

Abstract
Acetylation of lysine residues is an important post-translational protein modification. Lysine acetylation in histones and its crosstalk with other post-translational modifications in histone and non-histone proteins are crucial to DNA replication, DNA repair, and transcriptional regulation. We incorporated acetyl-lysine (AcK) and the non-hydrolyzable thioacetyl-lysine (ThioAcK) into full-length proteins in vitro, mediated by flexizyme. ThioAcK and AcK were site-specifically incorporated at different lysine positions into human histone H3 either individually or in pairs. We demonstrate that the thioacetyl group in histone H3 could not be removed by the histone deacetylase sirtuin type 1. This method provides a powerful tool to study protein acetylation and its role in crosstalk between posttranslational modifications.
Keywords: lysine acetylation, histone, thioacetyl-lysine, Flexizyme, post-translational modifications
Graphical abstract
Scheme represents the NH2 and COOH termini of the core histone, and its residue-specific epigenetic modifications.
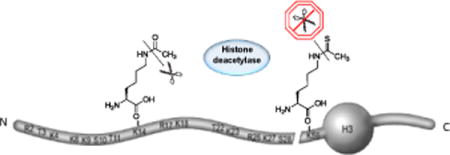
Posttranslational modifications (PTMs) of the N-terminal “tails” of histones serve as l epigenetic indicators of chromatin structure associated with genome regulation, DNA repair, and transcriptional silencing[1]. Lysine acetylation of histones is correlated with dynamic transitions in chromatin structure and function, transcriptional activation, and DNA replication[1a, 1c]. Crosstalk between lysine acetylation of histones and other PTMs is crucial in chromatin-based control and shaping inheritable epigenetic programs[1f, 2]. As histone acetyltransferases (HATs) and histone deacetylases (HDACs) modify several lysine residues and multiple enzymes target common sites in histones, it is thought that function and regulation of histone acetylation is obtained through the cumulative effects of HATs and HDACs[3]. Studies on the role of PTMs use non-hydrolysable analogs, which are novel tools in the analysis of enzymes that modulate modification in vivo. Nε-thioacetyl-lysine is a single-atom substituted acetyl-lysine derivative with the most similar chemical properties to AcK among the known analogs[4]. To date, solid-phase peptide synthesis was used to incorporate ThioAcK into short peptides derived p53 and α-tubulin. Such peptides were effective human sirtuin inhibitors and served as multifunctional probes owing to the high stability of ThioAcK[4–5]. However, ThioAcK has not been incorporated into a full-length protein, limiting its use in studies of histone modification.
Incorporation of non-canonical amino acids (ncAAs) into peptides and proteins was achieved by various methods, including protein semi-synthesis combined with chemical ligation, and by genetic code expansion using orthogonal aminoacyl-tRNA synthetase (aaRS)•tRNA pairs[6]. A combination of two pairs, pyrrolysyl-tRNA synthetase•tRNAPyl and M. jannaschii tyrosyl-tRNA synthetase•tRNATyr allowed the suppression of UAG and UGA codons simultaneously, while suppression of quadruplet codons was also utilized to incorporate different ncAAs into a single protein[7].
Acylation of tRNAs with ncAAs can be performed by orthogonal aaRS•tRNA pairs or through chemical aminoacylation of a dinucleotide and subsequent ligation to the tRNA body; however, both require multistep experiments involving substantial labor and materials[8]. An attractive, general method to form aminoacyl-tRNA was developed by Suga and collaborators[9]; a catalytic RNA (Flexizyme) recognizes and aminoacylates tRNA using an amino acid cyanomethyl ester with an aromatic side-chain as the amino acid donor. Further improvement led to dinitro-flexizyme (dFx), which utilizes amino acid 3,5-dinitrobenzyl esters (DBE) as the aminoacyl-donor without dependence on the amino acid side-chain[9a]. Flexizyme was used to produce N-terminal peptides of histone H3 containing methyl-lysine and acetyl-lysine residues; however, to date this has not been reported for full-length histone proteins[9b].
