

Abstract
Introduction
Hoyeraal-Hreidarsson syndrome is a dyskeratosis congenita-related telomere biology disorder that presents in infancy with intrauterine growth retardation immunodeficiency, and cerebellar hypoplasia in addition to the triad of nail dysplasia, skin pigmentation, and oral leukoplakia. Patients with Hoyeraal-Hreidarsson syndrome often develop bone marrow failure in early childhood. Germline mutations in DKC1, TERT, TINF2, RTEL1, ACD, and PARN cause about 60% of Hoyeraal-Hreidarsson syndrome cases.
Case Report
We report 14 years of follow-up for a patient with Hoyeraal-Hreidarsson syndrome who initially presented as an infant with intrauterine growth retardation, microcephaly, and central nervous system calcifications. He was diagnosed with Hoyeraal-Hreidarsson syndrome at age six and had a complicated medical history including severe developmental delay, cerebellar hypoplasia, esophageal and urethral stenosis, hip avascular necrosis, immunodeficiency, and bone marrow failure evolving to myelodysplastic syndrome requiring hematopoietic cell transplantation at age 14. He had progressive skin pigmentation changes, oral leukoplakia, and nail dysplasia leading to anonychia. Whole exome sequencing identified novel biallelic variants in PARN.
Conclusions
This case illustrates that the constellation of IUGR, central nervous system calcifications, and cerebellar hypoplasia, esophageal or urethral stenosis, and cytopenias, in the absence of congenital infection, may be due to Hoyeraal-Hreidarsson syndrome. Early diagnosis of Hoyeraal-Hreidarsson syndrome is important to optimize medical management and provide genetic counseling.
Keywords: dyskeratosis congenita, Hoyeraal-Hreidarsson syndrome, telomere, PARN, microcephaly, CNS calcification
Introduction
Dyskeratosis congenita is an inherited bone marrow failure syndrome caused by dysfunctional telomere maintenance1, 2. It is diagnosed by the presence of the classic triad of nail dysplasia, lacy skin pigmentation, and oral leukoplakia. Additional clinical features seen in dyskeratosis congenita and related telomere biology disorders are shown in Table 1. (Table 1)1, 3. Blood leukocyte telomere lengths less than the first percentile for age are consistent with the diagnosis of dyskeratosis congenita and a consequence of germline mutations in telomere biology genes4. X-linked recessive dyskeratosis congenita is caused by mutations in DKC1 (MIM 300126). Autosomal dominant dyskeratosis congenita can be caused by mutations in TERC (MIM 602322), TERT (MIM 187270), RTEL1 (MIM 608833), or TINF2 (MIM 604319). Autosomal recessive inheritance of mutations in TERT, RTEL1, CTC1 (MIM 613129), NOP10 (MIM 606471), NHP2 (MIM 606470), WRAP53 (MIM 612661), ACD (MIM 609377) or PARN (MIM 604212) also cause dyskeratosis congenita1, 5-7. Germline mutations in these genes account for approximately 70-80% of dyskeratosis congenita cases.
Table 1. Clinical Manifestations of Dyskeratosis Congenita, Hoyeraal-Hreidarsson Syndrome, and Related Disorders.
The + symbol indicates the feature has been reported in at least 50% of affected individuals; +/- denotes present in some affected individuals but absent in others and could develop with age; features with – have not yet reported in the disorder. Table derived from literature review1, 10, 26, 28-30 and unpublished data from the NCI cohort study of inherited bone marrow failure syndromes.
