

Abstract
Chronic progressive external ophthalmoplegia is a mitochondrial disorder usually caused by single or multiple mitochondrial DNA (mtDNA) deletions and, more rarely, by maternally inherited mtDNA point mutations, most frequently in tRNA genes (MTT).
We report on a patient presenting with a progressive eyelid ptosis with bilateral ophthalmoparesis, dysphagia, dysphonia and mild proximal limb weakness associate with a mild movement disorder characterized by abnormal involuntary movements involving head and limbs, imbalance and gait instability.
Muscle biopsy demonstrated the presence of ragged red fibers and several cytochrome-C-oxidase negative fibers. Molecular analysis showed the novel m.5613T > C heteroplasmic mutation in the mitochondrial tRNAAla gene (MTTA) which disrupts a conserved site and fulfills the accepted criteria of pathogenicity. Moreover, a 38 CAG trinucleotide repeat expansion was found on the huntingtin gene, thus configuring a singular CPEO/“reduced penetrance” Huntington disease “double trouble”.
With this novel MTTA point mutation, we extend the spectrum of provisional pathogenic changes in this gene, which is a very rare site of pathogenic mutation, and confirm that clinical expression of these mutations is hardly ever heterogeneous, including myopathy and CPEO.
Mitochondrial involvement is an emerging key determinant in the pathogenesis of Huntington disease and it is well known that mutant huntingtin influences the mitochondrial respiratory complexes II and III. A synergist effect of the HTT and MTTA mutations on respiratory chain function may be hypothesized in our patient and should be regarded as a spur for further studies on the mtDNA/HTT reciprocal interactions.
Keywords: Mitochondrial diseases, CPEO, Huntington disease, mtDNA, tRNA
1. Introduction
Mitochondrial DNA (mtDNA) mutations are associated with a wide spectrum of disorders involving different tissues, particularly brain and muscle [1], [2].
Chronic progressive external ophthalmoplegia (CPEO) is a classical mitochondrial disorder characterized by bilateral progressive ptosis and ophthalmoplegia. These ocular features can develop either in isolation or in association with other prominent neurological deficits. Molecularly, CPEO can be classified into three distinct genetic subgroups depending on whether patients harbour single large-scale mtDNA deletions or multiple mtDNA deletions secondary to a nuclear mutation disrupting mtDNA replication and repair or point mutations in the mitochondrial genome.
Patients harbouring a single mtDNA deletion are sporadic and the mutation is thought to arise in oogenesis or in early embryonic development. In contrast, multiple deletions of mtDNA are observed in autosomal dominant or recessive forms of CPEO [2], [3], [4], [5], [6], [7]. More rarely, CPEO is maternally inherited and due to mtDNA point mutations, most frequently in tRNA genes (MTT) [8].
Huntington disease (HD) is an autosomal dominant, adult-onset, progressive neurodegenerative disease characterized by abnormal movements, cognitive impairment and psychiatric disorders [9]. It is caused by an expanded CAG repeat in the gene HTT encoding the huntingtin on chromosome 4, resulting in an expanded N-terminal polyglutamine stretch in the protein, and it is characterized by a progressive atrophy of the striatum as well as cortical and other extrastriatal structures [10].
Here, we report on the identification of a novel point mutation in the MTTA gene in a patient affected with a sporadic CPEO. She also presents a “reduced penetrance” Huntington disease (HD). This very unusual “double trouble” condition represents the first reported case of a concomitance of these two disorders.
2. Patient and methods
2.1. Patient
A 70-year-old woman presented with progressive eyelid ptosis, bilateral ophthalmoparesis, dysphagia, dysphonia, mild proximal limb weakness, numbness and bilateral deafness since age of 55.
By the age of 64 she noticed some abnormal involuntary movements involving head and limbs and imbalance and gait instability. Mini Mental State Examination was rated 20/30. Family history was unremarkable.
Neurological examination showed bilateral eyelid ptosis, severe ophthalmoparesis in all the directions of gaze and mild involuntary choreiform movements of the head and distal limbs.
Blood and urine assays, including resting blood lactate, were normal except for serum CK levels that were repeatedly 2–3 times the normal values. Pure tone audiometry showed bilateral sensorineural deafness. EMG recording showed low-amplitude and short duration of motor unit potentials in the four limbs. Electroencephalography was normal as well as electrocardiography and echocardiography. Brain MRI showed cerebellar atrophy that affects more severely the superior and inferior semilunar lobules and the vermis. There is also a widespread mild brain cortical atrophy and an extrinsic ocular muscles atrophy.