To generate full-length H3 histones containing the non-deacylatable ThioAcK, we used dFx to acylate tRNA with the 3,5-dinitrobenzyl esters of Nε-acetyl-lysine (AcK-DBE, 3), Nε-thioacetyl-lysine (ThioAcK-DBE, 4), and Nε-selenoacetyl lysine (SeAcK-DBE, 5) (Figure 1). SeAcK was synthesized to show the effectiveness and general usefulness of our methods as AcK, ThioAck, and SeAck contain oxygen, sulfur, and selenium respectively; all of them belong to the same group of elements in the periodic table. The novel compounds ThioAcK-DBE and SeAcK-DBE were synthesized and further characterized by 1H-NMR, 13C-NMR and HPLC-MS (Supporting Information (SI)). As controls for dFx aminoacylation, Val-DBE (1), Tyr-DBE (2) and AcK-DBE (3) were synthesized as previously described with minor changes[9a].
Figure 1.
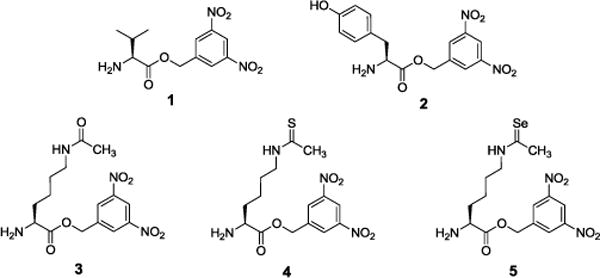
Structures of valine 3,5-dinitrobenzyl ester (Val-DBE, 1), tyrosine 3,5-dinitrobenzyl ester (Tyr-DBE, 2), acetyl-lysine 3,5-dinitrobenzyl ester (AcK-DBE, 3), thioacetyl-lysine 3,5-dinitrobenzyl ester (ThioAcK-DBE, 4), and selenoacetyl-lysine 3,5-dinitrobenzyl ester (SeAcK-DBE, 5).
The flexizyme and its derivatives recognize the 3′ end of tRNA, using the RCCA-3′ on the tRNA, where R is G or A at position 73. To match the 3′-end of dFx with ACCA-3′ in the dFx-based acylation system with our tRNA, we designed a stable U73A-tRNASepCUA derived from tRNASepCUA (UCCA-3′ end) by mutating the nucleotide in position 73 (Figure S1). We tested aminoacylation efficiency of tRNASepCUA or the U73A-tRNASepCUA variant with compounds 1 and 2 with the dFx system and all combinations showed charging in the 36–45% range (Figure S2). As previous investigations using the flexizyme system had utilized tRNAAsn, tRNAfMetE and Mj-tRNATyrCUA, we tested the aminoacylation efficiency of the dFx system using these tRNAs as controls to ensure aminoacylation activity with our synthesized amino acid benzyl esters (Figure S2).
To investigate the effect of altering anticodons on dFx activity, the U73A-tRNASepCUA anticodon was mutated to match UAA and UGA as well as quadruplet codons in coding sequences. All combinations of tRNASep mutants and amino acids resulted in significant charging, with efficiencies in the 26–38% range (Figure S2). Using the PURExpress (NEB) in vitro translation system lacking release factor 1 (PURE ΔRF1), we tested the ability of dFx aminoacylated U73A-tRNASepCUA to direct incorporation of AcK, ThioAcK, and SeAcK into proteins by UAG suppression. Suppression of UAG at position 151 of superfolder-GFP (GFP-Y151TAG) resulted in incorporation efficiencies of 15% for Val, 32% for Tyr, 41% for Ack, 37% for ThioAcK, and 26% for SeAcK compared to wild-type sfGFP when measured by fluorescence intensity (Figure S3–5 and Table S1–2).