| System | Feature | Dyskeratosis Congenita | Hoyeraal-Hreidarsson Syndrome | Revesz Syndrome | Coats plus |
|---|---|---|---|---|---|
| Neurologic | Intracranial calcifications | +/- | +/- | + | + |
| Cerebellar hypoplasia | +/-b | +a | +/- | +/- | |
| Microcephaly | +/- | +/- | +/- | +/- | |
| Developmental delay | +/- | + | +/- | +/- | |
| Progressive neurologic complications | +/- | + | +/- | +/- | |
| Leukodystrophy | - | - | - | + | |
| Parenchymal brain cysts | - | - | - | +/- | |
| Growth | Intrauterine growth retardation | +/- | +/- | +/- | + |
| Extrauterine growth retardation | +/- | +/- | +/- | + | |
| Dermatologic | Nail dysplasia | + | +/- | + | +/- |
| Lacy, reticular skin pigmentation | + | +/- | +/- | + | |
| Oral leukoplakia | + | + | +/- | - | |
| Early gray or sparse hair | +/- | +/- | +/- | + | |
| Nail dysplasia | + | +/- | + | +/- | |
| Ophthalmologic | Exudative retinopathy | - | - | +a | + |
| Retinal telangiectasias, hemorrhages | +/- | +/- | - | + | |
| Lacrimal duct stenosis | +/- | +/- | - | - | |
| Gastrointestinal | Gastrointestinal vascular ectasias | - | - | - | + |
| Esophageal stenosis | +/- | +/- | - | - | |
| Diarrhea, colitis | - | +/- | - | - | |
| Non-alcoholic liver disease | +/- | - | - | - | |
| Pulmonary | Pulmonary fibrosis | +/- | - | - | - |
| Hematopoietic | Immunodeficiency | +/- | + | - | - |
| Bone marrow failure | +/- | + | +/- | + | |
| Orthopedic | Osteopenia, fractures, poor bone healing | +/- | - | - | + |
| Avascular necrosis of hips or shoulders | +/- | - | - | - | |
| Urogenital | Urethral stenosis (males) | +/- | +/- | - | - |
| Other | Increased risk of cancer | +/- | - | - | - |
Required for diagnosis
Individuals reported as dyskeratosis congenita with cerebellar hypoplasia may have been misclassified and actually have Hoyeraal-Hreidarsson syndrome.
Hoyeraal-Hreidarsson syndrome is a form of dyskeratosis congenita with very early age at onset (Table 1). In addition to features of dyskeratosis congenita, patients with Hoyeraal-Hreidarsson syndrome have immunodeficiency8, 9, intrauterine growth retardation, developmental delay, and cerebellar hypoplasia; the latter is characteristic of Hoyeraal-Hreidarsson syndrome 10-12. Patients with Hoyeraal-Hreidarsson syndrome have extremely short leukocyte telomeres, even in comparison with other dyskeratosis congenita patients4. Mutations in a subset of dyskeratosis congenita-associated genes (DKC1, TINF2, TERT, RTEL1, ACD, and PARN) have been shown to cause Hoyeraal-Hreidarsson syndrome.
Mutations in PARN, which encodes poly(A)-specific ribonuclease, a deadenylase, have been linked with autosomal dominant familial pulmonary fibrosis13 and autosomal recessive Hoyeraal-Hreidarsson syndrome 5. Monoallelic deletions in PARN have recently been described in individuals with developmental delay or mental illness14. Mutations in PARN have been suggested to alter mRNA stability, DNA damage response, downregulate certain telomere biology proteins, and cause short telomeres5, 14.
We report 14 years of follow-up for a patient who initially presented as an infant with microcephaly, developmental delay, and central nervous system calcifications. The patient was diagnosed with Hoyeraal-Hreidarsson syndrome at six years of age and recently found to have AR mutations in the poly(A)-specific ribonuclease gene, PARN.
Methods
Family NCI-165 participated in an IRB-approved longitudinal cohort study at the National Cancer Institute (NCI) entitled “Etiologic Investigation of Cancer Susceptibility in Inherited Bone Marrow Failure Syndromes” (ClinicalTrials.gov Identifier: NCT00027274). This study includes comprehensive family history and individual history questionnaires, detailed medical record review, and biospecimen collection15. Telomere length was evaluated by flow cytometry with fluorescent in situ hybridization (flow FISH) in leukocytes 16. Comprehensive clinical evaluations were undertaken by the proband's clinical care team.
Whole exome sequencing (WES) was performed using DNA from the proband's brother (NCI-165-2), mother (NCI-165-3), and father (NCI-165-4) as previously described6, 17; there was insufficient DNA available from the proband for WES. As most cases of Hoyeraal-Hreidarsson syndrome are caused by biallelic mutations, we hypothesized that we could infer potential compound heterozygous or homozygous variants in the proband based on the genotypes of his first-degree relatives. We examined rare variants (MAF <0.7% in 1000 Genomes18 and the NHLBI ESP Exome Variant Server and prioritized them using in silico data using the Combined Annotation Dependent Depletion (CADD) scores19, PROVEAN20, SIFT21, PolyPhen-222, MutationTaster23, MutationAssessor24, FATHMM25, and the likelihood ratio test. All variants of interest were confirmed by Sanger sequencing in the proband's DNA.
