

Abstract
Recent breakthroughs in next-generation sequencing technologies have led to the discovery of several classes of non-coding RNAs (ncRNAs). It is now apparent that RNA molecules are not just carriers of genetic information, but are also key players in many cellular processes. While there has been a rapid increase in the number of ncRNA sequences deposited in various databases over the past decade, the biological functions of these ncRNAs are largely not well understood. Similar to proteins, RNA molecules carry out a function by forming specific three-dimensional structures. Understanding the function of a particular RNA therefore requires a detailed knowledge of its structure. However, determining experimental structures of RNA is extremely challenging. In fact, RNA-only structures represent just 1% of the total structures deposited in the PDB. Thus, computational methods that predict three-dimensional RNA structures are in high demand. Computational models can provide valuable insights into structure-function relationships in ncRNAs, and can aid in the development of functional hypotheses and experimental designs. In recent years, a set of diverse RNA structure prediction tools have become available, which differ in computational time, input data and accuracy. This review discusses the recent progress and challenges in RNA structure prediction methods.
Keywords: LncRNAs, RNA structure prediction, Group II introns, Homology modeling, RNA backbone refinement
Graphical abstract
Introduction
Large RNAs (>200 nts) have been implicated in a wide variety of biological processes [1]. The first example of large RNAs include ribosomal RNAs (rRNAs > 1 kb), which are key players in protein synthesis [2]. Other examples include ribozymes such as group I introns (~250–400 nts) [3] and group II introns (~400–1500 nts) [4], which are highly structured catalytic RNAs involved in RNA splicing. Beyond rRNAs and ribozymes, there is now increasing evidence suggesting that long non-coding RNAs (lncRNAs) are involved in many cellular processes [1]. For example, lncRNA HOTAIR (2.2 kb) silences the HOXD genes [5], RepA (1.6 kb) is involved in X-chromosome inactivation [6], and enhancer RNAs (~200–500 nts) promote neighboring gene transcription [7]. Thanks to the revolution in deep sequencing methods [8], the number of lncRNA sequences being discovered is currently in a phase of rapid growth. At present, the LNCipedia database holds 90,000 human lncRNAs sequences, which represents a five-fold increase in the number of sequences deposited within the last three years [9]. Despite increasing evidence suggesting that lncRNAs play myriad critical roles in biology, the molecular mechanisms by which these functions are accomplished remain poorly understood.
RNA molecules carry out various cellular functions that require static or dynamic structures. For example, ribozymes [10] form a discrete tertiary structure for catalysis and riboswitches toggle between structural states to regulate gene expression [1]. Understanding the function of a particular RNA, therefore, requires a detailed knowledge of its structure. RNA folding is generally considered a two-stage process [11], wherein the secondary structure forms first and then assembles into a compact tertiary structure. Secondary structure prediction has been a major area of research for many years [12], and with the advent of chemical probing techniques such as SHAPE [13] and DMS [14] it is now possible to determine secondary structures using a combination of experimental and computational methods [13]. Computational tools such as RNAstructure [15] can readily incorporate experimental data from SHAPE and DMS to aid in the assignment of RNA secondary structures. Progress has also been made in probing RNA secondary structures in living cells [16]. Recent publications reporting the secondary structures of the large lncRNAs SRA [17] and HOTAIR [18], as well as entire RNA genomes [19, 20] demonstrate that these techniques are robust and not limited by the size of an RNA target.
Compared to determining secondary structures, experimental determination of tertiary RNA structures is considerably more challenging and unlike secondary structure determination, is substantially complicated by increasing target size. As of July 2015, there are only 1114 RNA-only structures deposited in the PDB. Most of these structures comprise RNA molecules that are less than 200-nt in length, with the only exceptions being ribosomal RNAs [21–23], group I introns [3], group II introns [4] and RNaseP [24]. In the absence of empirical structural data, computational models provide valuable insights, guide experiments, and can be used to aid in solving tertiary structures experimentally [25]. RNA structure prediction is currently a major area of interest and has been described in several previous reviews [26–28]. This review addresses the current progress and challenges in modeling RNA structures, with a particular focus on large RNAs.
Principles of RNA structure
Much like proteins, RNA molecules are capable of forming complex globular structures required for their biological function. It is essential to understand the principles governing RNA tertiary structure in order to predict new target structures. Each experimentally determined RNA structure has enriched our knowledge of the principles governing the tertiary structure of RNAs. The crystal structures of tRNA solved in the 1970s demonstrated that RNA structures are stabilized by networks of hydrogen-bonds [29, 30]. These structures also revealed that coaxial stacking interactions have the greatest impact on dictating the three dimensional shape of tRNA [30]. Subsequent structural studies on diverse functional RNAs including the Hammerhead ribozyme [31], the HDV ribozyme [32], group I introns [3], group II introns [33] and ribosomal RNAs [21–23] revealed that these RNAs share many structural elements, and thus that the principles governing RNA tertiary structures are universal [34, 35]. Further, detailed biochemical experiments [33, 36, 37] combined with computational analysis of several RNA crystal structures led to the identification of the structural building blocks of RNA tertiary architecture, collectively known as RNA structural motifs [38–40]. A few examples of RNA motifs include kink-turns, S-turn, kissing loops, T-loops, pseudoknots, A-A platform, A-minor, base-triples and tetraloop-receptor interactions. These RNA motifs play a central role in assisting RNA folding and tertiary structure formation by mediating many long-range interactions [39, 40].
Many RNA motifs are recurrent, meaning that they appear in a number of different RNAs independent of sequence homology, and share similar three-dimensional structures across different sequences [38, 40]. For example, coaxial stacking is the most common tertiary element in structured RNAs, occurring in both simple systems like tRNA and in more elaborate structures such as those adopted by group I and group II introns (Fig. 1). However, large RNAs are complex because they may have multiple additional motifs such as tetraloop-receptor interactions (Fig. 1B) and kissing-loop interactions (Fig. 1C). Nonetheless, the fact that diverse RNA structures share many common structural elements suggests that principles learned from existing structures can be applied to model new target RNAs.
Fig. 1.