A biopsy of vastus lateralis muscle was performed and urine and blood samples were collected.
2.2. Methods
Morphological study of the muscle biopsy were done as described [11]. Mitochondrial respiratory chain complex activities were assayed according to established spectrophotometric methods and expressed as nmol/min/mg of protein [12]. DNA was extracted from muscle, blood and urine samples by Puregene DNA purification Kit (Gentra Systems, MN, USA).
Southern Blot was carried out with standardized procedures [8]. The sequence of the entire mtDNA was performed with suitable nucleotide primers as reported [13]. PCR-RFLP analysis was used to quantify the percentage of the m.5613T > C mutation. In brief, a fragment between mtDNA nucleotides 5544 and 5739 was amplified, digested with endonuclease SfaNI, and electrophoresis was performed on a 3% agarose gel. The enzyme cuts the wild-type DNA into 2 fragments, whereas the presence of the mutation abolishes the restriction site.
Isolation of single muscle fibers was performed on serial 10-μm thick transversal sections obtained from frozen muscle and stained for COX activity using standard methods; after fixation and dehydratation, two independent laser capture microdissection (LCM) of COX positive and COX negative muscle fibers were performed under direct microscopic visualization (PALM Robot Microbeam, PALM MicrolaserTechnologies AG, Munich, Germany). Isolated DNA (using QIAmp DNA Micro Kit, QIAGEN, Hilden, Germany) was used for PCR-RFLP as described above.
Molecular test for Huntington disease was performed as previously described [10].
3. Results
Muscle biopsy showed few atrophic fibers, many fibers with subsarcolemmal accumulation of mitochondria, some ragged red fibers (RRFs) (Fig. 1A) and several cytochrome C oxidase (COX)-negative fibers (Fig. 1B). All RRFs were COX-negative. No major rearrangements in mitochondrial DNA were detected by Southern Blot (data not shown).
Fig. 1.

Muscle biopsy showed mitochondrial abnormalities:
A: Modified Gomori's Trichrome staining showing ragged red fibers
B: COX-SDH double staining showing several COX-negative fibers
Sequencing of the whole mtDNA in muscle revealed a heteroplasmic m.5613T > C transition in the tRNA Alanine gene (MTTA) which disrupts a highly conserved residue through the species in the V-region of the molecule (Fig. 2A, C and D). This change was absent in one hundred and fifty unrelated muscle controls and in large mitochondrial variation databases (Mitomap, www.mitomap.org; Human Mitochondrial Genome Database, www.mtdb.igp.uu.se). The mutation, which accounted for 96% of the total mitochondrial genomes in muscle (Fig. 3A), was no detectable in blood and urine from the patient and her two sons (Fig. 2B).
Fig. 2.

In DNA from patient's muscle biopsy, electropherogram showed the heteroplasmic m.5613T > C transition located in the tRNA Alanine gene (MTTA) (A) which disrupt a highly conserved residue through the species (C) in the V-region of the molecule (D). Mutation is not detectable on patient's urine (B).
Fig. 3.
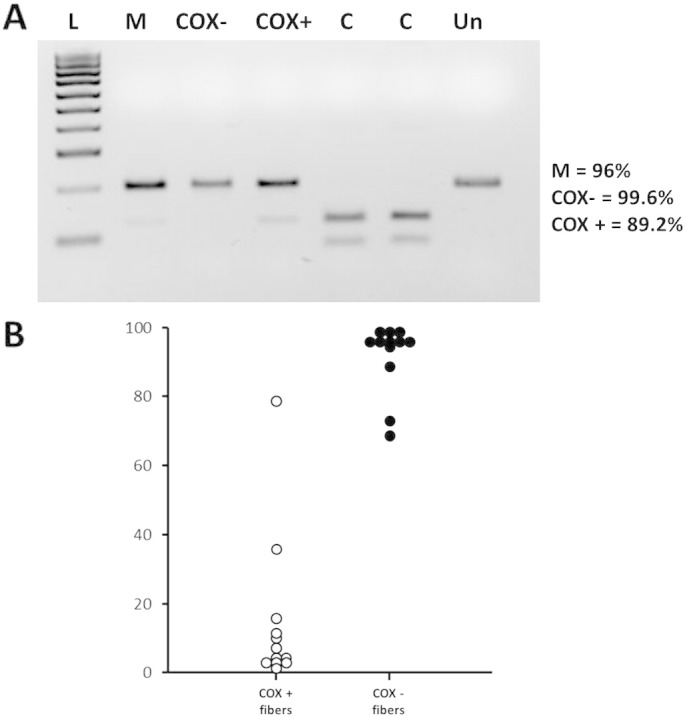
A: PCR-RFLP analysis showed that the proportion of mutant genomes was about 96%, in skeletal muscle and 99% and 89% in a representative set of COX negative andCOX positive muscle fiber, respectively (in control samples, the mutation load is < 0.5%). The endonuclease SfaNI cleaves the wild type molecules in two fragments sized 75 and120 base pairs (bp), respectively, whereas the presence of the m.5613T > C transition abolishes the single site of cleavage (PCR size 195 bp). L: DNA ladder; M: patient's skeletal muscle; C: control DNA; Un: uncut DNA.