Results
Clinical Case Report
The proband, NCI-165-1, was the first child of healthy, non-consanguineous parents, born at 39 weeks gestation, who weighed 2300 grams. He was noted to have microcephaly, hypotonia, and poor feeding as a neonate. A brain CT scan at three months of age showed thalamic and deep cerebral calcifications. Oral ulcers were noted before 12 months of age and his inability to tolerate oral feedings and failure to thrive prompted placement of a gastrostomy tube at 18 months. He was monitored by multiple pediatric subspecialties for severe developmental delay, esophageal stricture, undescended testes, microcephaly, seizures, and cerebellar ataxia. An MRI demonstrated cerebellar hypoplasia at age two years (Figure 1).
Figure 1. Clinical Features of the Proband.

The proband at age 6 years (left column) and 14 years (middle column). A) Facial features (top panels) include protuberant ears, broad nasal tip, broad prominent nasal root, full lips. Also noted are thin hair, microcephaly, and reticulated hyperpigmentation that have been progressive with age. B, C) Dry thickened skin of the hands and feet with progressive nail dystrophy leading to anonychia. D) Reticulated hyperpigmentation with areas of hypopigmentation, covering the entire skin surface became more pronounced with age. E) MRI illustrating cerebellar hypoplasia (yellow arrow) at age 2 years. F) Skin biopsy at 6 years of age shows mild epidermal atrophy with pigmentary incontinence and melanin-laden macrophages. G) Leukoplakia and ulcers of the tongue and small teeth at age 13 years. H) Colonoscopy at age 14 with multiple diffuse telangiectasias.
His family history was notable for a paternal aunt with lymphoma, paternal grandmother with breast cancer, paternal great uncle with leukemia, paternal great grandmother with breast cancer, maternal great aunt with ovarian cancer, and a maternal grandfather with emphysema or pulmonary fibrosis.
At five years of age, he was evaluated for persistent urethral meatus erosion. Physical examination revealed lingual and buccal leukoplakia, reticulated hyperpigmentation of the upper chest, and severe nail dystrophy and anonychia of several digits (Figure 1). Skin biopsy showed mild epidermal atrophy with pigmentary incontinence and melanin-laden macrophages. A complete blood count showed thrombocytopenia and mild macrocytosis but normal white blood cell count and hemoglobin. A bone marrow biopsy at age six showed 40% cellularity with left-shifted myeloid maturation. Laboratory testing of telomere length by flow FISH in leukocytes determined the patient's telomeres to be far below the 1st-percentile compared with age-matched controls (Figure 2). This constellation of findings is diagnostic of the Hoyeraal-Hreidarsson syndrome variant of dyskeratosis congenita.
Figure 2. Molecular Features of the Proband.
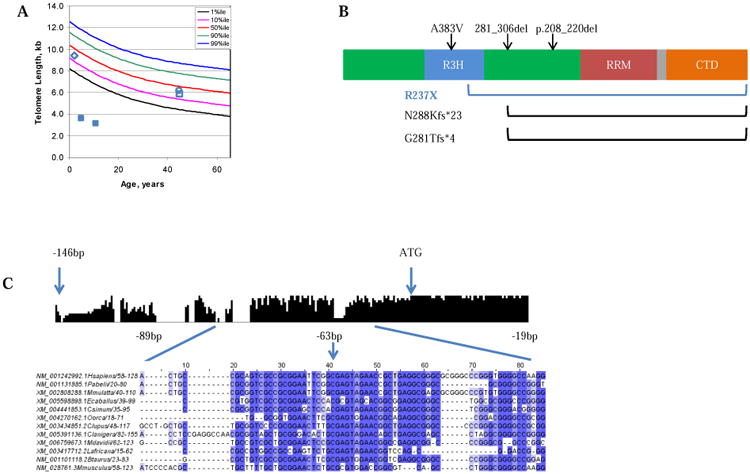
A) Lymphocyte telomere length, as measured by flow-FISH, for family NCI-165: proband, filled blue squares at age 5 and 13; brother, open blue diamond, father, open blue square, mother open blue circle. B) PARN contains three RNA-binding domains (the R3H domain [red], the RRM domain [RNA recognition motif, blue], two catalytic nuclease domains [green]), and a C-terminal domain (CTD, orange). A predicted bipartite NLS motif is located between the RRM domain and CTD (grey)27. Black arrows/brackets indicate previously reported PARN mutations associated with Hoyeraal-Hreidarsson syndrome 5, 14; The blue bracket indicates the deletion reported here. C) The histogram represents conservation amongst vertebrates across the PARN promoter region. Bases -89 to -19 (the region encompassing the 5′ UTR mutation detected in NCI-165-1) are shown in the detailed multiple sequence alignment below.