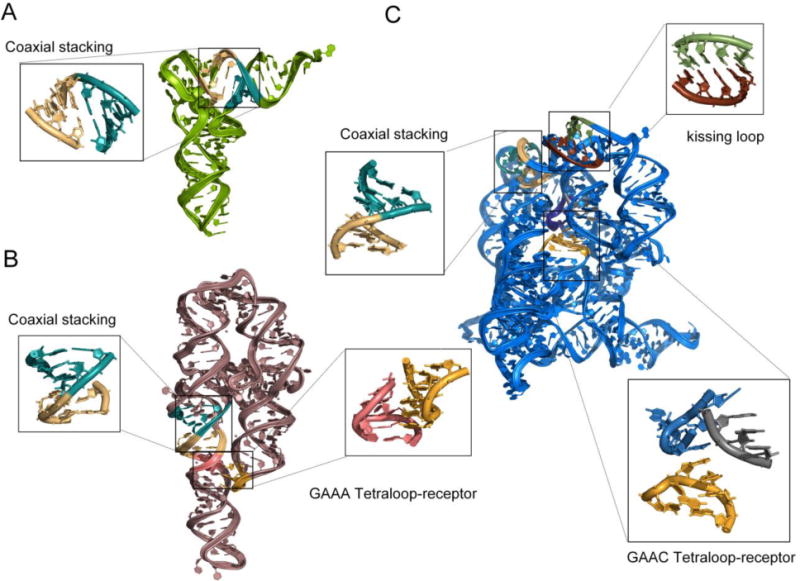
Comparison of structural motifs in crystal structures of three different RNAs A) tRNA (PDB ID: 1EHZ), B) P4-P6 domain of group I intron (PDB ID: 1GID), and C) Group IIC intron (PDB ID: 4FAW). RNA structures share many common structural elements such as coaxial stacking that is common in all the three RNAs shown here, yet large RNAs possess additional motifs, which make structure prediction a challenging problem.
Progress and Challenges in RNA 3D modeling methods
RNA 3D modeling methods can be classified into three main categories 1) Manual modeling 2) De novo modeling and 3) Homology modeling. In recent years, progress has been made to all these categories of RNA modeling. The following sections discuss the current advancements and challenges in RNA 3D modeling methods.
Manual modeling
Manual modeling is an interactive process wherein a user manually assembles individual nucleotides or structural motifs to build complete RNA models. The first RNA model was built manually in 1969 [41], and since then manual modeling methods have been employed to build many RNA models. A variety of programs are available that allow manual building of RNA models (see Table 1). ERNA-3D [42] provides a user-friendly graphical interface to build and edit RNA models including large ribosomal RNAs [42]. MANIP [43] allows real-time assembly of known RNA motifs and builds the RNA model interactively on the computer screen. RNA2D3D [44] uses a pre-determined secondary structure as an initial input to build a first order approximation of the 3D structure. This approximation requires manual manipulation to refine the initial model with various built-in interactive tools. ASSEMBLE [45] provides two synchronized windows to build and edit both secondary and tertiary structures.
Table 1.
List of computational tools for RNA structure prediction, model quality evaluation and refinement.
These tools allow modeling of large RNAs > 200 nts.
These tools are optimal for modeling mid-sized RNAs <150 nts.
These tools are optimal for modeling small RNAs < 50 nts.
Although manual modeling methods have been employed to model large RNAs, such an approach is time consuming and requires expert knowledge of the target RNA. Moreover, building the entire molecule manually could result in steric clashes, or assignments of the wrong sugar pucker and backbone conformations. A more effective use of manual RNA modeling in the context of large RNAs involves first building a model using automated methods, and then manually adjusting or rebuilding nucleotides at problematic positions.
De novo modeling
The goal of De novo modeling methods is to predict three dimensional RNA structures directly from the sequence. De novo modeling methods can be categorized into three groups including 1) all-atom based 2) coarse-grained and 3) fragment assembly techniques (Table 1). All-atom based methods predict structure by simulating the folding process using force-fields and molecular mechanics. These simulations are very detailed and in principle can explain the folding process as a function of real kinetics and thermodynamics at work in the molecule. Various software packages such as AMBER [46] and CHARMM [47] have force fields optimized to run RNA simulations. The Šponer group benchmarked these force fields and other parameters – such as salt conditions and water models – for RNA by performing simulations on RNA A-form helices and tetraloops [48, 49]. Recently, Chen and Garcia [50] recapitulated the conformations of three RNA tetraloops starting from the unfolded state. However, these simulations are computationally expensive and therefore limited to small RNAs.
Coarse-grained methods reduce computational time by simplifying how the nucleotides are represented in the model. For example, NAST [51] and YUP [52] represent each nucleotide as a single pseudo atom, Vfold [53], SimRNA [54] and iFoldRNA [55] define each nucleotide using three pseudo atoms, CG [56] represents each nucleotide with five pseudo atoms, and HiRE-RNA [57] uses six pseudo atoms to represent purines and seven for pyrimidines. In addition, there are methods that represent RNA as graphs, which further reduces the sampling space by representing each helix as a stick or a cylinder [58, 59]. The accuracy of coarse-grained methods depends on the choice of representation and the scoring function. Coarse-grained methods have been employed to model various RNA targets such as the P4-P6 domain of the group I intron [51], the HCV IRES pseudoknot domain [60] and Domain 3 of the foot-and-mouth-disease virus IRES element [61].
Fragment-assembly methods build RNA models by assembling short fragments extracted from structural databases. FARNA is a fragment assembly method built upon the ROSETTA package that has been adapted to model RNA [62]. MC-Sym [63] assembles structures using a library of nucleotide cyclic motifs and Monte Carlo sampling. Recently, Popenda et al, developed a novel method, RNAComposer [64], that can build large RNAs using a fragment database (RNA FRABASE) [64]. Both MC-Sym and RNAComposer are fully automated and available as webservers. Fragment based methods have been employed to model and study many RNAs including the TPP riboswitch [65], the SAM-I riboswitch [66], different states of P4-P6 domain of group I intron [67], and various domains of group II intron [68].
