

Abstract
The Wnt pathway is an evolutionarily conserved and tightly regulated signaling network with important roles in embryonic development and adult tissue regeneration. Impaired Wnt pathway regulation, arising from mutations in Wnt signaling components, such as Axin, APC, and β-catenin, results in uncontrolled cell growth and triggers oncogenesis. To explore the reported link between CK2 kinase activity and Wnt pathway signaling, we sought to identify a potent, selective inhibitor of CK2 suitable for proof of concept studies in vivo. Starting from a pyrazolo[1,5-a]pyrimidine lead (2), we identified compound 7h, a potent CK2 inhibitor with picomolar affinity that is highly selectivity against other kinase family enzymes and inhibits Wnt pathway signaling (IC50 = 50 nM) in DLD-1 cells. In addition, compound 7h has physicochemical properties that are suitable for formulation as an intravenous solution, has demonstrated good pharmacokinetics in preclinical species, and exhibits a high level of activity as a monotherapy in HCT-116 and SW-620 xenografts.
Keywords: CK2 kinase; pyrazolo[1,5-a]pyrimidine; Wnt; β-catenin
The serine/threonine protein kinase CK2 is a constitutively active heterotetrameric complex composed of two catalytic (α or α′) and two regulatory (β) subunits,1 which has emerged as an attractive drug discovery target in oncology.2 Researchers from Cylene have recently advanced CX-4945, a selective, orally available inhibitor of CK2 into the clinic for treatment of patients with solid tumors and hematological malignancies.3
Among its diverse functions, CK2 interacts with and regulates multiple components of the Wnt pathway, an evolutionarily conserved signaling network that regulates embryonic development and the regeneration of intestinal epithelial cells.4 Certain cancers, including colorectal carcinoma (CRC), arise due to gene mutations among constituents of the Wnt pathway, including the CK2 substrates dishevelled (Dvl), APC, and β-catenin.5 Inhibition of CK2, either by RNA knockdown or with small molecules, decreases β-catenin–Tcf-mediated transcription of Wnt target genes such as survivin and leads to cell death and apoptosis in a range of CRC lines.6,7 In addition, elevated levels of CK2 activity have been reported in CRC tissue samples and expression levels correlate with poor prognosis in CRC patients.8,9 Taken together, these data illustrate the potential utility of CK2 inhibitors in CRC and other cancers characterized by aberrant Wnt pathway activity.
We sought to identify a potent, selective inhibitor of CK2 kinase for hypothesis testing in vivo using preclinical models of CRC. An early probe from our previously described series of ATP-competitive pyrazolo[1,5-a]pyrimidine-derived inhibitors of CK2 (1, 2; Figure 1) was used to assess the link between CK2 inhibition and Wnt signaling.10
Figure 1.

Early pyrazolo[1,5-a]pyrimidine leads (1 and 2).
Treatment of DLD-1(APC mutant) cells with 2 inhibits β-catenin phosphorylation and decreases Wnt-mediated gene transcription as shown in a Luciferase reporter assay in APC mutant DLD-1 cells (IC50 = 0.06 μM).11 In an acute dose pharmacokinetic/pharmacodynamic (PK/PD) study, treatment of DLD-1/AKT1 overexpressing murine xenografts with 2 (10 mg/kg, PO) resulted in the 20% inhibition of Wnt-mediated luciferase gene transcription at 8 h, and coincided with an unbound drug concentration at the level of the Wnt reporter IC50. When tested in disease model studies using a murine DLD-1(APCmut) xenograft, compound 2 showed limited tumor growth inhibition.10
Our medicinal chemistry strategy subsequently focused on achieving potent inhibition of the Wnt pathway, as demonstrated using a Wnt luciferase reporter assay in DLD-1 cells,11 while seeking to improve physicochemical properties, enhance target coverage, and potentially deliver increased in vivo efficacy in preclinical models characterized by aberrant Wnt signaling.