The proband developed worsening thrombocytopenia at eight years of age. A repeat bone marrow examination identified continued hypocellularity of 30% with left-shifted myeloid maturation and worsening reduction of erythrocytic and megakaryocytic progenitors. Cytogenetic analysis was normal (46XY[20]) and FISH for monosomy 7 and for trisomy 8 was negative. He was managed with periodic blood and platelet transfusions in addition to the androgen, oral oxymetholone, resulting in gradual improvement of his blood counts and transfusion independence.
Over the next several years, he experienced several significant medical events, including hip dislocation that required surgical correction and was complicated by delayed wound closure. He was treated for esophageal stenosis with serial dilations; however he continued to require all feedings via gastrostomy tube. His mucocutaneous findings progressed to include lacrimal duct stenosis with chronic epiphora, persistent urethral meatal stenosis, progressive hair loss with brittle thin hair, increasing skin hyperpigmentation, and advancement of nail dystrophy to complete anonychia (Figure 1).
By age 13, he had developed pancytopenia; bone marrow biopsies showed myelodysplastic changes with cellularity of 10%. Karyotyping revealed a clonal abnormality in all analyzed metaphases: 47, XY, +der(1)i(1)(q10)inv(q10;q25) in which there were two normal chromosomes 1 and an isochromosome of the long arm of chromosome 1 with an inversion. Progressive immunodeficiency was noted and he had prolonged pneumonitis due to Pneumocystis jiroveci. Severe, recurrent hematochezia developed requiring platelet and red blood cell transfusions. MRI enterography showed normal bowel wall thickness, but colonoscopy revealed multiple vascular telangiectatic lesions in the cecum and ascending colon (Figure 1). Oxymetholone was discontinued due to loss of hematologic response. Interestingly, after that, the hematochezia improved and repeat colonoscopy demonstrated resolution of telangiectasias.
The patient's cerebellar function was poor and he had only nonverbal communication and was in special education programs. Ophthalmologic examinations at age 13 years were normal with no cataracts, vitritis, retinopathy, or chorioretinitis. Thoracic CT found no evidence of pulmonary fibrosis at age 13, but a CT the following year showed interstitial thickening with associated bronchiectasis.
He underwent a nonmyeloablative hematopoietic cell transplant at age 14 because of life-threatening pancytopenia and high risk of developing acute myeloid leukemia associated with the myelodysplastic bone marrow changes with clonal abnormality. His donor was his 9/10 HLA-matched sibling. The preparative regimen consisted of Campath (3 mg test dose then 10 mg on day -21, 15 mg on day -20, and 20 mg on day -19), fludarabine 30 mg/m2/dose once daily on days -6 to -2, and total body irradiation of 200 cGy on day -1. Graft versus host disease prophylaxis consisted of cyclophosphamide 50 mg/kg on day +3, mycophenolate mofetil, and tacrolimus. Neutrophil engraftment occurred on day +26 and platelets were sustained at over 50,000 without transfusion on day +31. His post-transplant course was complicated by two episodes of E. coli bacteremia. Subsequently, he developed coagulase negative Staphlococcus bacteremia, cytomegalovirus reactivation, posterior reversible leukoencephalopathy syndrome complicated by a seizure, upper and lower gastrointestinal bleeding with duodenal hematoma, and acute graft versus host disease of the gastrointestinal tract and liver that were refractory to steroids. He developed respiratory failure requiring intubation due to pulmonary hemorrhage and died on day +119 post transplant.