Despite considerable progress, there are many bottlenecks that make de novo modeling of large RNAs an exceptionally challenging task. For example, all-atom simulations are not only computationally expensive but often require optimization of force-field parameters. In fact, it was essential to reparametrize the force-field and energy function to fold even small RNA tetraloops [50]. On the other hand, coarse-grained methods are relatively fast, but suffer from low accuracy, as RNA structures are stabilized by many non-canonical interactions, such as tetra-loop receptor interactions [39] and base-triples [40], which are hard to recapitulate using reduced representations [50]. Finally, fragment-assembly methods extensively rely on existing structural data and may not be able to predict structures of RNA molecules that contain novel motifs. Nevertheless, de novo modeling can be especially useful in modeling large RNAs in conjunction with manual and homology modeling methods.
Homology modeling
Homology modeling, also known as comparative modeling or template-based modeling, builds a model using a closely related experimental structure. Homology modeling is among the most widely used methods for predicting protein structures [69]. Over the past few decades, there has been continuous progress and improvement in protein homology modeling methods [69, 70]. Protein homology models are now routinely used in various bio-medical applications, including drug discovery [71]. Similar to proteins, homologous RNAs also frequently share similar tertiary structures, and therefore can be modelled using homology methods. In recent years, various tools have been developed to build RNA homology models (Table 1).
ModeRNA [72] is a homology modeling tool, which builds the RNA model by copying the coordinates from the template structure by analyzing the target-template sequence alignment. Both template structure and target-template sequence alignment must be provided by the user. One main advantage of ModeRNA is that it allows modeling of modified nucleotides. Further, ModeRNA is now available as a webserver that automates several steps, including identifying the template and analyzing the model structure [73].
RNAbuilder [74], also known as MacroMoleculeBuilder, is a homology modeling tool built upon the SimTK.org package. RNAbuilder defines the nucleotides using internal coordinates, such as bond angles and dihedral angles, which may be constrained by the user. When a template structure is available, the user-provided sequence alignment is used to place constraints on regions that are common to the target and template structures. RNAbuilder also allows coarse-grained level modeling, where RNA bases are modeled as rigid bodies with forces and torques applied to the base instead of individual atoms. In theory, this functionality can be employed to build regions that are not present in the template onto the homology model, making it an attractive method to model large RNAs [75].
Although homology modeling is rapid, there are two main requirements which limit its use. First, it is necessary that similar structures are available in the PDB. Next, a proper alignment between template and target sequences is critical in order to build accurate models. Both identifying the homolog structures for the given RNA sequence and building the target-template alignment are non-trivial tasks, as RNAs can exhibit higher conservation at the secondary structure level than at the sequence level [76]. Therefore, sequence based search tools (such as BLAST) may not always identify a structural homolog. In such cases, it is possible to search for templates using co-variation models built with computational tools such as Infernal [77]. Covariation models can also be used to build the target-template alignment [77]. Alternately, alignments can be obtained from the Rfam database [78], or built using tools that combine sequence and secondary structure such as LocaRNA [79] or R-Coffee [80]. Additionally, manual adjustments may still be necessary to build an accurate alignment [72]. Once the template has been identified and proper alignment has been built, homology modeling tools can be readily applied to even large RNAs, including ribosomal RNAs [81].
Model Evaluation and Refinement
After building a model, it is essential to evaluate the quality, find any errors and refine the accordingly. First, it is important to make sure that all the base-pairs and the overall secondary structure is maintained correctly in the model. Tools such as RNAview [82], MC-Annotate [83], and DSSR [84] can calculate the secondary structure from a given 3D structure and thereby allow identification of problematic base-pairs. Recently, Antczak et al [85], developed a web server, RNApdbee, which integrates RNAview, MC-Annotate and DSSR, and extracts not only secondary structures but also kissing-loops and pseudoknots from a target tertiary model. Problematic base pairs can be fixed or rebuilt using interactive tools such as S2S/ASSEMBLE [45].
Next, the model has to be checked for any steric clashes and incorrect backbone conformations or sugar puckers. Molprobity [86] server can readily identify and list any steric clashes or problems with backbone conformations and sugar puckers. In general, simple errors in the RNA structural geometry can be fixed by energy minimization using simulation packages such as AMBER [46] or CHARMM [47]. However, large steric clashes and errors in the backbone conformations require molecular dynamics (MD) based refinement. MD refinement is computationally expensive and may not be applicable for large RNAs. Moreover, RNA backbones are negatively charged, and are stabilized by mono or divalent cations. Performing MD simulations without proper placement of cations may result in local unfolding of the structure. Thus, there is need for methods that can refine the RNA backbone without unfolding the local structure.
Several methods are available to refine the RNA backbone conformations such as RNABC [87], RCrane [88] and ERASSER [89], but they require an electron-density map. Recently, RCrane has been modified to correct backbone conformations even before an electron-density map is available [68]. Originally developed for building RNA into an electron density map, RCrane is a bioinformatics based approach that uses pseudotorsions and RNA backbone conformers to model RNA [90]. For each nucleotide in a given RNA structure, RCrane predicts the backbone conformation based on the positions of the base and the phosphate group, and rebuilds the entire backbone (Fig. 2). In cases where no electron density map is available, RCrane can refine the backbone if the base and phosphates are modelled correctly. The main advantage of RCrane is that it keeps the position of the base constant throughout the refinement process and therefore preserves the secondary structure (Fig. 2).
Fig. 2.