To aid our design efforts we obtained the X-ray crystallograhic structure of human CK2α at 2.55 Å resolution in complex with compound 2 (Figure 2).12 The inhibitor occupies the ATP-binding cleft and is anchored to the hinge region via a pair of hydrogen bonds. The C7 aminocyclopropane group is directed toward solvent and forms a hydrogen bond with the main-chain carbonyl oxygen of V116, while the N1 position of the pyrazolopyrimidine core interacts with the amide NH of the same residue. An additional interaction is observed between an ordered water molecule and the C5 NH of the inhibitor. The ortho-methyl substituent enforces an energetically disfavored cisoid configuration of the acetamide that permits coordination of the carbonyl group with a nearby water molecule. This bound water, adjacent to the gatekeeper residue F113, also interacts with the cyano group of the pyrazolo[1,5-a]pyrimidine core and the main-chain amide of D175. In addition, the solved structure suggested that substitution of the ortho-position (R1 in Figure 1, Scheme 1, and Table 1) with polar functionality could enable additional energetically favorable interactions with the protein.
Figure 2.
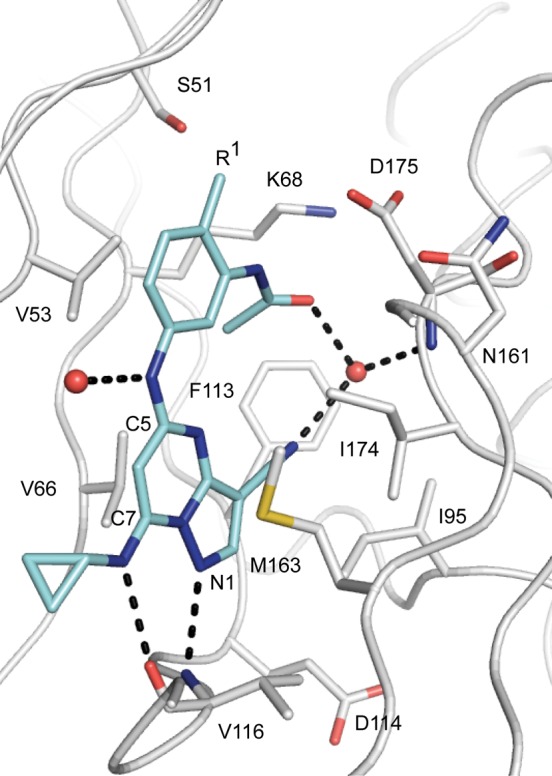
X-ray crystallographic structure of human CK2α in complex with compound 2 determined at 2.55 Å resolution (PDB accession code: 5H8B).12,13 Water molecules are shown as red spheres. Hydrogen bonds to the inhibitor are shown as black dashed lines.14
Scheme 1. Synthesis of Compounds 7a–q.

Reagents and conditions: (a) R,R′NH, Cs2CO3, DMF, 80 °C; (b) H2, Pd/C, MeOH, 25 °C; (c) 6, 5 mol % Xantphos, 10 mol % Pd2(dba)3, Cs2CO3, DMA, 150 °C in a microwave; (d) 6, KF, NMP, or DMSO, 150 °C; (e) 6, 20 mol % tBuXphos, 10 mol % Pd2(dba)3, Cs2CO3, NMP/dioxane, 100 °C; (f) TFA, CH2Cl2, 25 °C; (g) 7f, pyridine, MsCl, 0 °C; (h) R,R′NH, MeCN, 65 °C.
Table 1. Optimization of the ortho-Substituent Improves Cellular Potency, Solubility, and Metabolic Stability.

Mean value of two experiments. Deviations were within <±25%.
Intrinsic clearance (CLint) determined from human liver microsome incubations (μL/min/mg) or rat hepatocyte incubations (μL/min/106 cells); ND = not determined.