Whole Exome Sequencing
WES identified 14 genes for which both parents carried one heterozygous rare variant and the brother inherited at most one of the two variants (Supplementary Table 1). Each parent had a different variant in PARN, a gene recently found to be mutated in patients with Hoyeraal-Hreidarsson syndrome5. Sanger sequencing revealed that the proband inherited a maternal nonsense mutation (chr16:14698077G>A [hg19], c.C709T [NM_002582.3], p.R237X [NP_002573.1]) and a paternal variant in the 5′ untranslated region (UTR, chr16:14724045G>A, c.-63C>T [NM_002582.3]); the healthy brother inherited neither variant.
The transcript bearing the premature stop codon, p.R237X, is predicted to be degraded by the nonsense mediated decay pathway. This mutation is extremely rare, with a MAF of 0.002% in the Exome Aggregation Consortium (ExAC) database (Exome Aggregation Consortium (ExAC), Cambridge, MA; http://exac.broadinstitute.org [accessed July 2015]); it is not present in either the ESP6500 dataset from the Exome Variant Server or the 1000 Genomes database. It is predicted to be deleterious by in silico algorithms, and its CADD score places it in the top 10% most deleterious mutations. If translated, this mutation would encode a protein lacking 402 amino acids (over 60% of the protein NP_002573.1), including part of an R3H domain, a ribonuclease domain, an RNA recognition motif, a nuclear localization sequence, and a C-terminal domain (Figure 2B).
The 5′ UTR variant, c.-63C>T, is in a highly conserved block upstream of the PARN start codon, and therefore may alter transcription of PARN (Figure 2C). The affected nucleotide is in a region of H3K27 acetylation as reported by the ENCODE consortium, adding further support to its role in transcriptional regulation of PARN. Additionally, in one of the three PARN transcript variants reported in the RefSeq database (NM_001134477), this variant is within two base pairs of the intron-exon border, and is therefore predicted to affect splicing of this transcript. This variant is not covered by the exome sequencing data in the ExAC database, and is not present in the 1000 Genomes data; the CADD score ranks this variant among the 10% most deleterious.
Discussion
This case illustrates the diagnostic and clinical management challenges often seen in Hoyeraal-Hreidarsson syndrome. In early infancy, the proband was thought to have congenital cytomegalovirus infection, although the antibody titers were not entirely consistent with that diagnosis. He was thoroughly evaluated for IUGR, congenital infections, severe developmental delay, failure to thrive, seizures, and esophageal and urethral stenosis by numerous experts. The diagnosis of Hoyeraal-Hreidarsson syndrome was not made until age six when his medical problems had progressed and the constellation of findings, including thrombocytopenia, prompted further evaluation.
In addition to advancing skin pigmentation abnormalities, nail dysplasia, and oral leukoplakia, our patient had progressive urethral and esophageal stenosis that required intervention. He had significant developmental delay associated with cerebellar hypoplasia and CNS calcifications. His cytopenias worsened with age and corresponded with a reduction in bone marrow cellularity. Notably, his platelet and red blood cell counts improved with oxymetholone but declined again with the onset of myelodysplasia.
Gastrointestinal ectasias are a common feature of the related telomere biology disorder, Coats plus, but this finding has not been widely reported in Hoyeraal-Hreidarsson syndrome. Our patient had widespread ectasias of the large bowel with resultant severe hematochezia. He also had CNS periventricular calcifications, another feature of the Coats-plus syndrome26, but he did not have the characteristic exudative retinopathy, retinitis or retinal vascular malformation when examined at age 13.
Germline variants in PARN were first linked to human disease with the discovery of autosomal dominant mutations in pulmonary fibrosis patients with short telomeres13. Subsequently, autosomal recessive PARN mutations were connected with dyskeratosis congenita/Hoyeraal-Hreidarsson syndrome and shown to likely affect telomere biology through widespread effects on mRNA stability, DNA damage response defects, and an association with down regulation of telomere biology proteins5. PARN deficiency also appears to be associated with loss of specific H/ACA box RNAs, snoRNAs and TERC14.
Tummala et al described two cases with novel biallelic PARN variants; in both cases, one copy of PARN was severely truncated5. Two patients with developmental delay and one with mental illness were reported to have monoalleleic deletions of part of PARN by Dhanraj et al14; all had normal brain MRIs and no known hematopoietic phenotype. Notably, patient 1 in the same report had a deletion and a missense variant of PARN and features consistent with Hoyeraal-Hreidarsson syndrome, including very short lymphocyte telomeres, global developmental delay, and small cerebellum.