To study the incorporation of AcK and ThioAcK modifications in histone proteins, we directed AcK, ThioAcK, and SeAcK to positions K9 and K56 in the human histone H3 protein by UAG suppression with dFx aminoacylated U73A-tRNASepCUA in the PURE ΔRF1 system. Detection of incorporation by site-specific anti-acetyl histone H3K9 and H3K56 antibodies showed high efficiency of ThioAcK incorporation at K9, and K56 (Figure 2A). Further experiments showed that AcK and ThioAcK have similar intensities against AcK-specific antibodies (Figure S14). Incorporation of AcK and ThioAcK at position K56 was confirmed by MS/MS (Figure 2B and 2C). However, we were unable to detect peptides from H3 that contained AcK or ThioAcK at position 9. Position 9 is in a lysine rich sequence that gives rise to short, polar peptides after trypsin digestion; such peptides would not be detectable by MS[6b]. Furthermore, we did not detect peptides containing SeAcK, but found that the corresponding SeAcK modifications were chemically changed into AcK. While we found good incorporation of ThioAcK in response to UAG and UGA codons, incorporation of ThioAcK at position K56 in response to the UAA codon was unsuccessful (Figure 2A). This is in agreement with incorporation of ThioAcK at other positions, which shows incorporation at UAA codons to be context dependent and weak overall.
Figure 2.
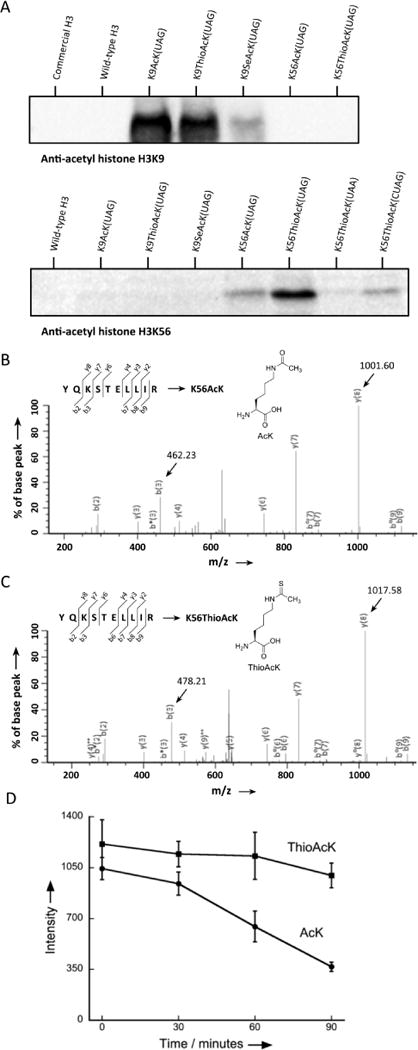
Incorporation of ncAAs into human histone H3 (A) western blots of H3K9AcK and H3K56AcK variants with anti-histone H3K9AcK and H3K56AcK antibodies. 2.5 μl of the corresponding PURE reaction, or 1 μg of commercial H3, was loaded into each lane. MS/MS spectrum of the (B) H3K56AcK(YQ(AcK)STELLIR) and (C)H3K56ThioAcK(YQ(ThioAcK)STELLIR) tryptic peptides. (D) HDAC assays performed on histone H3K56AcK and H3K56ThioAcK. Deacetylation and de(thio)-acetylation reactions were quantified by western blots of histone H3 with anti-acetyl histone H3K56 antibody. Non-enzymatic reactions were used as time zero. Data points are the average of three experiments, with error bars representing ±1 SD.
Peptides prepared by SPPS that contain ThioAcK have been used as a mimetic of p53 in a HDAC assay, inhibiting human SIRT1-catalyzed deacetylation[4, 10]; however, few HDAC studies have been successfully performed directly on full-length proteins containing ncAAs due to the difficulty in synthesis of full-length proteins. Furthermore, although chemically modified peptides have been used as mimetic histones, so far only weak interactions have been detected between PTM and PTM reading domains[11]. Having incorporated both AcK and ThioAcK into the H3 protein, we sought to examine the ability of HDACs to act on AcK residues in full-length proteins. Here we chose human SIRT1, a class III enzyme that catalyzes the NAD+ dependent conversation of acetyl-lysine residues. We compared the activity of SIRT1 on full-length H3 containing the ThioAcK at position K56 (K56ThioAcK) and with that of H3 containing AcK at position K56 (K56AcK). Analysis of a 90-minute time course at 37°C showed that treatment with SIRT1 resulted in a 70% loss in AcK at position K56, while SIRT1 treatment of K56ThioAcK containing H3 resulted in insignificant loss in histone K56ThioAcK (Figure 2D). These results demonstrate that the new K56ThioAcK modified H3 at position K56 afforded H3 the ability to resist SIRT1 deacetylation.
