION
Our previous study demonstrated that VNS paired with forelimb training enhanced map reorganization in the motor cortex [5]. The cholinergic nucleus basalis is a key substrate in training-dependent motor cortex map reorganization [36, 47, 48], and several lines of evidence suggest that VNS activates cholinergic circuitry in the basal forebrain [20, 29]. In this study, we examined whether the NB is required for the plasticity enhancing effects of VNS paired with motor training. VNS paired with motor training increased the proximal forelimb representation area in the motor cortex in rats with an intact NB. NB lesions prevent VNS-dependent expansion of proximal forelimb representation, demonstrating that the NB is required for the plasticity enhancing effects of VNS. No differences in forelimb performance were observed in either NB+ or NB- groups before or after lesion, excluding the possibility that differences in map representation may arise from differences in behavior. Together, the findings from this study demonstrate that the NB is required for VNS-dependent enhancement of plasticity in motor cortex.
VNS paired with training on a lever task in rats with an intact NB results in a larger representation of the proximal forelimb in the motor cortex, replicating the findings of a previous study [5]. Area corresponding to proximal forelimb is increased without altering the representation of the distal forelimb. This likely reflects the greater dependence on the proximal musculature compared to distal musculature to perform this lever pressing task [33]. Our previous study using a similar design demonstrated that lever training without VNS did not result in a measurable increase in proximal forelimb representation compared to untrained controls, indicating the importance of VNS paired with training to drive increased proximal representations [5]. The current study does not incorporate this lever-trained control group that did not receive VNS; therefore, while unlikely, we cannot directly rule out the possibility that proximal forelimb expansion was a result of training alone. NB lesions prevented VNS-dependent expansion of proximal forelimb representations. The lesion method employed in this study using 192-IgG-saporin selectively lesions cortically-projecting cholinergic neurons and leaves surrounding neurons intact [44, 45], resulting in a specific depletion of cortical cholinergic innervation in the NB- group. This suggests that the cortical cholinergic innervation from the NB is required for VNS-dependent enhancement of plasticity, and that other neuromodulatory systems engaged by VNS cannot substitute for the loss of cholinergic input.
The proximal forelimb representations observed in this study are slightly smaller than those reported in some previous studies [49-51]. Differences in representational area observed across studies could arise from a variety of sources. The lower frequency stimulation train used to evoke movement during ICMS in the present study could in part account for disparity in proximal forelimb area. Additionally, differences in the amount of time between the beginning of behavioral training and ICMS and the specific behavioral training paradigm used may contribute. The proximal representations observed in this study are similar those reported in our previous study using the same design, suggesting the experimental conditions specified in this design yield consistent representations [5].
Map expansion in rats with an intact NB was not totally restricted to proximal forelimb area. We observed a significant increase in the representation of the jaw in rats with an intact NB that received VNS. As the timing between an event and VNS pairing is critical for map reorganization [2], the increase in jaw representation may arise from the fact that rats would receive VNS during a lever press and immediately afterwards (<2 seconds, on average) eat a reward pellet. It is likely that the close temporal approximation of chewing the pellet and VNS was sufficient to enhance the representation of the jaw. The absence of an increase in jaw area in rats with NB lesions provides further support that cholinergic innervation is required for VNS-dependent map expansion.
The absence of differences in behavioral performance between the NB+ and NB- groups before and after VNS in the present study excludes the possibility that alterations in behavior arising from NB lesion or imbalanced experimental groups could account for differences in map plasticity. Lesion of the NB did not affect performance of the trained task, as demonstrated previously [34, 47]. Moreover, VNS paired with training did not enhance or impair task performance. Map reorganization despite an absence of change in motor performance is consistent with the notion that map plasticity may support learning but is unnecessary in the performance of a learned task [52-54]. It remains to be determined whether pairing VNS with motor training during early stages of acquisition of a motor task could speed learning. Future studies should evaluate the role of VNS-dependent enhancement of map plasticity in the context of known behavioral changes, such as improvement of motor recovery after brain injury driven by VNS paired with rehabilitative training [8-12].