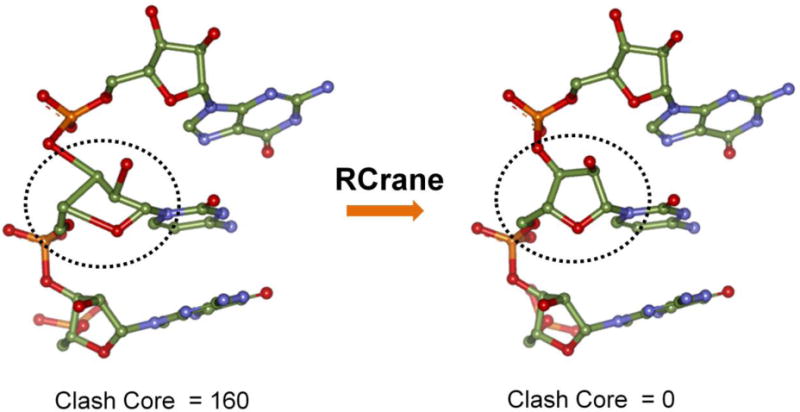
RNA backbone refinement with RCrane. RCrane can refine the backbone without disrupting the secondary structure. It uses the phosphate and base positions to predict the sugar conformations and rebuilds the backbone.
Current strategies for modeling large RNAs
Modeling large RNAs using remote templates
Currently, the most straightforward method to model large RNAs is by using related structures from the PDB. However, given the limited number of RNA structures in the PDB, it is probable that the available experimentally determined structures are only remotely homologous to the target RNA with very low sequence-similarity. Predicting structures using a remote template is a challenging problem both in the protein and RNA modeling worlds [91]. For proteins, there is evidence showing that structures are conserved even with low sequence similarity [92], thus methods exist that can predict structures even with remote templates [93]. Conversely, when sequence similarity decreases, RNA structures tend to diverge quickly [94]. However, many of the structural differences that appear in homologous or remote templates tend to lay in peripheral domains [95], with the core structure conserved even in very remote RNA homologs [94, 95]. In such cases, where only remote structures are available, models can be predicted using a combination of homology and de novo methods. This modeling pipeline first employs homology modeling to build the core structure (Fig. 3). All additional domains are then sampled as an ensemble using de novo methods, followed by docking, filtering and refinement. A short region present in the homology model should be included in the input provided for the de novo modeling tool, which can later serve as an anchor to align de novo modeled domains onto the homology model.
Fig. 3.
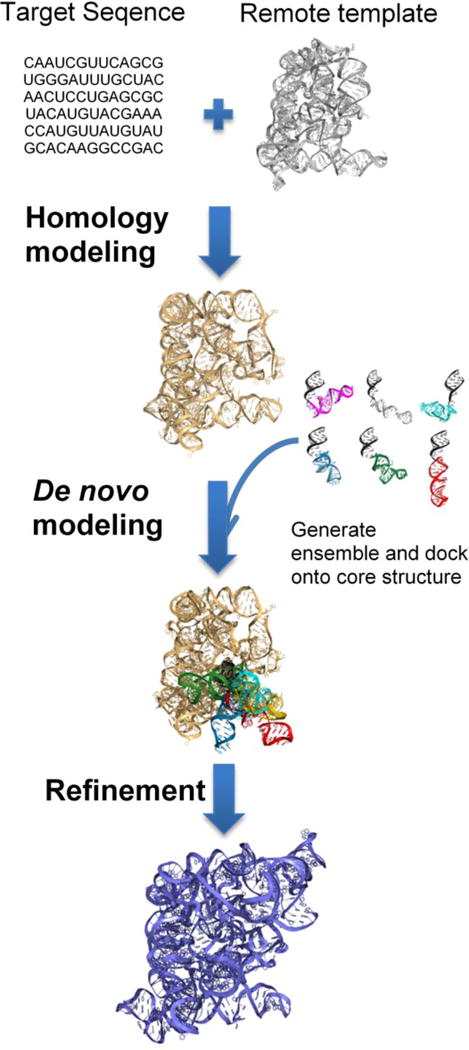
Modeling large RNA structures using remote homologs. This pipeline involves three steps. First, the structurally conserved core regions are built using homology modeling. Next, all additional domains are modelled using de novo methods and docked on to the core structure. Finally, the models are filtered and refined.
Using this pipeline, the Pyle lab has recently modelled the all-atom three-dimensional structure of the group IIB intron ai5γ from the budding yeast Saccharomyces cerevisiae (ai5γIIB) [68]. Group II introns are self-splicing ribozymes thought to be ancestors of the human spliceosome [96, 97]. They are excellent model systems to study RNA structure-function relationships [96]. Despite their importance, crystal structures were available only for a group IIC intron from Oceanobacillus iheyensis [10]. Group IIC introns (~400 nts) are a primitive class of introns that lack many of the domains and functional capabilities of the structurally evolved IIA and IIB introns (>800 nts) [96]. Therefore, it was essential to combine homology and de novo methods to build the model of ai5γ group IIB intron. Thanks to the Toor group, who recently solved the crystal structure of a group IIB intron from Pylaiella littoralis (P.li.LSUI2) [98], we can now compare the model to the structure, and evaluate the current state of modeling large RNAs from remote homologs.
The overall topology and domain locations are strikingly similar in both the crystal structure of P.li.LSUI2 and the ai5γ model (Fig. 4). Most importantly, both the model and the crystal structure provide similar insights into the mechanism of group II introns. First, they both show that the second exon binding site helix (a motif specific to IIA and IIB introns) is not coaxial with the first exon binding site helix (Fig. 4C). Additionally, ai5γ model suggested that the Domain 6 does not undergo large-scale conformational change between the two steps of splicing, which is now also implicated by the crystal structures of P.li.LSUI2. However, crystal structures of P.li.LSUI2 revealed new interaction between Domain 6 and Domain 2 that had not been visualized in the ai5γ model. The other main difference between the structure and the model is in the peripheral subdomains of Domain 1. This is expected because although both P.li.LSUI2 and the ai5γ belong to the same IIB class, ai5γ possesses an additional subdomain (D1d2b) and a long-range interaction (β-β′ kissing loop between D1c2 and D1d2a) that are lacking in P.li.LSUI2. Conversely, domain 5, the most conserved region in both the introns, is superimposable in these two structures (see Fig. 4D). This comparison demonstrates the feasibility of modeling large RNAs from remote homologs with reasonable accuracy.
Fig. 4.
