

Abstract

Beginning with promiscuous COT inhibitors, which were found to inhibit CDK8, a series of 6-aza-benzothiophene containing compounds were developed into potent, selective CDK8 inhibitors. When cocrystallized with CDK8 and cyclin C, these compounds exhibit an unusual binding mode, making a single hydrogen bond to the hinge residue A100, a second to K252, and a key cation−π interaction with R356. Structure-based drug design resulted in tool compounds 13 and 32, which are highly potent, kinase selective, permeable compounds with a free fraction >2% and no measurable efflux. Despite these attractive properties, these compounds exhibit weak antiproliferative activity in the HCT-116 colon cancer cell line. Further examination of the activity of 32 in this cell line revealed that the compound reduced phosphorylation of the known CDK8 substrate STAT1 in a manner identical to a CDK8 knockout clone, illustrating the complex effects of inhibition of CDK8 kinase activity in proliferation in these cells.
Keywords: CDK8, cyclin C, kinase inhibitor
CDK8 is a cyclin-dependent kinase that forms part of the Mediator complex, which itself regulates the transcriptional activity of RNA polymerase II. A number of studies have shown that CDK8 modulates the transcriptional output from distinct transcription factors involved in oncogenic control.1 These factors include the Wnt/β-catenin pathway, Notch, p53, and TGF-β.2,3
While the means by which CDK8 activity accomplishes the regulation of these various pathways remains an active area of investigation, CDK8 has been found to be amplified and overexpressed in colon cancer. In this context, CDK8 has been reported to act as a colon cancer oncogene,4 driving Wnt pathway activity by mediating β-catenin transcriptional output.5 Suppression of CDK8 expression in colon cancer cell lines expressing high levels of both CDK8 and β-catenin led to reduced proliferation of these cell lines and induced differentiation.6 Furthermore, in a survey of colorectal cancer tissue samples, CDK8 was associated with β-catenin activation and with reduced rates of survival.7 More recently, it has been shown that suppression of CDK8 expression in melanoma cells reduces proliferation.8 Interestingly, the reverse has also been reported; mutations in MED12, which disrupt the association of CDK8 with the mediator complex are associated with uterine leiomyomas.9 Additionally, the conditional knockout of CDK8 in young adult mice produced no gross abnormalities, while CDK8 deletion in the ApcMin tumor model reduced survival and increased tumor burden.10
To date, the investigations of the role of CDK8 in both cellular signaling and colon cancer have relied upon RNAi mediated suppression of CDK85 and on the use of a kinase dead mutant CDK8.4 In order to more fully investigate the role of CDK8 in colon cancer, we aimed to develop a potent and selective small molecule inhibitor of CDK8. In order for such a molecule to serve as a useful tool compound in understanding CDK8 activity in colon cancer cell lines, we required that it also be cell permeable with minimal efflux and have a high free fraction in plasma.
The two small molecule inhibitors of CDK8 reported at the start of our investigations, sorafenib11 and Senexin A,12 exhibited insufficient kinase selectivity and potency, respectively, to serve our purposes. We therefore examined our in-house database of kinase inhibition data and looked for compounds that might have been prepared for a different target, but serendipitously inhibited CDK8.
We were pleased to find that compound 2, which has been reported to be a COT inhibitor,13,14 potently inhibited CDK8. As part of our standard follow-up for a lead, we resynthesized the material and assayed intermediates in the synthesis in a fluorescence polarization binding assay. We were surprised to discover that compound 1 was a still more potent inhibitor of CDK8 than our initial lead and exhibited superior selectivity as well (Figure 1 and Table 1).
Figure 1.