The attenuation of map reorganization resulting from NB lesions observed in this study suggests that cortical cholinergic innervation is required for VNS-dependent enhancement of plasticity. However, these findings do not rule out the importance of other neuromodulatory systems in VNS-dependent plasticity. Several previous studies have indicated that the noradrenergic system is involved in the effects of VNS [23-28, 55]. Norepinephrine and acetylcholine often act synergistically to influence plasticity [32]; therefore, it is possible that both the noradrenergic and cholinergic systems contribute to VNS-dependent plasticity. Delineation of the complex interaction of neuromodulatory pathways engaged by VNS may be required to understand the effect of VNS therapies.
The neuronal mechanisms engaged by VNS to promote plasticity are unknown. VNS and acetylcholine have been implicated in a variety of synaptic changes associated with cortical map reorganization [56]. LTP of connections in motor cortex is believed to underlie in part the increase in representational area resulting from training dependent plasticity [57, 58]. Several studies report that VNS enhances the induction of LTP [59-61]. Similarly, activation of muscarinic acetylcholine receptors facilitates induction of LTP [62, 63]. While these studies are restricted to hippocampus, it is possible that similar mechanisms are activated in the cortex in response to VNS. This convergent control of LTP may in part underlie VNS-dependent enhancement of map plasticity.
Based on the robust, specific enhancement of plasticity driven by VNS, a number of targeted plasticity therapies using VNS paired with rehabilitative training have been developed to support recovery after neurological injury and disease. VNS paired with rehabilitative regimens significantly improves recovery in animal models of chronic tinnitus, ischemic stroke, intracerebral hemorrhage, and traumatic brain injury [2, 8-12]. Moreover, pilot trials evaluating VNS therapies in patients have demonstrated reduced handicap in chronic tinnitus patients [64-66]. As these therapies are translated to the broader clinical population in larger trials, it is critical to identify conditions that interfere with the efficacy of VNS. The cholinergic system is affected by a number of common pharmaceuticals and pathologies. As such, cholinergic transmission will likely be at least partially compromised in many patients, which may consequently occlude the benefits of VNS therapy. Indeed, in a pilot study evaluating VNS therapy for chronic tinnitus, a subset of patients were taking drugs that in part altered cholinergic and noradrenergic transmission. Patients on the drugs failed to improve, while those who were not on drugs demonstrated a significant reduction of tinnitus intensity and distress [66]. While more testing is required to provide a direct demonstration, these findings suggest that alterations of neuromodulatory transmission may occlude the effect of VNS therapy. The absence of VNS-dependent plasticity after NB lesion in this study suggests that further studies should evaluate whether cholinergic lesions prevent the benefits of VNS therapy in models of neurological disease. The delineation of the neuromodulatory pathways engaged by VNS therapy will provide insight into the mechanisms that underlie the benefits of VNS therapy and is critical to the successful translation of VNS therapy.
Supplementary Material
Highlights.
Vagus nerve stimulation (VNS) paired with forelimb training drives reorganization of movement representations in motor cortex
Lesions of cortical cholinergic projections from the nucleus basalis prevent this VNS-dependent enhancement of plasticity
Differences in forelimb use cannot account for the observed differences in map plasticity
Acknowledgments
We would like to thank Andrew Sloan for engineering support and Reema Casavant for assistance with surgeries. We thank Michael Borland, Aisha Khan, John Buell, and Mark Lane for help with electronics construction. Additionally, we thank Elizabeth Nutting, Xavier Carrier, Meera Iyengar, and Virginia Land for assistance with behavioral testing and ICMS.
SOURCES OF FUNDING
This project was supported in part by funding from the Defense Advanced Research Projects Agency, NIH NINDS R01 NS085167, NIH NIDCD R01 DC010433, and the Texas Biomedical Device Center.
Footnotes
DISCLOSURES
MPK is a consultant for and has a financial interest in MicroTransponder, Inc., a company which is developing VNS-based therapies. RLR owns Vulintus, Inc.