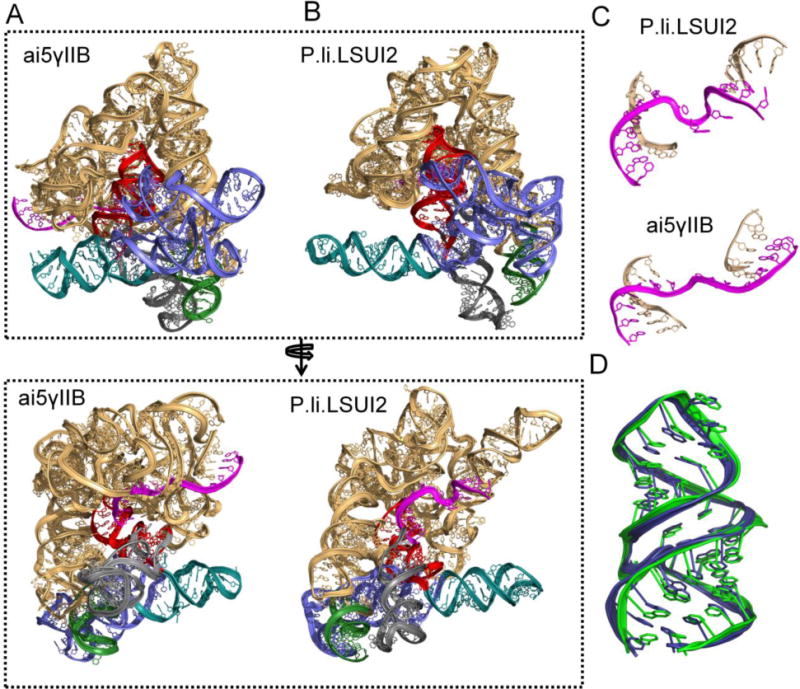
Comparison of ai5γIIB model with crystal structure of P.li.LSUI2. A) Three-dimensional model of ai5γIIB intron, color coded as follows; domain 1 in gold, domain 2 in green, domain 3 in blue, domain 4 in teal, domain 5 in red and domain 6 in grey. B) Crystal structure of P.li.LSUI2 (PDB ID: 4R0D), color coded same as above. C) Comparison of two exon-binding site helices in the model and the crystal structure. D) Alignment of domain 5 in the model (green) with the domain 5 in the crystal structure of P.li.LSUI2 (blue).
Modeling with the aid of experimental data
One main advantage of modelling RNA is that there are experimental techniques available to probe the RNA structure at single nucleotide resolution, thus providing valuable information for model generation. For example, SHAPE [13] and DMS [14] can determine whether a particular nucleotide is involved in a base pair. Hydroxyl radical footprinting can provide valuable information on solvent accessibility of each nucleotide, helping to constrain its position in the context of a globular structure [99]. Methods such as cross-linking [100] and MOHCA [101] can identify nucleotides in the proximity of a target nucleotide. In addition, strategies such as mutate and map [102] can inform on tertiary contacts. As reviewed recently [103] and discussed above, several tools can readily incorporate experimental data to obtain a modeled RNA structure. Integrating the experimental data has shown to achieve reasonable accuracies (RMSD ~10 Å) for mid-sized RNAs (~150 nts) [101].
Critical assessment of RNA 3D structure prediction
Critical assessment and “benchmarking the prediction” methods not only improve current tools, but also allow development of new tools, as is evident in the case of CASP (Critical Assessment of protein Structure Prediction), which played a crucial role in advancing the field of protein structure prediction by identifying the progress and highlighting the focus of future research. Recently, a CASP-like effort has been undertaken to benchmark RNA structure prediction methods, known as RNA-Puzzles [104]. RNA-Puzzles is a blind experiment where the computational labs are asked to solve ‘puzzles’, in order to predict the structures of RNAs that are recently solved but not released in the PDB. However, they are allowed to utilize available structures in the PDB and data from chemical probing experiments such as SHAPE to aid in modeling of the RNAs. There were three puzzles in the first round, 1) RNA Dimer (46-nt) 2) RNA Square (100-nts) and 3) riboswitch domain [104]. Twelve to fourteen models were submitted for each ‘puzzle’ and the RMSD’s for the top ranking models were within the range of 2.4Å – 7.3Å [104]. The second round of RNA-puzzles has already increased the difficulty level of the challenge by introducing large RNAs (>160 nts) [105]. Despite the increase in the size of the ‘puzzles’, the results were still promising, with RMSDs of top ranking models in the range of 6.8Å – 11.7Å [105], demonstrating the progress in RNA structure prediction. Thus, critical assessment of structure prediction methods plays a crucial role in advancing the field by identifying the progress and highlighting the challenges.
Conclusions
The ability to accurately predict RNA tertiary structure is a topic of major interest as it would aid in rapid mechanistic understanding and hypothesis generation in the study of large RNAs. In recent years, progress has been made in all areas of RNA tertiary structure prediction; it is now feasible to model even large RNAs with the aid of experimental data.
However, structure prediction of large RNAs using only sequence or secondary structure is still very limited. This is because large RNAs form complex tertiary structures that are stabilized by many long-range interactions, non-canonical base pairs and structural motifs. There are many bottlenecks that make structure prediction of large RNAs from sequence a challenging problem. For example, at present there are no tools that can accurately predict long-range interactions directly from an RNA sequence. Also, as assessed by RNA-puzzles [105], current tools achieve good accuracy in predicting the right topologies and Watson-Crick base pairs, but cannot predict non-canonical base pairs. Finally, there are no methods that can predict structures with novel motifs. The limitations in current methods underscore the major gaps in our understanding of RNA structures and can be improved only with the availability of more empirically determined, high resolution structures.
Future Prospects
In the short term, RNA structure prediction can be accelerated by combining experimental data and computational methods. For instance, integrating data from chemical probing experiments with computational methods is shown to be promising for modeling intermediate-sized RNAs such as the lariat-capping ribozyme [101, 105]. Certainly, if the experimental structure of a related homolog is available, homology modeling can be readily applied to large RNAs. Nevertheless, there is still considerable room for improvement in these approaches. Homology modeling methods need to be better automated to identify and build structures from remote homologs. Further, even the models built from close homologs often result in steric clashes. Therefore, fast and automated refinement methods to fix RNA models are required. Finally, development of experimental techniques to identify nucleotides involved in non-canonical or tertiary interactions will greatly advance the RNA structure prediction methods.