A comparison of the effects of directing ThioAcK insertion into H3 with differing stop codons and positions (K9, K14, K36, K56) was undertaken and western blots performed with a histone H3 antibody. ThioAcK was most efficiently incorporated in response to an UAG codon at position K14 and an UGA codon at K56 (Figure 3A, S7 and S8). Further validation of AcK and ThioAcK incorporation was performed by western blotting with position-specific anti-AcK histone H3K14 and H3K56 antibodies (Figure 3A and S9). To incorporate simultaneously AcKs or ThioAcKs into H3 we tested insertion of AcK and ThioAcK at positions K14/K56 and K36/K56. We found that incorporation of the same ncAA into multiple positions of a single protein was highest when using UGA codons as opposed to UAG codons (Figure 3 and Figure S7). Analysis by MS/MS showed the correct multiple incorporation of ThioAcK at K36/K56 or K14/K56 (Figure S12–13 and Table S5–6).
Figure 3.
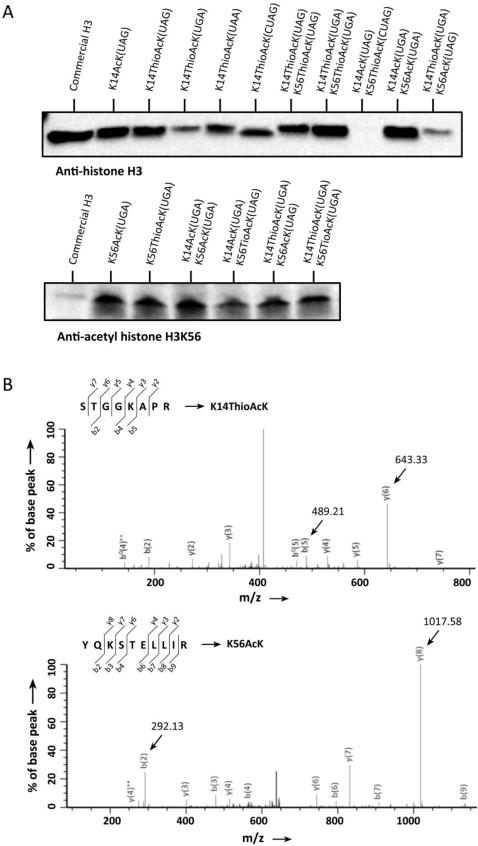
Incorporation of single or multiple ThioAcK residues into histone H3K14 or H3K14/K56. (A) Western blots of single or multiple ThioAcKs on H3 with anti-histone H3 antibodies. 5 μl of the corresponding PURE reaction was loaded into each lane. Lanes 1 and 12, commercial histone H3 (1 μg) as antibiody control. (B) MS/MS spectra of two tryptic peptides STGG(ThioAcK)APR at the site of K14 and YQ(AcK)STELLIR at K56 of the K14ThioAcK/K56AcK digestion.
After successful incorporation of multiple AcKs and ThioAcKs into full-length H3, we attempted to simultaneously incorporate both AcK and ThioAcK. Because insertion efficiency at K14 was higher than that at K36 (Figure S7), we chose K14/K56 in histone H3 to incorporate AcK and ThioAcK simultaneously by a combination of UAG and UGA codon suppression (Figure 3A). We randomized stop codons and positions for AcK/ThioAcK incorporation into full-length histone H3: ThioAcK with UGA at position K14 and AcK with UAG at position K56 (K14ThioAcK(UGA)/K56AcK(UAG)), AcK with UGA at position K14 and ThioAcK with UAG at position K56 (K14AcK(UGA)/K56ThioAcK(UAG)), ThioAcK with UAG at position K14 and AcK with UGA at position K56 (K14ThioAcK(UAG)/K56AcK(UGA)), AcK with UAG at position K14 and ThioAcK with UGA at position K56 (K14AcK(UAG)/K56ThioAcK(UGA)). Western blots showed observable signals in nearly all of the above samples (Figure 3A and S8–9). MS/MS analyses gave the correct mass spectra for two digested peptides containing ThioAcK at K14 and AcK at the site of K56 of histone H3K14ThioAcK/K56AcK, respectively (Figure 3B).