Analogues of 2 were synthesized by adapting the convergent approach described in our earlier work (Scheme 1).10 Treatment of N-(2-fluoro-5-nitro-phenyl)acetamide (3) with secondary amines, followed by reduction of the nitro group in the resulting products (4), afforded anilines of general structure 5. Palladium-catalyzed or KF-promoted coupling of these intermediates (5) with 5-chloro-7-(cyclopropylamino) pyrazolo[1,5-a]pyrimidine-3-carbonitrile (6) provided the desired analogues (Table 1) either directly (7a–c, f, g, j, k), via the methansulfonyl derivative of 7f or, as in the case of 7h–i, l, and q, following TFA deprotection of the corresponding Boc derivatives (Scheme 1)
Introduction of an N-methylpiperazinyl substituent (7a) enhanced solubility, but reduced enzymatic and cellular potency relative to 2. However, the acyclic 1,2-diaminoethyl substituent of 7b imparted high solubility in addition to potent enzymatic (CK2 IC50 < 3 nM) and cellular activity (Wnt DLD-1 Luciferase IC50 = 75 nM). Further substitution of the terminal amino group, as in N,N-diethyl analogue 7c, reduced the biochemical and cellular potency. Weakly basic or nonbasic groups at the terminal position (7d–7f) contributed to a reduction in Wnt reporter activity. Extension of the linker segment in 7b by an additional methylene unit, producing analogue 7g, results in a greater than 10-fold reduction in enzyme activity and suggested that optimal positioning of the charged dimethylamino group was achieved with a three atom linker. Pharmacokinetics for 7b in the rat following oral administration of 10 mg/kg dose are characterized by low exposure (AUC = 0.36 μM·h) and high clearance (CL = 65 mL/min/kg). In vitro metabolite identification studies using rat hepatocytes identified a series of products derived from 7b that likely arise via oxidative demethylation of the side chain nitrogen atoms. Incubations in the presence of ABT in rat and human microsomes give less extensive metabolite formation and suggest a predominantly oxidative mechanism of clearance for 7b. We subsequently synthesized the didemethylated analogue 7h, which exhibits improved cellular activity (Wnt DLD-1 Luciferase IC50 = 50 nM), high solubility, and reduced intrinsic clearance in rat hepatocytes and human microsomes relative to 7b. In addition, 7h showed greatly reduced activity (IC50 > 100 μM) compared to 7b (IC50 = 4 μM) in our hERG ion channel assay. Interestingly, rigidification of the linker segment through the use of 3-aminopyrrolidino- (7l, m) and 3-aminopiperidino- substituents (7n, o), despite contributing to an overall increase in lipophilicity, afforded compounds with reduced turnover in rat hepatocyte and human microsome preparations but led to a reduction in Wnt reporter assay potency (Table 1). Substitution adjacent to the primary amine, as in 7p and 7q, led to further improvement in the in vitro metabolic stability while preserving cellular activity.
To confirm our initial design hypothesis, we determined the X-ray crystallographic structures of CK2α with 7b and 7h (Figure 3).12 Both inhibitors are bound in the ATP-binding site in a manner analogous to that of 2. However, while the side chain of 7b positions the terminal dimethylamino group for an electrostatic interaction with D175, the unsubstituted amine of 7h is able to directly coordinate an ordered water molecule and the side-chain carbonyl group of N161.
Figure 3.
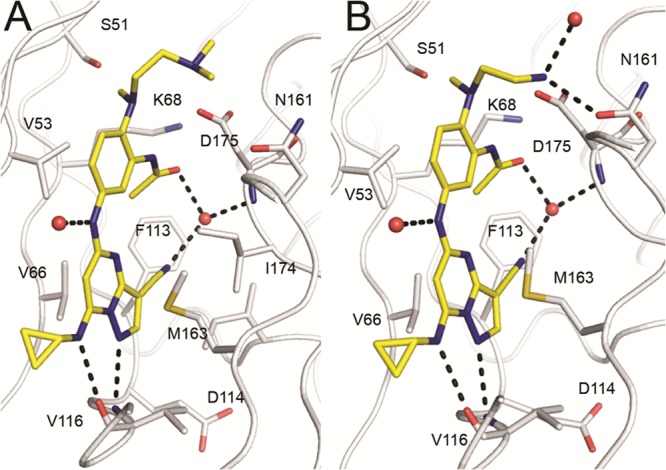
X-ray crystallographic structure of human CK2α in complex with compounds 7b (A) and 7h (B) determined at 2.00 and 2.15 Å resolution (PDB accession codes: 5H8G and 5H8E).12,13 Water molecules are shown as red spheres, and hydrogen bonds to the inhibitor are shown as black dashed lines.