Other authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hays SA, Rennaker RL, II, Kilgard MP. Targeting plasticity with vagus nerve stimulation to treat neurological disease. Prog Brain Res. 2013;207:275–299. doi: 10.1016/B978-0-444-63327-9.00010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engineer ND, Riley JR, Seale JD, Vrana WA, Shetake JA, Sudanagunta SP, Borland MS, Kilgard MP. Reversing pathological neural activity using targeted plasticity. Nature. 2011;470:101–104. doi: 10.1038/nature09656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shetake JA, Engineer ND, Vrana WA, Wolf JT, Kilgard MP. Pairing tone trains with vagus nerve stimulation induces temporal plasticity in auditory cortex. Exp Neurol. 2011;233:342–49. doi: 10.1016/j.expneurol.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Engineer CT, Engineer ND, Riley JR, Seale JD, Kilgard MP. Pairing Speech Sounds With Vagus Nerve Stimulation Drives Stimulus-specific Cortical Plasticity. Brain Stimulation. 2015 doi: 10.1016/j.brs.2015.01.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter BA, Khodaparast N, Fayyaz T, Cheung RJ, Ahmed SS, Vrana WA, Rennaker RL, II, Kilgard MP. Repeatedly Pairing Vagus Nerve Stimulation with a Movement Reorganizes Primary Motor Cortex. Cerebral Cortex. 2011;22:2365–2374. doi: 10.1093/cercor/bhr316. [DOI] [PubMed] [Google Scholar]
- 6.De Ridder D, Vanneste S, Engineer ND, Kilgard MP. Safety and Efficacy of Vagus Nerve Stimulation Paired With Tones for the Treatment of Tinnitus: A Case Series. Neuromodulation: Technology at the Neural Interface. 2013 doi: 10.1111/ner.12127. [DOI] [PubMed] [Google Scholar]
- 7.De Ridder D, Kilgard M, Engineer N, Vanneste S. Placebo-Controlled Vagus Nerve Stimulation Paired with Tones in a Patient with Refractory Tinnitus: A Case Report. Otol Neurotol. 2015 doi: 10.1097/MAO.0000000000000704. [DOI] [PubMed] [Google Scholar]
- 8.Khodaparast N, Hays SA, Sloan AM, Hulsey DR, Ruiz A, Pantoja M, Rennaker RL, II, Kilgard MP. Vagus nerve stimulation during rehabilitative training improves forelimb strength following ischemic stroke. Neurobiol Dis. 2013;60:80–88. doi: 10.1016/j.nbd.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Khodaparast N, Hays SA, Sloan AM, Fayyaz T, Hulsey DR, Rennaker RL, II, Kilgard MP. Vagus nerve stimulation delivered during motor rehabilitation improves recovery in a rat model of stroke. Neurorehabil Neural Repair. 2014;28:698–706. doi: 10.1177/1545968314521006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hays SA, Khodaparast N, Ruiz A, Sloan AM, Hulsey DR, Rennaker RL, Kilgard MP. The timing and amount of vagus nerve stimulation during rehabilitative training affect post-stroke recovery of forelimb strength. NeuroReport. 2014;25(9):676–682. doi: 10.1097/WNR.0000000000000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hays SA, Khodaparast N, Hulsey DR, Ruiz A, Sloan AM, Rennaker RL, II, Kilgard MP. Vagus Nerve Stimulation during Rehabilitative Training Improves Functional Recovery after Intracerebral Hemorrhage. Stroke. 2014;45:10–3097. 3100. doi: 10.1161/STROKEAHA.114.006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pruitt D, Schmid A, Kim L, Abe C, Trieu J, Choua C, Hays S, Kilgard M, Rennaker RL., II Vagus nerve stimulation delivered with motor training enhances recovery of function after traumatic brain injury. J Neurotrauma. 2015 doi: 10.1089/neu.2015.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Microtransponder. ClinicalTrials gov. Bethesda (MD): National Library of Medicine (US); 2014. Paired Vagus Nerve Stimulation (VNS) With Rehabilitation for Upper Limb Function Improvement After Stroke. NCT01669161: https://clinicaltrials.gov/ct2/show/NCT01669161. [Google Scholar]
- 14.Microtransponder. ClinicalTrials gov. Bethesda (MD): National Library of Medicine (US); 2014. VNS During Rehabilitation for Improved Upper Limb Motor Function After Stroke. NCT02243020: https://clinicaltrials.gov/ct2/show/study/NCT02243020. [Google Scholar]
- 15.Morris GL, Mueller WM. Long-term treatment with vagus nerve stimulation in patients with refractory epilepsy. Neurology. 1999;53:1731–1731. doi: 10.1212/wnl.53.8.1731. [DOI] [PubMed] [Google Scholar]