B: Single-fiber PCR analysis for m.5613T > C mutation showing an higher rate of mutant load in COX negative fibers (n = 12) compared to COX positive fibers (n = 12).
PCR-RFLP analysis on COX-positive and COX-deficient single fibers clearly showed highest levels of the mutation segregating with the biochemical defect (Fig. 3B). Biochemical assay showed a partial reduction of complexes I and IV (30% and 24% respectively) and a severe impairment of complex II + III activity (80%) (Table 1); citrate synthase was increased, suggesting mitochondrial proliferation. An expansion of 38 CAG repeats was detected in the coding region of HTT gene, which falls in the range of intermediate/reduced penetrance alleles (data not shown).
Table 1.
Mitochondrial enzyme assay showed a partial reduction of complexes I and IV (30% and 24%, respectively) and a severe impairment of the activity of complexes II + III (80%). Activity is expressed as mmol/min/g tissue; in parenthesis, the activity value compared to citrate synthase.
| Enzymes | Enzyme activity | Normal values |
|---|---|---|
| NADH dehydrogenase | 20.13 (1.08) | 27.5–39.5 (1.56–2.60) |
| Succinate dehydrogenase | 2.06 (0.11) | 1.2–2.0 (0.05–0.105) |
| NADH cytochrome C reductase (I + III) | 2.56 (0.14) | 0.65–1.50 (0.11–0.25) |
| Succinate cytochrome c reductase (II + III) | 0.15 (0.01) | 0.45–0.90 (0.05–0.08) |
| Cytochrome C oxidase | 2.51 (0.13) | 1.80–2.45 (0.17–0.28) |
| Citrate synthase | 18.70 | 8.91–15.00 |
4. Discussion
Both large rearrangements and point mutations in mtDNA have been described in association with sporadic CPEO [1], [8]. In our patient we identified the novel heteroplasmic m.5613T > C mutation in the MTTA gene that we consider to be pathogenic for several reasons: first, it is consistent with the histochemical and biochemical findings; second, the mutation disrupts a strongly conserved base pair site in the molecule; third, it is heteroplasmic and the mutational load is related to COX deficiency, as shown by single-fiber PCR; fourth, it was absent in 150 unrelated control subjects. Moreover, the m.5613T > C mutation results “definitely pathogenetic” by applying the pathogenicity scoring system for mitochondrial tRNA variations (which summarize functional and biochemical studies, heteroplasmy, segregation, conservation) [14].
The MTTA gene is a rare site of pathogenic mutation. The Mitomap dataset (www.mitomap.org) reports four pathogenic mutations in the tRNAAla gene, two of them associated with CPEO and two with myopathy, all of them having high mtDNA heteroplasmy thresholds [15], [16], [17], [18]. More recently, two other mutations in the tRNAAla gene (m.5631G > A and m.5610G > A) was reported, again associated with isolated myopathy despite very high-mutation levels in all the tissues [19]. The CPEO mutation m.5628T > C and two other changes (m.5587T > C and m.5655T > C) are described to have a modifying role in the phenotypic manifestation in families with hearing loss [20], [21], [22].
By reporting this new mutation, we extend the spectrum of provisional pathogenic changes in the tRNAAla gene and confirm that the heteroplasmy threshold for impairment of respiratory chain function is usually very high for mutations in this gene and that their clinical expression is hardly ever heterogeneous, including substantially myopathy and CPEO.
Noteworthy, our patient presents with the uncommon association of CPEO and a late-onset HD whose diagnosis has been established by the presence of a mild movement disorder and an expansion of CAG repeats in the range of “reduced penetrance” with a count of 38 triplets.