A number of compounds in this series possess half-maximum inhibitory potency below the lower limit of detection (IC50 < 3 nM) in our enzymatic assay, which measures the inhibition of recombinant human full-length CK2α mediated phosphorylation of a synthetic peptide substrate at Km ATP concentration. To better understand the contribution of the side-chain interactions on ligand binding we developed a surface plasmon resonance (SPR) assay to determine the binding affinities of 2, 7b, and 7h.14 The results (Table 2) indicate that 2 and 7h possess similar affinity and are approximately 10-fold more potent than 7b. Comparison of 7b and 7h indicate that the dimethylamino side-chain of 7b is disfavored compared to the primary amine of 7h.
Table 2. CK2α SPR Data.
| compd | KD (pM) |
|---|---|
| 2 | 4.91 ± 1.64 |
| 7b | 48.2 ± 5.78 |
| 7h | 6.33 ± 0.054 |
Kinase selectivity profiling of 7h at a concentration of 0.1 μM against a panel of 402 kinases revealed a high degree of selectivity (Figure 4).15 The limited off-target activity (12 kinases with >50% inhibition) was restricted to members of the CMGC family, including isoforms of the dual-specificity tyrosine-regulated kinases (Dyrk) and CAMK kinases such as the death-associated protein kinases (Dapk) and homeodomain interacting protein kinases (Hipk). Moderate inhibition (74%) of bone morphogenetic protein receptor type-1B kinase (BMPR1b) was also observed. Biochemical IC50 determinations ([ATP] = Km) revealed that 7h exhibits moderate-to-weak activity against the Hipk and Dyrk isoforms (IC50s = 0.04–1.3 μM) and is active in the 10–20 nM range against Dapk2 and Dapk3.14
Figure 4.
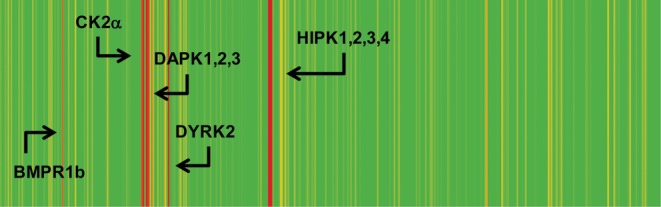
Kinase selectivity profile for 7h when tested at a concentration of 0.1 μM against a panel of 402 kinases.
The ability of our compounds to inhibit the Wnt pathway, as measured using the DLD-1 Topflash reporter assay, correlates well with inhibition of a direct CK2 substrate, pAKTS129, in cells.16 These data are consistent with the finding that CK2α-dependent up-regulation of β-catenin driven transcriptional activity requires phosphorylation of AKT.17 In addition, Wnt pathway inhibition correlates with antiproliferative effects in DLD-1 (APC mutant) cells (Figure 5). Similar levels of activity are observed in other CRC cell lines with constitutively activated Wnt signaling that is driven either by β-catenin (HCT-116) or APC mutations (SW620) (Table 3).
Figure 5.
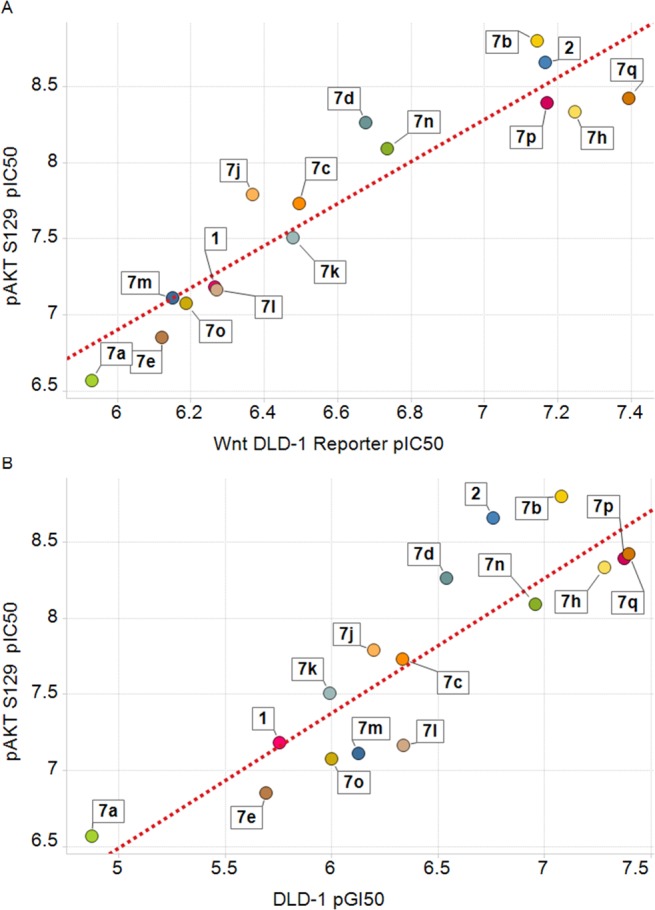
Relationship between pAKTS129 depletion and Wnt Topflash reporter inhibition in DLD-1 cells (A). Relationship between pAKTS129 depletion and growth inhibition in DLD-1 cells (B).