- 16.George MS, Sackeim HA, Rush AJ, Marangell LB, Nahas Z, Husain MM, Lisanby S, Burt T, Goldman J, Ballenger JC. Vagus nerve stimulation: a new tool for brain research and therapy. Biol Psychiatry. 2000;47:287–295. doi: 10.1016/s0006-3223(99)00308-x. [DOI] [PubMed] [Google Scholar]
- 17.Sackeim HA, Rush AJ, George MS, Marangell LB, Husain MM, Nahas Z, Johnson CR, Seidman S, Giller C, Haines S. Vagus nerve stimulation (VNS™) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology. 2001;25:713–728. doi: 10.1016/S0893-133X(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 18.Englot DJ, Chang EF, Auguste KI. Vagus nerve stimulation for epilepsy: a meta-analysis of efficacy and predictors of response: A review. J Neurosurg. 2011;115:1248–1255. doi: 10.3171/2011.7.JNS11977. [DOI] [PubMed] [Google Scholar]
- 19.Groves DA, Brown VJ. Vagal nerve stimulation: a review of its applications and potential mechanisms that mediate its clinical effects. Neuroscience & Biobehavioral Reviews. 2005;29:493–500. doi: 10.1016/j.neubiorev.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Detari L, Juhasz G, Kukorelli T. Effect of stimulation of vagal and radial nerves on neuronal activity in the basal forebrain area of anaesthetized cats. Acta Physiol Hung. 1983;61:147–154. [PubMed] [Google Scholar]
- 21.Groves DA, Bowman EM, Brown VJ. Recordings from the rat locus coeruleus during acute vagal nerve stimulation in the anaesthetised rat. Neurosci Lett. 2005;379:174–179. doi: 10.1016/j.neulet.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 22.Manta S, Dong J, Debonnel G, Blier P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psy Neurosci. 2009;34:272. [PMC free article] [PubMed] [Google Scholar]
- 23.Hassert D, Miyashita T, Williams C. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity on norepinephrine output in the basolateral amygdala. Behav Neurosci. 2004;118:79. doi: 10.1037/0735-7044.118.1.79. [DOI] [PubMed] [Google Scholar]
- 24.Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. 2006;1119:124–132. doi: 10.1016/j.brainres.2006.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L, Puligheddu M, Marrosu F, Biggio G. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007;1179:28–34. doi: 10.1016/j.brainres.2007.08.045. [DOI] [PubMed] [Google Scholar]
- 26.Landau AM, Dyve S, Jakobsen S, Alstrup AK, Gjedde A, Doudet DJ. Acute Vagal Nerve Stimulation Lowers α2 Adrenoceptor Availability: Possible Mechanism of Therapeutic Action. Brain Stimulation. 2015 doi: 10.1016/j.brs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Krahl SE, Clark KB, Smith DC, Browning RA. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39:709–714. doi: 10.1111/j.1528-1157.1998.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 28.Grimonprez A, Raedt R, Portelli J, Dauwe I, Larsen LE, Bouckaert C, Delbeke J, Carrette E, Meurs A, De Herdt V. The antidepressant-like effect of vagus nerve stimulation is mediated through the locus coeruleus. J Psychiatr Res. 2015 doi: 10.1016/j.jpsychires.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Nichols J, Nichols A, Smirnakis S, Engineer N, Kilgard M, Atzori M. Vagus nerve stimulation modulates cortical synchrony and excitability through the activation of muscarinic receptors. Neuroscience. 2011;189:207–214. doi: 10.1016/j.neuroscience.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 30.Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience. 2002;111:815–835. doi: 10.1016/s0306-4522(02)00026-x. [DOI] [PubMed] [Google Scholar]