HD is transmitted with an autosomal dominant pattern with a prevalence of 5–10 per 100,000 individuals in the Caucasian population [9], [10]. Subjects with more than 40 CAG-repeats in the first exon of the huntingtin gene show a complete penetrance of the disease, with an inverse correlation between number of triplets and age of onset. CAG-repeat expansion ranging between 36 and 39 shows reduced penetrance [9].
Huntingtin is a 350 KDa protein ubiquitously expressed in many tissues in the body, with the highest levels in the brain [23], [24]. The exact function of this protein is not known, but it seems involved in signaling, transporting materials, binding proteins and protecting against programmed cell death in neurons [23], [24], [25], [26], [27]. Since it is primarily associated with vesicles and microtubules, it appears to play a role in cytoskeletal anchoring and transporting mitochondria [23]. It has been recently showed that mutant huntingtin could be implicated in mtDNA damage and mitochondrial DNA impairment could be an early biomarker for HD neurodegeneration, supporting the hypothesis that mtDNA dysfunction might contribute to the pathogenesis of the disorder [27].
Significant abnormalities on muscle biopsy are uncommon in HD patients, but some cases displaying mitochondrial pathology have been reported [28], [29]. Interestingly, mutant huntingtin influences the mitochondrial complex II/III function in neuronal and non-neuronal tissue as skeletal muscle and enhances the expression of Drp1 and Fis1, two GTPases that increase mitochondrial fission with subsequent risk for mtDNA integrity [30], [31], [32]. In our patient, we confirmed the presence of a severe impairment of complexes II/III, as already reported in muscles form HD patients, in addition to demonstrating a less prominent deficiency of complexes I and IV.
Finally, we reported the unusual association of two clinical conditions which both affect the function of mitochondrial respiratory chain, thus an additive role of huntingtin and MTTA mutation in determining oxidative phosphorylation damage in our patient could be supposed.
To our knowledge, this is the first description in the literature of such “double trouble”.
Acknowledgements
Authors thank Doctors Chiara Mazzanti and Michele Menicagli for help in single muscle fibre studies.
References
- 1.Zeviani M., Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 2.Lightowlers R.N., Taylor R.W., Turnbull D.M. Mutations causing mitochondrial disease: what is new and what challenges remain? Science. 2015;349:1494–1499. doi: 10.1126/science.aac7516. [DOI] [PubMed] [Google Scholar]
- 3.Greaves L.C., Reeve A.K., Taylor R.W., Turnbull D.M. Mitochondrial DNA and disease. J. Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 4.Kaukonen J., Juselius J.K., Tiranti V., Kyttala A., Zeviani M., Comi G.P. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 5.Spelbrink J.N., Li F.Y., Tiranti V., Nikali K., Yuan Q.P., Tariq M. Human mitochondrial DNA deletions associated with mutations in the gene encoding twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 6.Van Goethem G., Dermaut B., Lofgren A., Martin J.J., Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 7.Filosto M., Mancuso M., Nishigaki Y. Clinical and genetic heterogeneity in progressive external ophthalmoplegia due to mutations in polymerase gamma. Arch. Neurol. 2003;60:1279–1284. doi: 10.1001/archneur.60.9.1279. [DOI] [PubMed] [Google Scholar]
- 8.Chinnery P.F., Johnson M.A., Taylor R.W., Durward W.F., Turnbull D.M. A novel mitochondrial tRNA isoleucine gene mutation causing chronic progressive external ophtalmoplegia. Neurology. 1997;49:1166–1168. doi: 10.1212/wnl.49.4.1166. [DOI] [PubMed] [Google Scholar]
- 9.Ha A.D., Fung V.S.C. Huntington's disease. Curr. Opin. Neurol. 2012;25:491–498. doi: 10.1097/WCO.0b013e3283550c97. [DOI] [PubMed] [Google Scholar]
- 10.The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 11.Bancroft J.D., Cook H.C. Churchill Livingstone; Edimburgh: 1984. Manual of histological techniques. [Google Scholar]
- 12.Spinazzi M., Casarin A., Pertegato V. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 13.Andreu A.L., Marti R., Hirano M. Analysis of human mitochondrial DNA mutations. In: Potter N.T., editor. Methods in Molecular Biology, Neurogenetics: Methods and Protocols. Vol. 217. Humana Press Inc; Totowa NJ: 2002. pp. 185–197. [DOI] [PubMed] [Google Scholar]