Table 3. Selected Growth Inhibition Data.
| compd | HCT-116 GI50 (μM)a | DLD-1 GI50 (μM)a | SW620 GI50 (μM)a |
|---|---|---|---|
| 1 | 0.7 | ND | 0.7 |
| 2 | 0.08 | 0.17 | 0.1 |
| 7b | 0.03 | 0.08 | 0.03 |
| 7h | 0.01 | 0.05 | 0.005 |
Mean value of at least three experiments; ND = not determined.
To further strengthen the mechanistic data, we subsequently demonstrated that 7h induced a concentration-dependent decrease in the active form of β-catenin in Wnt3a expressing mouse fibroblast L-cells,14 an in vitro system in which pathway up-regulation is triggered by constitutive Wnt3a expression (Figure S1). In addition, the mouse L S/L line contains a TCF4 driven luciferase construct and the degree of active β-catenin inhibition seen with 7h correlates with the potency of the compound in the corresponding Wnt3a L S/L reporter assay (IC50 = 0.05 μM).14
Based on the in vitro biomarker, pathway and growth inhibition data of 7h, characterization of this compound in vivo was undertaken. Oral dosing of 7h resulted in low bioavailability and limited the unbound drug concentration to levels expected to be subtherapeutic. However, intraperitoneal (IP) or intravenous (IV) dosing regimens delivered sustained free drug concentrations above efficacious levels.
The ability of compound 7h to inhibit substrate and downstream marker phosphorylation in an APC mutant CRC model was evaluated in SW620 tumor-bearing murine xenografts. Administration of 7h induced dose-dependent modulation of the downstream markers pAKTS129 and β-catenin, as determined by Western blot analysis of tumor cell lysates (Figure 6A).14 Similarly, DLD-1 TOPflash luciferase (APC mutant) xenografts were utilized to assess the effect of the compound on Wnt-associated gene transcription. In this model, treatment with a single dose (10 mg/kg, IV) of 7h led to 40–50% inhibition of Tcf4-luciferase signal at the 8 h time point with suppression of this signal, and AKTS129 phosphorylation (not shown), still evident at 24 h, by which time no detectable drug remained in plasma (Figure 6B).
Figure 6.