Highlights.
Large RNAs are implicated as key players in many cellular processes.
Current progress in RNA tertiary structure prediction methods is reviewed.
Strategies for modeling large RNAs are discussed.
Acknowledgments
I would like to thank Prof. Anna Pyle for guidance, support and constructive discussions. I acknowledge members of the Pyle lab for helpful discussions, and in particular Megan Fitzgerald, David Rawling, Thayne Dickey and Nathan Pirakitikulr for critical review of this manuscript. I apologize to all our colleagues whose work could not be extensively cited in this paper.
Abbreviations
- LncRNA
long non-coding RNA
- SHAPE
selective 2′-hydroxyl acylation analyzed by primer extension
- DMS
dimethyl sulfate
- MOHCA
multiplexed hydroxyl radical cleavage analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Moore PB, Steitz TA. The roles of RNA in the synthesis of protein. Cold Spring Harbor perspectives in biology. 2011;3:a003780. doi: 10.1101/cshperspect.a003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–85. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 4.Marcia M, Somarowthu S, Pyle AM. Now on display: a gallery of group II intron structures at different stages of catalysis. Mobile DNA. 2013;4:14. doi: 10.1186/1759-8753-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Sun BK, Erwin JA, Song J-J, Lee JT. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science. 2008;322:750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas BJ, Zody MC. Advancing RNA-Seq analysis. Nat Biotech. 2010;28:421–3. doi: 10.1038/nbt0510-421. [DOI] [PubMed] [Google Scholar]
- 9.Volders PJ, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J, et al. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic acids research. 2015;43:4363–4. doi: 10.1093/nar/gkv295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nature reviews Molecular cell biology. 2004;5:908–19. doi: 10.1038/nrm1497. [DOI] [PubMed] [Google Scholar]
- 12.Mathews DH. Revolutions in RNA secondary structure prediction. Journal of molecular biology. 2006;359:526–32. doi: 10.1016/j.jmb.2006.01.067. [DOI] [PubMed] [Google Scholar]
- 13.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cordero P, Kladwang W, VanLang CC, Das R. Quantitative dimethyl sulfate mapping for automated RNA secondary structure inference. Biochemistry. 2012;51:7037–9. doi: 10.1021/bi3008802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloma MF, Mathews DH. Improving RNA secondary structure prediction with structure mapping data. Methods in enzymology. 2015;553:91–114. doi: 10.1016/bs.mie.2014.10.053. [DOI] [PubMed] [Google Scholar]
- 16.Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–90. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novikova IV, Hennelly SP, Sanbonmatsu KY. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic acids research. 2012;40:5034–51. doi: 10.1093/nar/gks071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, Pyle AM. HOTAIR forms an intricate and modular secondary structure. Molecular cell. 2015;58:353–61. doi: 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–5. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–6. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 22.Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, et al. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–39. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 23.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, et al. High Resolution Structure of the Large Ribosomal Subunit from a Mesophilic Eubacterium. Cell. 2001;107:679–88. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 24.Krasilnikov AS, Yang X, Pan T, Mondragon A. Crystal structure of the specificity domain of ribonuclease P. Nature. 2003;421:760–4. doi: 10.1038/nature01386. [DOI] [PubMed] [Google Scholar]
- 25.Marcia M, Humphris-Narayanan E, Keating KS, Somarowthu S, Rajashankar K, Pyle AM. Solving nucleic acid structures by molecular replacement: examples from group II intron studies. Acta crystallographica Section D, Biological crystallography. 2013;69:2174–85. doi: 10.1107/S0907444913013218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rother K, Rother M, Skiba P, Bujnicki JM. Automated modeling of RNA 3D structure. Methods in molecular biology. 2014;1097:395–415. doi: 10.1007/978-1-62703-709-9_18. [DOI] [PubMed] [Google Scholar]
- 27.Laing C, Schlick T. Computational approaches to RNA structure prediction, analysis, and design. Current opinion in structural biology. 2011;21:306–18. doi: 10.1016/j.sbi.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sripakdeevong P, Beauchamp K, Das R. Why Can’t We Predict RNA Structure At Atomic Resolution? In: Leontis N, Westhof E, editors. RNA 3D Structure Analysis and Prediction. Springer Berlin Heidelberg; 2012. pp. 43–65. [Google Scholar]
- 29.Robertus JD, Ladner JE, Finch JT, Rhodes D, Brown RS, Clark BF, et al. Structure of yeast phenylalanine tRNA at 3 A resolution. Nature. 1974;250:546–51. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 1974;185:435–40. doi: 10.1126/science.185.4149.435. [DOI] [PubMed] [Google Scholar]
- 31.Pley HW, Flaherty KM, McKay DB. Three-dimensional structure of a hammerhead ribozyme. Nature. 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 32.Ferre-D’Amare AR, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–74. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 33.Boudvillain M, de Lencastre A, Pyle AM. A tertiary interaction that links active-site domains to the 5[prime] splice site of a group II intron. Nature. 2000;406:315–8. doi: 10.1038/35018589. [DOI] [PubMed] [Google Scholar]
- 34.Ferré-D’Amaré AR, Doudna JA. RNA FOLDS: Insights from Recent Crystal Structures. Annual Review of Biophysics and Biomolecular Structure. 1999;28:57–73. doi: 10.1146/annurev.biophys.28.1.57. [DOI] [PubMed] [Google Scholar]
- 35.Doherty EA, Batey RT, Masquida B, Doudna JA. A universal mode of helix packing in RNA. Nat Struct Mol Biol. 2001;8:339–43. doi: 10.1038/86221. [DOI] [PubMed] [Google Scholar]
- 36.Szewczak AA, Ortoleva-Donnelly L, Ryder SP, Moncoeur E, Strobel SA. A minor groove RNA triple helix within the catalytic core of a group I intron. Nat Struct Mol Biol. 1998;5:1037–42. doi: 10.1038/4146. [DOI] [PubMed] [Google Scholar]
- 37.Costa M, Michel F. Frequent use of the same tertiary motif by self-folding RNAs. The EMBO Journal. 1995;14:1276–85. doi: 10.1002/j.1460-2075.1995.tb07111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4899–903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Accounts of chemical research. 2011;44:1302–11. doi: 10.1021/ar200098t. [DOI] [PubMed] [Google Scholar]