- 31.Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and Noradrenaline. Nature. 1986:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- 32.Seol GH, Ziburkus J, Huang SY, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hays SA, Khodaparast N, Sloan AM, Fayyaz T, Hulsey DR, Ruiz AD, Pantoja M, Kilgard MP, Rennaker RL., II The bradykinesia assessment task: an automated method to measure forelimb speed in rodents. J Neurosci Methods. 2013;214:52–61. doi: 10.1016/j.jneumeth.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Conner JM, Chiba AA, Tuszynski MH. The basal forebrain cholinergic system is essential for cortical plasticity and functional recovery following brain injury. Neuron. 2005;46:173–179. doi: 10.1016/j.neuron.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 35.Ramanathan D, Tuszynski MH, Conner JM. The basal forebrain cholinergic system is required specifically for behaviorally mediated cortical map plasticity. J Neurosci. 2009;29:5992–6000. doi: 10.1523/JNEUROSCI.0230-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conner J, Kulczycki M, Tuszynski M. Unique contributions of distinct cholinergic projections to motor cortical plasticity and learning. Cerebral Cortex. 2010;20:2739–2748. doi: 10.1093/cercor/bhq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleim JA, Bruneau R, Calder K, Pocock D, VandenBerg PM, MacDonald E, Monfils MH, Sutherland RJ, Nader K. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron. 2003;40:167–176. doi: 10.1016/s0896-6273(03)00592-0. [DOI] [PubMed] [Google Scholar]
- 38.Neafsey E, Sievert C. A second forelimb motor area exists in rat frontal cortex. Brain Res. 1982;232:151–156. doi: 10.1016/0006-8993(82)90617-5. [DOI] [PubMed] [Google Scholar]
- 39.Neafsey E, Bold E, Haas G, Hurley-Gius K, Quirk G, Sievert C, Terreberry R. The organization of the rat motor cortex: a microstimulation mapping study. Brain Res Rev. 1986;11:77–96. doi: 10.1016/s0006-8993(86)80191-3. [DOI] [PubMed] [Google Scholar]
- 40.Gioanni Y, Lamarche M. A reappraisal of rat motor cortex organization by intracortical microstimulation. Brain Res. 1985;344:49–61. doi: 10.1016/0006-8993(85)91188-6. [DOI] [PubMed] [Google Scholar]
- 41.Di Patre PL, Mathes CW, Butcher LL. Differential visualization of cholinesterasic neuronal somata and fibers by use of modifications of acetylcholinesterase pharmacohistochemistry. J Histochem Cytochem. 1993;41:129–135. doi: 10.1177/41.1.7678024. [DOI] [PubMed] [Google Scholar]
- 42.Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb Cortex. 1996;6:165–177. doi: 10.1093/cercor/6.2.165. [DOI] [PubMed] [Google Scholar]
- 43.Conner JM, Darracq MA, Roberts J, Tuszynski MH. Nontropic actions of neurotrophins: subcortical nerve growth factor gene delivery reverses age-related degeneration of primate cortical cholinergic innervation. PNAS. 2001;98:1941–1946. doi: 10.1073/pnas.98.4.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berger-Sweeney J, Heckers S, Mesulam MM, Wiley RG, Lappi DA, Sharma M. Differential effects on spatial navigation of immunotoxin-induced cholinergic lesions of the medial septal area and nucleus basalis magnocellularis. J Neurosci. 1994;14:4507–4519. doi: 10.1523/JNEUROSCI.14-07-04507.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiba AA, Bucci DJ, Holland PC, Gallagher M. Basal forebrain cholinergic lesions disrupt increments but not decrements in conditioned stimulus processing. J Neurosci. 1995;15:7315–7322. doi: 10.1523/JNEUROSCI.15-11-07315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waite JJ, Wardlow ML, Chen AC, Lappi DA, Wiley RG, Thal LJ. Time course of cholinergic and monoaminergic changes in rat brain after immunolesioning with 192 IgG-saporin. Neurosci Lett. 1994;169:154–158. doi: 10.1016/0304-3940(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 47.Conner JM, Culberson A, Packowski C, Chiba AA, Tuszynski MH. Lesions of the basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron. 2003;38:819–829. doi: 10.1016/s0896-6273(03)00288-5. [DOI] [PubMed] [Google Scholar]
- 48.Ramanathan D, Tuszynski MH, Conner JM. The basal forebrain cholinergic system is required specifically for behaviorally mediated cortical map plasticity. The Journal of Neuroscience. 2009;29:5992–6000. doi: 10.1523/JNEUROSCI.0230-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleim JA, Barbay S, Nudo RJ. Functional reorganization of the rat motor cortex following motor skill learning. J Neurophysiol. 1998;80:3321–3325. doi: 10.1152/jn.1998.80.6.3321. [DOI] [PubMed] [Google Scholar]
- 50.Kleim JA, Cooper NR, VandenBerg PM. Exercise induces angiogenesis but does not alter movement representations within rat motor cortex. Brain Res. 2002;934:1–6. doi: 10.1016/s0006-8993(02)02239-4. [DOI] [PubMed] [Google Scholar]