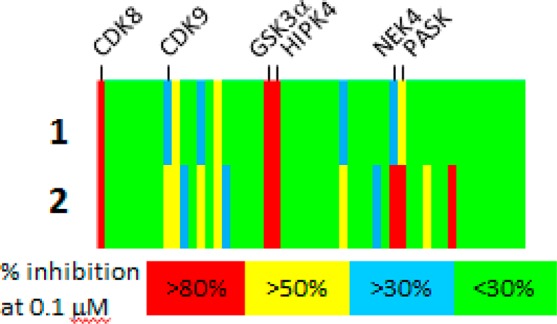
Kinase selectivity for compounds 1 and 2 measured at a concentration of 0.1 μM in a panel of 52 kinases. Each vertical line represents a single kinase and is colored by the percent inhibition seen. Selected potently inhibited kinases are highlighted. Complete data is in the Supporting Information.
Table 1. Potency, Permeability, and Plasma Protein Binding Data for 2-Tetrazolyl-thieno[2,3-c]pyridines 1–10.
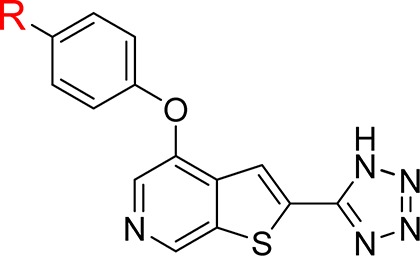

Having determined that the second aryl ring in the biphenyl ether provided no advantage, we prepared and evaluated compounds 2–10 in an effort to understand the limitations of this chemotype. Despite the presence of the highly polar tetrazole group, compounds with a topological polar surface area (TPSA) lower than approximately 100 Å2 were permeable as measured in a standard MDCK assay. None of the compounds examined exhibited any significant level of efflux.
As might be expected from a set of lipophilic compounds containing a tetrazole, which would be negatively charged at physiological pH, the compounds were found to be highly protein bound, with all compounds >99.7% bound in human, rat, and mouse serum. In an effort to reduce plasma protein binding, polar functionality was introduced at the 4-position of our aryl ether. These modifications did not adversely affect the potency of these compounds for CDK8, but as the TPSA went above 100 Å2, the MDCK permeability dropped dramatically, making them unlikely to be active in a cellular context. We therefore initiated an examination of what functional groups would be tolerated in place of the tetrazole.
We began our exploration of substitution at the 2-position of the thieno[2,3-c]pyridines by examining the carboxylate 11 (Table 2).15 As expected, the activity of the compound was only slightly diminished, and the plasma protein binding was reduced only very slightly. To determine if an anion was required at physiological pH, we prepared inactive ester 12 and amides 13–18. With the smallest amide substituents, the inhibition of CDK8 remained potent, but alkyl groups larger than methyl and particularly disubstitution of the amide resulted in 10- to 100-fold losses in activity. More promisingly, the plasma protein binding was seen to diminish in both the caboxamide 13 and N-methyl carboxamide 14.
Table 2. Alternatives to the Tetrazolea.
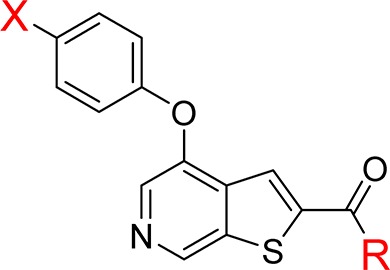
| X | R | CDK8 IC50 (nM) | PPB % bound (H/R/M) | |
|---|---|---|---|---|
| 11 | Cl | OH | 2.8 | 99.8/99.7/99.7 |
| 12 | Cl | OMe | 62 | n/d |
| 13 | Cl | NH2 | 6.2 | 98.0/97.0/98.0 |
| 14 | Cl | NHMe | 8.2 | 98.5/99.0/97.7 |
| 15 | Cl | NMe2 | 75 | n/d |
| 16 | Cl | NHEt | 16 | 99.3/99.6/98.8 |
| 17 | Cl | NHC3H5 | 24 | 99.2/99.3/99.1 |
| 18 | Cl | NHiPr | 100 | n/d |
| 19 | Cl | Me | 32 | 99.7/99.8/99.7 |
| 20 | Cl | Et | 29 | >99.9/>99.9/>99.9 |
| 21 | Cl | C3H5 | 59 | n/d |
| 22 | I | NHMe | 5.3 | 99.8/99.9/99.8 |
PPB figures in human, rat, and mouse plasma (H/R/M).