Similarly, the patient described here bears a variant (R237X) resulting in a significant PARN truncation. Unlike the variants described previously, the second PARN allele in our patient is affected by a potential regulatory variant in the 5′ UTR. This mutation affects a highly conserved sequence in the PARN promoter region (Figure 2C), and may also alter splicing between the first and second exons of PARN transcript NM_001134477.
The cases reported by Tummala et al have marked similarity to our patient. All patients had significant developmental delay, with microcephaly and cerebellar hypoplasia, and developed bone marrow failure5. Additionally, DCR380 case 1, DCR380 case 2, DCR270 case 3, and our patient, NCI-165-1, had the dyskeratosis congenita mucocutaneous triad. The facial features shown in DCR 380 case 2 are remarkably similar to those in our patient, suggesting that recognition of similar features could be helpful in making future diagnoses.
In summary, this case illustrates that the constellation of IUGR, CNS calcifications, and cerebellar hypoplasia, esophageal or urethral stenosis, and cytopenias, in the absence of congenital infection, may be due to Hoyeraal-Hreidarsson syndrome. Early diagnosis of this complex disorder is important to optimize medical management and provide genetic counseling and testing.
Supplementary Material
Supplementary Table 1: Variants identifed by whole exome sequencing in the proband's first-degree relatives. There was insuffucient DNA for whole exome sequencing of the proband. Variants were evaluated as potential compound heterozygous or homozygous mutations in the proband based on the genotypes of his first-degree relatives. Variants in biologically plausible genes, PARN and MCPH1, were evaluated by Sanger sequencing in the proband, parents, and sibling. MCPH1 encodes microcephalin which is associated with primary microcephaly. However, the proband had neither of the variants carried by the parents. PARN encodes poly(A)-specific ribonuclease which was recently associated with Hoyeraal-Hreidarsson syndrome and pulmonary fibrosis and has newly discovered telomeric functions.
Acknowledgments
We are grateful to the family for their valuable contributions to our understanding of Hoyeraal-Hreidarsson syndrome. Dr. Lisa Cartwright, Pediatric Urology, Walter Reed National Military Medical Center (WRNNMC) is acknowledged for recognizing the atypical features of this patient and placing the initial referral to dermatology in 2006. Dr. Daniel Cordaro, Hematopathology, WRNNMC, is acknowledged for evaluation of bone marrow biopsies. Outstanding study support was provided by Lisa Leathwood, RN, Maureen Risch, RN, and Ann Carr, CGC, MS, Westat, Inc. The Bone Marrow Transplant Program at the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University Hospital, is acknowledged for their participation in the care of this patient.
This work was supported, in part, by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, and contract HHSN2612006550018C with Westat, Inc. (Rockville, MD, USA).
Disclaimer: The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army, U.S. Navy or the Department of Defense.
Funding: Intramural Research Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH
Footnotes
NCI DCEG Cancer Genomics Research Laboratory: Sara Bass, Joseph Boland, Laurie Burdett, Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. Hutchinson, Kristine Jones, Sally Larson, Kerrie Lashley, Hyo Jung Lee, Wen Luo, Michael Malasky, Jason Mitchell, David Roberson, Aurelie Vogt, Mingyi Wang, Meredith Yeager, Xijun Zhang
NCI DCEG Cancer Sequencing Working Group: Neil E. Caporaso, Stephen J. Chanock, Mark H. Greene, Lynn R. Goldin, Alisa M. Goldstein, Allan Hildesheim, Nan Hu, Maria Teresa Landi, Jennifer T. Loud, Phuong L. Mai, Mary L. McMaster, Lisa Mirabello, Lindsay Morton, Melissa Rotunno, Douglas R. Stewart, Phil Taylor, Geoffrey S. Tobias, Margaret A. Tucker, Xiaohong R. Yang, Guoqin Yu
Conflict of Interest Statement: The authors have no conflicts of interest relevant to this article to disclose.