- 51.Vanden Berg PM, Hogg M, Kleim A, Whishaw IQ. Long–Evans rats have a larger cortical topographic representation of movement than Fischer-344 rats: A microstimulation study of motor cortex in naïve and skilled reaching-trained rats. Brain Res Bull. 2002;59:197–203. doi: 10.1016/s0361-9230(02)00865-1. [DOI] [PubMed] [Google Scholar]
- 52.Molina-Luna K, Hertler B, Buitrago MM, Luft AR. Motor learning transiently changes cortical somatotopy. Neuroimage. 2008;40:1748–1754. doi: 10.1016/j.neuroimage.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 53.Reed A, Riley J, Carraway R, Carrasco A, Perez C, Jakkamsetti V, Kilgard MP. Cortical map plasticity improves learning but is not necessary for improved performance. Neuron. 2011;70:121–131. doi: 10.1016/j.neuron.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 54.Kilgard MP. Harnessing plasticity to understand learning and treat disease. Trends Neurosci. 2012;35:715–22. doi: 10.1016/j.tins.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raedt R, Clinckers R, Mollet L, Vonck K, El Tahry R, Wyckhuys T, De Herdt V, Carrette E, Wadman W, Michotte Y. Increased hippocampal noradrenaline is a biomarker for efficacy of vagus nerve stimulation in a limbic seizure model. J Neurochem. 2011;117:461–469. doi: 10.1111/j.1471-4159.2011.07214.x. [DOI] [PubMed] [Google Scholar]
- 56.Buonomano DV, Merzenich MM. Cortical plasticity: from synapses to maps. Annu Rev Neurosci. 1998;21:149–186. doi: 10.1146/annurev.neuro.21.1.149. [DOI] [PubMed] [Google Scholar]
- 57.Rioult-Pedotti MS, Friedman D, Hess G, Donoghue JP. Strengthening of horizontal cortical connections following skill learning. Nat Neurosci. 1998;1:230–234. doi: 10.1038/678. [DOI] [PubMed] [Google Scholar]
- 58.Rioult-Pedotti M, Friedman D, Donoghue JP. Learning-Induced LTP in Neocortex. Science. 2000;290:533–536. doi: 10.1126/science.290.5491.533. [DOI] [PubMed] [Google Scholar]
- 59.Zuo Y, Smith DC, Jensen RA. Vagus nerve stimulation potentiates hippocampal LTP in freely-moving rats. Physiol Behav. 2007;90:583–589. doi: 10.1016/j.physbeh.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen H, Fuchino Y, Miyamoto D, Nomura H, Matsuki N. Vagus nerve stimulation enhances perforant path-CA3 synaptic transmission via the activation of β-adrenergic receptors and the locus coeruleus. The International Journal of Neuropsychopharmacology. 2012;15:523–530. doi: 10.1017/S1461145711000708. [DOI] [PubMed] [Google Scholar]
- 61.Ura H, Sugaya Y, Ohata H, Takumi I, Sadamoto K, Shibasaki T, Maru E. Vagus nerve stimulation induced long-lasting enhancement of synaptic transmission and decreased granule cell discharge in the hippocampal dentate gyrus of urethane-anesthetized rats. Brain Res. 2012 doi: 10.1016/j.brainres.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 62.Burgard EC, Sarvey JM. Muscarinic receptor activation facilitates the induction of long-term potentiation (LTP) in the rat dentate gyrus. Neurosci Lett. 1990;116:34–39. doi: 10.1016/0304-3940(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 63.Shinoe T, Matsui M, Taketo MM, Manabe T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci. 2005;25:11194–11200. doi: 10.1523/JNEUROSCI.2338-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Microtransponder. ClinicalTrials gov. Bethesda (MD): National Library of Medicine (US); 2010. Proof-of-Concept Study Assessing VNS Paired with Tones for Tinnitus. NCT01253616: Available from: http://clinicaltrials.gov/ct2/show/NCT01253616. [Google Scholar]
- 65.Microtransponder. ClinicalTrials gov. Bethesda (MD): National Library of Medicine (US); 2013. Vagus Nerve Stimulation (VNS) Paired with Tones for Tinnitus. NCT01962558: http://clinicaltrials.gov/ct2/show/NCT01962558. [Google Scholar]
- 66.De Ridder D, Vanneste S, Engineer ND, Kilgard MP. Safety and Efficacy of Vagus Nerve Stimulation Paired With Tones for the Treatment of Tinnitus: A Case Series. Neuromodulation: Technology at the Neural Interface. 2013 doi: 10.1111/ner.12127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.