- 40.Leontis NB, Westhof E. Analysis of RNA motifs. Current opinion in structural biology. 2003;13:300–8. doi: 10.1016/s0959-440x(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 41.Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature. 1969;224:759–63. doi: 10.1038/224759a0. [DOI] [PubMed] [Google Scholar]
- 42.Mueller F, Döring T, Erdemir T, Greuer B, Jünke N, Osswald M, et al. Getting closer to an understanding of the three-dimensional structure of ribosomal RNA. Biochemistry and Cell Biology. 1995;73:767–73. doi: 10.1139/o95-085. [DOI] [PubMed] [Google Scholar]
- 43.Massire C, Westhof E. MANIP: an interactive tool for modelling RNA1. Journal of Molecular Graphics and Modelling. 1998;16:197–205. doi: 10.1016/s1093-3263(98)80004-1. [DOI] [PubMed] [Google Scholar]
- 44.Martinez HM, Maizel JV, Jr, Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. Journal of biomolecular structure & dynamics. 2008;25:669–83. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jossinet F, Ludwig TE, Westhof E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010;26:2057–9. doi: 10.1093/bioinformatics/btq321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Iii, DeBolt S, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Computer Physics Communications. 1995;91:1–41. [Google Scholar]
- 47.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. Journal of computational chemistry. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Besseova I, Banas P, Kuhrova P, Kosinova P, Otyepka M, Sponer J. Simulations of A-RNA duplexes. The effect of sequence, solute force field, water model, and salt concentration. The journal of physical chemistry B. 2012;116:9899–916. doi: 10.1021/jp3014817. [DOI] [PubMed] [Google Scholar]
- 49.Ditzler MA, Otyepka M, Šponer J, Walter NG. Molecular Dynamics and Quantum Mechanics of RNA: Conformational and Chemical Change We Can Believe In. Accounts of chemical research. 2010;43:40–7. doi: 10.1021/ar900093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen AA, Garcia AE. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simulations. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16820–5. doi: 10.1073/pnas.1309392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, et al. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. Rna. 2009;15:189–99. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan RKZ, Petrov AS, Harvey SC. YUP: A Molecular Simulation Program for Coarse-Grained and Multi-Scaled Models. Journal of chemical theory and computation. 2006;2:529–40. doi: 10.1021/ct050323r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu X, Zhao P, Chen SJ. Vfold: a web server for RNA structure and folding thermodynamics prediction. PloS one. 2014;9:e107504. doi: 10.1371/journal.pone.0107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rother K, Rother M, Boniecki M, Puton T, Tomala K, Łukasz P, et al. Template-Based and Template-Free Modeling of RNA 3D Structure: Inspirations from Protein Structure Modeling. In: Leontis N, Westhof E, editors. RNA 3D Structure Analysis and Prediction. Springer Berlin Heidelberg; 2012. pp. 67–90. [Google Scholar]
- 55.Sharma S, Ding F, Dokholyan NV. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008;24:1951–2. doi: 10.1093/bioinformatics/btn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia Z, Gardner DP, Gutell RR, Ren P. Coarse-Grained Model for Simulation of RNA Three-Dimensional Structures. The Journal of Physical Chemistry B. 2010;114:13497–506. doi: 10.1021/jp104926t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasquali S, Derreumaux P. HiRE-RNA: a high resolution coarse-grained energy model for RNA. The journal of physical chemistry B. 2010;114:11957–66. doi: 10.1021/jp102497y. [DOI] [PubMed] [Google Scholar]
- 58.Kim N, Laing C, Elmetwaly S, Jung S, Curuksu J, Schlick T. Graph-based sampling for approximating global helical topologies of RNA. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4079–84. doi: 10.1073/pnas.1318893111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kerpedjiev P, Höner zu Siederdissen C, Hofacker IL. Predicting RNA 3D structure using a coarse-grain helix-centered model. Rna. 2015;21:1110–21. doi: 10.1261/rna.047522.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lavender CA, Ding F, Dokholyan NV, Weeks KM. Robust and Generic RNA Modeling Using Inferred Constraints: A Structure for the Hepatitis C Virus IRES Pseudoknot Domain. Biochemistry. 2010;49:4931–3. doi: 10.1021/bi100142y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung S, Schlick T. Candidate RNA structures for domain 3 of the foot-and-mouth-disease virus internal ribosome entry site. Nucleic acids research. 2013;41:1483–95. doi: 10.1093/nar/gks1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14664–9. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–5. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 64.Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, et al. Automated 3D structure composition for large RNAs. Nucleic acids research. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ali M, Lipfert J, Seifert S, Herschlag D, Doniach S. The Ligand-Free State of the TPP Riboswitch: A Partially Folded RNA Structure. Journal of molecular biology. 2010;396:153–65. doi: 10.1016/j.jmb.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang W, Kim J, Jha S, Aboul-ela F. The impact of a ligand binding on strand migration in the SAM-I riboswitch. PLoS computational biology. 2013;9:e1003069. doi: 10.1371/journal.pcbi.1003069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das R, Kudaravalli M, Jonikas M, Laederach A, Fong R, Schwans JP, et al. Structural inference of native and partially folded RNA by high-throughput contact mapping. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4144–9. doi: 10.1073/pnas.0709032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Somarowthu S, Legiewicz M, Keating KS, Pyle AM. Visualizing the ai5gamma group IIB intron. Nucleic acids research. 2014;42:1947–58. doi: 10.1093/nar/gkt1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang YJ, Mao B, Aramini JM, Montelione GT. Assessment of template-based protein structure predictions in CASP10. Proteins. 2014;82(Suppl 2):43–56. doi: 10.1002/prot.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kryshtafovych A, Fidelis K, Moult J. CASP10 results compared to those of previous CASP experiments. Proteins. 2014;82(Suppl 2):164–74. doi: 10.1002/prot.24448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwede T, Sali A, Honig B, Levitt M, Berman HM, Jones D, et al. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17:151–9. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rother M, Rother K, Puton T, Bujnicki JM. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic acids research. 2011;39:4007–22. doi: 10.1093/nar/gkq1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rother M, Milanowska K, Puton T, Jeleniewicz J, Rother K, Bujnicki JM. ModeRNA server: an online tool for modeling RNA 3D structures. Bioinformatics. 2011;27:2441–2. doi: 10.1093/bioinformatics/btr400. [DOI] [PubMed] [Google Scholar]