- 14.Yarham J.W., Al-Dosary M., Blakely E.L. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum. Mutat. 2011;32:1319–1325. doi: 10.1002/humu.21575. [DOI] [PubMed] [Google Scholar]
- 15.Swalwell H., Deschauer M., Hartl H. Pure myopathy associated with a novel mitochondrial tRNA gene mutation. Neurology. 2006;66:447–449. doi: 10.1212/01.wnl.0000196490.36349.83. [DOI] [PubMed] [Google Scholar]
- 16.McFarland R., Swalwell H., Blakely E.L. The m.5650G N a mitochondrial tRNA(Ala) mutation is pathogenic and causes a phenotype of pure myopathy. Neuromuscul. Disord. 2008;18:63–67. doi: 10.1016/j.nmd.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Pinos T., Marotta M., Gallardo E. A novel mutation in the mitochondrial tRNA(Ala) gene (m.5636T N C) in a patient with progressive external ophthalmoplegia. Mitochondrion. 2011;11:228–233. doi: 10.1016/j.mito.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Spagnolo M., Tomelleri G., Vattemi G., Filosto M., Rizzuto N., Tonin P. A new mutation in the mitochondrial tRNA(Ala) gene in a patient with ophthalmoplegia and dysphagia. Neuromuscul. Disord. 2001;11:481–484. doi: 10.1016/s0960-8966(01)00195-x. [DOI] [PubMed] [Google Scholar]
- 19.Lehmann D., Schubert K., Joshi P.R. Pathogenic mitochondrial mt-tRNA Ala variants are uniquely associated with isolated myopathy. Eur. J. Hum. Genet. 2015;23(12):1735–1738. doi: 10.1038/ejhg.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han D., Dai P., Zhu Q. The mitochondrial tRNA(Ala) T5628C variant may have a modifying role in the phenotypicmanifestation of the 12S rRNA C1494T mutation in a large Chinese family with hearing loss. Biochem. Biophys. Res. Commun. 2007;357:554–560. doi: 10.1016/j.bbrc.2007.03.199. [DOI] [PubMed] [Google Scholar]
- 21.Tang X., Li R., Zheng J. Maternally inherited hearing loss is associated with the novel mitochondrial tRNA Ser(UCN) 7505T N C mutation in a Han Chinese family. Mol. Genet. Metab. 2010;100:57–64. doi: 10.1016/j.ymgme.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R.A., Guan M.X. Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 2004;32:867–877. doi: 10.1093/nar/gkh226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirendeb U.P., Calkins M.J., Manczak M. Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum. Mol. Genet. 2012;21:406–420. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherzinger E., Lurz R., Turmaine M. Huntingtin-encoded polyglutamine expansions from amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 25.Reddy P.H., Shirendeb U. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim. Biophys. Acta. 2012;1822:101–110. doi: 10.1016/j.bbadis.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirendeb U., Reddy A.P., Manczak M., Calkins M.J., Mao P., Tagle D.A., Reddy P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum. Mol. Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acevedo-Torres K., Berríos L., Rosario N. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease. DNA Repair. 2009;8:126–136. doi: 10.1016/j.dnarep.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kosinski C.M., Schlangen C., Gellerich F.N. Myopathy as a first symptom of Huntington's disease in a marathon runner. Mov. Disord. 2007;22:1637–1640. doi: 10.1002/mds.21550. [DOI] [PubMed] [Google Scholar]
- 29.Zielonka D., Piotrowska I., Marcinkowski J.T., Mielcarek M. Skeletal muscle pathology in Huntington's disease. Front. Physiol. 2014;5 doi: 10.3389/fphys.2014.00380. 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner C., Cooper J.M., Schapira A.H. Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov. Disord. 2007;22:1715–1721. doi: 10.1002/mds.21540. [DOI] [PubMed] [Google Scholar]
- 31.Mancuso M., Filosto M., Orsucci D., Siciliano G. Mitochondrial DNA sequence variation and neurodegeneration. Hum. Genomics. 2008;3:71–78. doi: 10.1186/1479-7364-3-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Correia S.C., Santos R.X., Perry G., Zhu X., Moreira P.I., Smith M.A. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Adv. Exp. Med. Biol. 2012;724:205–221. doi: 10.1007/978-1-4614-0653-2_16. [DOI] [PubMed] [Google Scholar]