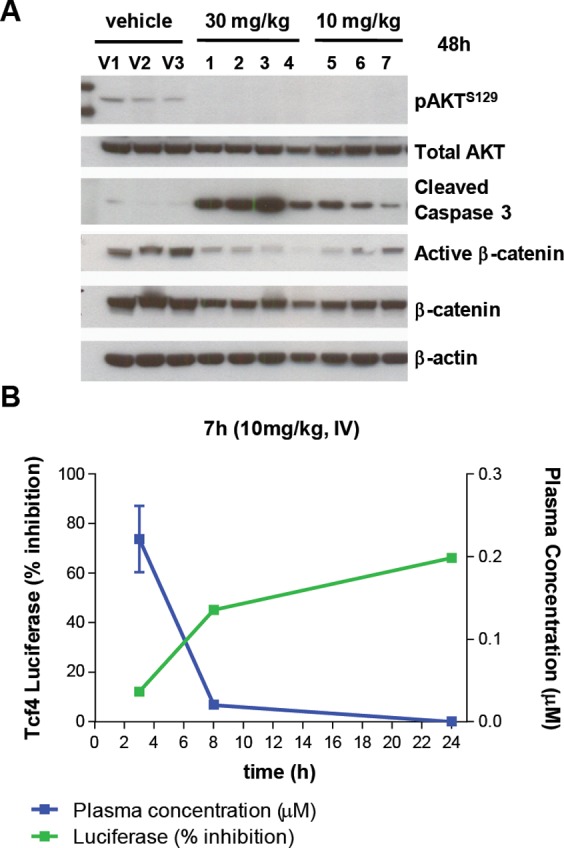
Inhibition of substrate (AKTS129) and downstream marker (active β-catenin) phosphorylation in SW620 xenografts by 7h (10, 30 mg/kg, IV). Individual lanes correspond to vehicle or compound treated animals and are numbered (A). Inhibition of Wnt/Tcf4 Topflash luciferase reporter activity in DLD-1 xenografts following treatment with 7h (10 mg/kg, IV) (B).
The durable substrate and pathway suppression observed with 7h following its clearance from plasma may be due, in part, to the high (pM) affinity of the compound (Table 2) and its associated slow dissociation rate (kd = 0.00025 s–1). These and other data18 suggested the potential for an intermittent dosing schedule as a means to achieve activity in disease model studies while minimizing tolerability issues.
Compound 7h showed a dose-dependent tumor growth inhibition, achieving 94% TGI in a HCT-116 (β-catenin mutant/model) and 74% TGI in a SW620 (APC mutant) model at a 30 mg/kg dose given weekly for 3 cycles. Reversible dose-proportional body weight loss was observed in both experiments; in the SW620 study, mean body weight changes observed on day 25 in treated animals ranged from −0.9 to −6.8%.
In conclusion, we have identified a series of potent and selective CK2 kinase inhibitors that decrease AKTS129 phosphorylation in cells and whose antiproliferative effects correlate with inhibition of Wnt luciferase reporter gene transcription. Using the in vivo probe 7h, we have demonstrated a reduction in the downstream biomarkers pAKTS129 and β-catenin and a high level of activity as a monotherapy in HCT-116 and SW-620 xenografts.18 Further studies, using 7h and related analogues, are planned to more fully understand the dependence of CRC on CK2-mediated Wnt pathway inhibition and will be reported in future communications.
Acknowledgments
The authors wish to thank Mei Su and Helen Xiaomei Feng for compound synthesis, and Vicki Racicot and Zhong-Ying Liu for in vitro and in vivo biology support, respectively.
Glossary
ABBREVIATIONS USED
- heps
hepatocytes
- CL
clearance
- PK
pharmacokinetics
- PD
pharmacodynamics:
- aq
aqueous
- sol
solubility
- PPB
plasma protein binding
- CLint
intrinsic clearance
- IP
intraperitoneal
- IV
intravenous
- Hu
human
- ABT
aminobenzotriazole
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00452.