The work presented here is the first example of lysine modifications in full-length human histone H3 with combinations of ThioAcK and AcK. The use of these and similar histone variants should lead to a deeper understanding of the important processes of histone acetylation and deacetylation[4, 9b, 12].
Supplementary Material
Acknowledgments
We thank the members of the Söll lab for helpful comments. This work was supported grants from the National Institutes of Health (GM22854 to DS; GM68649 to SJM; AI119813 to CF), and the Defense Advanced Research Projects Agency (contract N66001-12-C-4211) to DS.
References
- 1.a) Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. Mol Cell. 2007;25:703–712. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]; d) Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Söll D. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Duan MR, Smerdon MJ. J Biol Chem. 2014;289:8353–8363. doi: 10.1074/jbc.M113.540732. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yang XJ, Seto E. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JS, Smith E, Shilatifard A. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lee KK, Workman JL. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]; b) Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 4.Fatkins DG, Monnot AD, Zheng W. Bioorg Med Chem Lett. 2006;16:3651–3656. doi: 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 5.a) Mellini P, Kokkola T, Suuronen T, Salo HS, Tolvanen L, Mai A, Lahtela-Kakkonen M, Jarho EM. J Med Chem. 2013;56:6681–6695. doi: 10.1021/jm400438k. [DOI] [PubMed] [Google Scholar]; b) Kiviranta PH, Suuronen T, Wallen EAA, Leppanen J, Tervonen J, Kyrylenko S, Salminen A, Poso A, Jarho EM. J Med Chem. 2009;52:2153–2156. doi: 10.1021/jm801401k. [DOI] [PubMed] [Google Scholar]
- 6.a) Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]; b) Gattner MJ, Vrabel M, Carell T. Chem Commun (Camb) 2013;49:379–381. doi: 10.1039/c2cc37836a. [DOI] [PubMed] [Google Scholar]; c) Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Nat Chem Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kim CH, Kang M, Kim HJ, Chatterjee A, Schultz PG. Angew Chem Int Ed Engl. 2012;51:7246–7249. doi: 10.1002/anie.201203349. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wan W, Tharp JM, Liu WR. Biochim Biophys Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Sachdeva A, Wang K, Elliott T, Chin JW. J Am Chem Soc. 2014;136:7785–7788. doi: 10.1021/ja4129789. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang K, Sachdeva A, Cox DJ, Wilf NM, Lang K, Wallace S, Mehl RA, Chin JW. Nat Chem. 2014;6:393–403. doi: 10.1038/nchem.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. Angew Chem Int Ed Engl. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]; d) Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 8.a) Hecht SM, Alford BL, Kuroda Y, Kitano S. J Biol Chem. 1978;253:4517–4520. [PubMed] [Google Scholar]; b) Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 9.a) Murakami H, Ohta A, Ashigai H, Suga H. Nat Methods. 2006;3:357–359. doi: 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]; b) Kang TJ, Yuzawa S, Suga H. Chem Biol. 2008;15:1166–1174. doi: 10.1016/j.chembiol.2008.09.014. [DOI] [PubMed] [Google Scholar]; c) Goto Y, Katoh T, Suga H. Nat Protoc. 2011;6:779–790. doi: 10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]; d) Bessho Y, Hodgson DR, Suga H. Nat Biotechnol. 2002;20:723–728. doi: 10.1038/nbt0702-723. [DOI] [PubMed] [Google Scholar]; e) Terasaka N, Hayashi G, Katoh T, Suga H. Nat Chem Biol. 2014;10:555–557. doi: 10.1038/nchembio.1549. [DOI] [PubMed] [Google Scholar]
- 10.a) Hirsch BM, Zheng W. Mol Biosyst. 2011;7:16–28. doi: 10.1039/c0mb00033g. [DOI] [PubMed] [Google Scholar]; b) Vaziri H, Dessain SK, Eagon EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 11.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gut P, Verdin E. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.