- 74.Flores SC, Altman RB. Turning limited experimental information into 3D models of RNA. Rna. 2010;16:1769–78. doi: 10.1261/rna.2112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flores SC, Wan Y, Russell R, Altman RB. Predicting RNA structure by multiple template homology modeling. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2010:216–27. doi: 10.1142/9789814295291_0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic acids research. 1994;22:2079–88. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–5. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, et al. Rfam 12.0: updates to the RNA families database. Nucleic acids research. 2015;43:D130–7. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic acids research. 2010;38:W373–7. doi: 10.1093/nar/gkq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moretti S, Wilm A, Higgins DG, Xenarios I, Notredame C. R-Coffee: a web server for accurately aligning noncoding RNA sequences. Nucleic acids research. 2008;36:W10–3. doi: 10.1093/nar/gkn278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tung CS, Joseph S, Sanbonmatsu KY. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nature structural biology. 2002;9:750–5. doi: 10.1038/nsb841. [DOI] [PubMed] [Google Scholar]
- 82.Yang H, Jossinet F, Leontis N, Chen L, Westbrook J, Berman H, et al. Tools for the automatic identification and classification of RNA base pairs. Nucleic acids research. 2003;31:3450–60. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures1. Journal of molecular biology. 2001;308:919–36. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 84.Lu X-J, Bussemaker HJ, Olson WK. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic acids research. 2015 doi: 10.1093/nar/gkv716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Antczak M, Zok T, Popenda M, Lukasiak P, Adamiak RW, Blazewicz J, et al. RNApdbee—a webserver to derive secondary structures from pdb files of knotted and unknotted RNAs. Nucleic acids research. 2014;42:W368–W72. doi: 10.1093/nar/gku330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X, Kapral G, Murray L, Richardson D, Richardson J, Snoeyink J. RNABC: forward kinematics to reduce all-atom steric clashes in RNA backbone. J Math Biol. 2008;56:253–78. doi: 10.1007/s00285-007-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keating KS, Pyle AM. RCrane: semi-automated RNA model building. Acta crystallographica Section D, Biological crystallography. 2012;68:985–95. doi: 10.1107/S0907444912018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chou F-C, Sripakdeevong P, Dibrov SM, Hermann T, Das R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Meth. 2013;10:74–6. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Keating KS, Pyle AM. Semiautomated model building for RNA crystallography using a directed rotameric approach. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8177–82. doi: 10.1073/pnas.0911888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tramontano A. Homology modeling with low sequence identity. Methods. 1998;14:293–300. doi: 10.1006/meth.1998.0585. [DOI] [PubMed] [Google Scholar]
- 92.Kinch LN, Grishin NV. Evolution of protein structures and functions. Current opinion in structural biology. 2002;12:400–8. doi: 10.1016/s0959-440x(02)00338-x. [DOI] [PubMed] [Google Scholar]
- 93.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids research. 2005;33:W244–W8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Capriotti E, Marti-Renom MA. Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinformatics. 2010;11:322. doi: 10.1186/1471-2105-11-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krasilnikov AS, Xiao Y, Pan T, Mondragón A. Basis for Structural Diversity in Homologous RNAs. Science. 2004;306:104–7. doi: 10.1126/science.1101489. [DOI] [PubMed] [Google Scholar]
- 96.Pyle AM. The tertiary structure of group II introns: implications for biological function and evolution. Critical Reviews in Biochemistry and Molecular Biology. 2010;45:215–32. doi: 10.3109/10409231003796523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lambowitz AM, Belfort M. Mobile Bacterial Group II Introns at the Crux of Eukaryotic Evolution. Microbiology spectrum. 2015;3 doi: 10.1128/microbiolspec.MDNA3-0050-2014. MDNA3-0050-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robart AR, Chan RT, Peters JK, Rajashankar KR, Toor N. Crystal structure of a eukaryotic group II intron lariat. Nature. 2014;514:193–7. doi: 10.1038/nature13790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tullius TD, Greenbaum JA. Mapping nucleic acid structure by hydroxyl radical cleavage. Current opinion in chemical biology. 2005;9:127–34. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 100.Harris ME, Christian EL. RNA crosslinking methods. Methods in enzymology. 2009;468:127–46. doi: 10.1016/S0076-6879(09)68007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu Cheng C, Chou F-C, Kladwang W, Tian S, Cordero P, Das R. Consistent global structures of complex RNA states through multidimensional chemical mapping. 2015 doi: 10.7554/eLife.07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kladwang W, VanLang CC, Cordero P, Das R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nature chemistry. 2011;3:954–62. doi: 10.1038/nchem.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Magnus M, Matelska D, Lach G, Chojnowski G, Boniecki MJ, Purta E, et al. Computational modeling of RNA 3D structures, with the aid of experimental restraints. RNA biology. 2014;11:522–36. doi: 10.4161/rna.28826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cruz JA, Blanchet MF, Boniecki M, Bujnicki JM, Chen SJ, Cao S, et al. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. Rna. 2012;18:610–25. doi: 10.1261/rna.031054.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miao Z, Adamiak RW, Blanchet M-F, Boniecki M, Bujnicki JM, Chen S-J, et al. RNA-Puzzles Round II: assessment of RNA structure prediction programs applied to three large RNA structures. Rna. 2015;21:1066–84. doi: 10.1261/rna.049502.114. [DOI] [PMC free article] [PubMed] [Google Scholar]