Experimental details and characterization data for key compounds, crystallographic and biophysical protocols, and active β-catenin Western blot protocol (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Niefind K.; Guerra B.; Ermakowa I.; Issinger O.-G. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. 10.1093/emboj/20.19.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzene M.; Pinna L. A. Addiction to protein kinae CK2: A common denominator of diverse cancer cells?. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 499–504. 10.1016/j.bbapap.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Pierre F.; Chua P. C.; O’Brien S. E.; Siddiqui-Jain A.; Bourbon P.; Haddach M.; Michaux J.; Nagasawa J.; Schwaebe M. K.; Stefan E.; Vialettes A.; Whitten J. P.; Chen T. K.; Darjania L.; Stansfield R.; Anderes K.; Bliesath J.; Drygin D.; Ho C.; Omori M.; Proffitt C.; Streiner N.; Trent K.; Rice W. G.; Ryckman D. M. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c] [2,6] naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–54. 10.1021/jm101251q. [DOI] [PubMed] [Google Scholar]
- Clevers H.; Nusse R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Dominguez I.; Sonenshein G. E.; Seldin D. C. CK2 and its role in Wnt and NF-kappa B signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Wang H.-Y. Casein Kinase 2 Is Activated and Essential for Wnt/β-Catenin Signaling. J. Biol. Chem. 2006, 281, 18394–18400. 10.1074/jbc.M601112200. [DOI] [PubMed] [Google Scholar]
- Tapia J. C.; Torres V. A.; Rodriguez D. A.; Leyton L.; Quest A. F. G. Casein kinase 2 (CK2) increases survivin expression via enhanced β-catenin–T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. U. S. A. 2006, 41, 15079–15084. 10.1073/pnas.0606845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J.; Luo H.; Zeng Q.; Dong Z.; Wu D.; Liu L. Protein kinase CK2a is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J. Transl. Med. 2011, 9, 97–108. 10.1186/1479-5876-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.-Y.; Tai C.; Hsu J.-C.; Li C.-F.; Fang C.-L.; Lai H.-C.; Hseu Y.-C.; Lin Y.-F.; Uen Y.-H. Overexpression of Nuclear Protein Kinase CK2 α Catalytic Subunit (CK2α) as a Poor Prognosticator in Human Colorectal Cancer. PLoS One 2011, 6 (2), e17193. 10.1371/journal.pone.0017193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. E.; Bao L.; Brassil P.; Chen H.; Chuaqui C.; Cooke E. L.; Denz C. R.; Larsen N. A.; Lyne P. D.; Peng B.; Pontz T. W.; Racicot V.; Russell D.; Su N.; Thakur K.; Wu A.; Ye Q.; Zhang T. Potent and selective inhibitors of CK2 kinase identified through structure-guided hybridization. ACS Med. Chem. Lett. 2012, 3, 278–283. 10.1021/ml200257n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes J. W.; Almeida L.; Barlaam B.; Boriack-Sjodin P. A.; Casella R.; Croft R. A.; Dishington A. P.; Gingipalli L.; Gu C.; Hawkins J. L.; Holmes J. L.; Howard T.; Huang J.; Ioannidis S.; Kazmirski S.; Lamb M. L.; McGuire T. M.; Moore J. E.; Ogg D.; Patel A.; Pike K. G.; Pontz T.; Robb G. R.; Su N.; Wang H.; Wu X.; Zhang H. J.; Zhang Y.; Zheng X.; Wang T. Pyrimidinone nicotinamide mimetics as selective tankyrase and Wnt pathway inhibitors suitable for in vivo pharmacology. ACS Med. Chem. Lett. 2015, 6, 254–259. 10.1021/ml5003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDB deposition codes for the cocrystal structures are 5H8B (2), 5H8G (7b), and 5H8E (7h).
- Figure produced using Pymol. DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2002; http://www.pymol.org. [Google Scholar]
- See Supporting Information.
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Di Maira G.; Brustolon F.; Pinna L. A.; Ruzzene M. Dephosphorylation and inactivation of Akt/PKB is counteracted by protein kinase CK2 in HEK 293T cells. Cell. Mol. Life Sci. 2009, 66, 3363–3373. 10.1007/s00018-009-0108-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce D. P.; Maturana J. L.; Cabello P.; Yefi R.; Niechi I.; Silva E.; Armisen R.; Galindo M.; Antonelli M.; Tapia J. C. Phosphorylation of Akt/PKB by CK2 is necessary for the Akt-dependent up-regulation of β-catenin transcriptional activity. J. Cell. Physiol. 2011, 226, 1953–1959. 10.1002/jcp.22527. [DOI] [PubMed] [Google Scholar]
- Dowling J. E.; Alimzhanov M.; Bao L.; Chuaqui C.; Denz C.; Ferguson A.; Graff C.; Liu Z.-Y.; Lyne P.; Racicot V.; Wu A.; Wu J.; Ye Q.; Cooke E. L. Discovery and characterization of AZ968, a potent and selective inhibitor of CK2 kinase with effects on AKT signaling in vivo. Poster presented at the 103rd Annual Meeting of the American Association for Cancer Research, March 31–April 4, 2012. Chicago, IL. Abstract No. 3907.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.