Contributors' Statement: Dr. Ashley M. Burris evaluated clinical data, drafted the initial manuscript and reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Bari J. Ballew analyzed exome sequencing data, performed bioinformatic analyses, and reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Joshua B. Kentosh evaluated clinical data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Clesson E. Turner evaluated clinical data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Scott A. Norton evaluated clinical data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
NCI DCEG Cancer Genomics Research Laboratory
NCI DCEG Cancer Sequencing Working Group
Dr. Neelam Giri evaluated clinical and genetic data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Blanche P. Alter evaluated clinical and genetic data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Anandani Nellan evaluated clinical data, drafted the initial manuscript and reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Christopher Gamper evaluated clinical data, drafted the initial manuscript and reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Kip R. Hartman conceptualized and designed the study, evaluated clinical data, drafted the initial manuscript and reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Sharon A. Savage conceptualized and designed the study, evaluated clinical data, drafted the initial manuscript and reviewed and revised the manuscript, and approved the final manuscript as submitted.
NCI DCEG Cancer Genomics Research Laboratory: Performed exome and Sanger sequencing.
NCI DCEG Cancer Sequencing Working Group: Contributed expertise in genomics and internal control samples for exome sequencing.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013;6(3):327–337. doi: 10.1586/ehm.13.23. [DOI] [PubMed] [Google Scholar]
- 2.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 3.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–359. doi: 10.3324/haematol.2011.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151–2160. doi: 10.1172/JCI78963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kocak H, Ballew BJ, Bisht K, Eggebeen R, Hicks BD, Suman S, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014;28(19):2090–2102. doi: 10.1101/gad.248567.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo Y, Kartawinata M, Li J, Pickett HA, Teo J, Kilo T, et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124(18):2767–2774. doi: 10.1182/blood-2014-08-596445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jyonouchi S, Forbes L, Ruchelli E, Sullivan KE. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum--a single-center pediatric experience. Pediatr Allergy Immunol. 2011;22(3):313–319. doi: 10.1111/j.1399-3038.2010.01136.x. [DOI] [PubMed] [Google Scholar]
- 9.Cossu F, Vulliamy TJ, Marrone A, Badiali M, Cao A, Dokal I. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br J Haematol. 2002;119(3):765–768. doi: 10.1046/j.1365-2141.2002.03822.x. [DOI] [PubMed] [Google Scholar]
- 10.Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015 doi: 10.1111/bjh.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borggraefe I, Koletzko S, Arenz T, Fuehrer M, Hoffmann F, Dokal I, et al. Severe variant of x-linked dyskeratosis congenita (Hoyeraal-Hreidarsson Syndrome) causes significant enterocolitis in early infancy. J Pediatr Gastroenterol Nutr. 2009;49(3):359–363. doi: 10.1097/MPG.0b013e3181a15b94. [DOI] [PubMed] [Google Scholar]
- 12.Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999;107(2):335–339. doi: 10.1046/j.1365-2141.1999.01690.x. [DOI] [PubMed] [Google Scholar]
- 13.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–517. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhanraj S, Gunja SM, Deveau AP, Nissbeck M, Boonyawat B, Coombs AJ, et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN) J Med Genet. 2015 doi: 10.1136/jmedgenet-2015-103292. [DOI] [PubMed] [Google Scholar]
- 15.Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150(2):179–188. doi: 10.1111/j.1365-2141.2010.08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nature protocols. 2006;1(5):2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 17.Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132(4):473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7(8):575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 24.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44(3):338–342. doi: 10.1038/ng.1084. [DOI] [PubMed] [Google Scholar]
- 27.He GJ, Yan YB. Self-association of poly(A)-specific ribonuclease (PARN) triggered by the R3H domain. Biochim Biophys Acta. 2014;1844(12):2077–2085. doi: 10.1016/j.bbapap.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–764. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polvi A, Linnankivi T, Kivela T, Herva R, Keating JP, Makitie O, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet. 2012;90(3):540–549. doi: 10.1016/j.ajhg.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Variants identifed by whole exome sequencing in the proband's first-degree relatives. There was insuffucient DNA for whole exome sequencing of the proband. Variants were evaluated as potential compound heterozygous or homozygous mutations in the proband based on the genotypes of his first-degree relatives. Variants in biologically plausible genes, PARN and MCPH1, were evaluated by Sanger sequencing in the proband, parents, and sibling. MCPH1 encodes microcephalin which is associated with primary microcephaly. However, the proband had neither of the variants carried by the parents. PARN encodes poly(A)-specific ribonuclease which was recently associated with Hoyeraal-Hreidarsson syndrome and pulmonary fibrosis and has newly discovered telomeric functions.