Finally, we examined the necessity of having a hydrogen bond donor as part of the 2-substituent and prepared compounds 19–21. As with the amides, the smaller substituents were more potent, but we observed a 5-fold or greater loss in affinity relative to the carboxamide, and plasma protein binding was greater than 99% for all the ketones examined.
With the knowledge that carboxamides combined the best potency and the lowest PPB yet obtained, we prepared another series of analogues to determine whether we could continue to reduce PPB while maintaining potency and permeability by incorporating polar functional groups at the 4-position of our aryl ether (Table 3).
Table 3. Incorporation of Polar Functional Groups on Aryl Ethera.
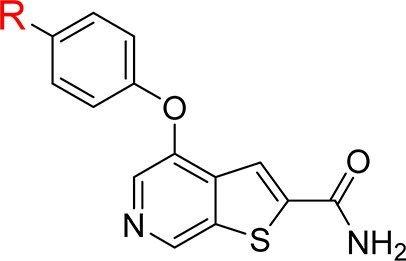
| R | CDK8 IC50 (nM) | PPB % bound (H/R/M) | kinetic solubility (μM) | |
|---|---|---|---|---|
| 23 | Me | 6.0 | 97.2/95.9/96.1 | <1 |
| 24 | OMe | 25 | 93.6/94.0/90.2 | 10 |
| 25 | OCF3 | 12 | 97.9/96.8/97.2 | <1 |
| 26 | CN | 5.1 | 84.7/87.3/89.1 | 8 |
| 27 | CONH2 | 21 | 77.3/76.8/78.3 | 7 |
| 28 | NHAc | 29 | 91.0/84.8/79.6 | 18 |
PPB figures in human, rat, and mouse plasma (H/R/M).
In preparing these analogues we discovered that alkyl ethers at this position led to decreases in potency, as seen in compounds 24 and 25, perhaps due to the incorporation of the polar oxygen atom, which can be partially compensated for by the increased lipophilicity of the trifluoromethyl group in 25. Addition of a further heteroatom in amide 27 and reverse amide 28 led to potency very similar to that of the methyl ether. Nitrile 26 pushes its heteroatom further away from the ring and offered good potency, reduced PPB, and moderate kinetic solubility.
Having generated SAR data at both of the two most easily modified regions of our molecules, we determined the 2.2 Å resolution crystal structure (PDB: 5CEI and Supporting Information) of the CDK8/cyclin C complex with 22 bound to facilitate design. The structure contains a single monomer of the CDK8/cyclin C complex in the asymmetric unit, with CDK8 stabilized in the “active” conformation (helix C “in” and the side chain of Glu66 of the helix coordinated to the active site Lys252). Prior structures of sorafenib and other inhibitors bound the kinase with both DMG-in and DMG-out binding modes.11,16 The complex with 22 elicited the DMG-in conformation in the enzyme and interactions between the protein and 22 resemble those proposed for molecules of this type binding to COT (Figure 2). A cation−π interaction is seen in the close approach of Arg356 to the iodophenyl ring, stabilizing a 65° rotation of the iodophenyl ring out of the plane of the heterocyle. Hydrogen bonds are formed between the pyridyl nitrogen of 22 and the backbone nitrogen of Ala100 of the hinge as well as from the side chain of Lys52 to the amide carbonyl. While the binding mode of the compound is clear from the electron density (Table S1), the temperature factors of the ligand suggest its presence at 50–75% occupancy; however, the intermediate resolution of the data precluded definitive refinement of that parameter. Interestingly, a formate ion is observed at partial occupancy in the region underneath the P loop, adjacent to the inhibitor, raising the possibility of building into that region to increase the buried surface area. Examination of the structure prompted the design of a few targeted analogues in an effort to maximize potency.
Figure 2.
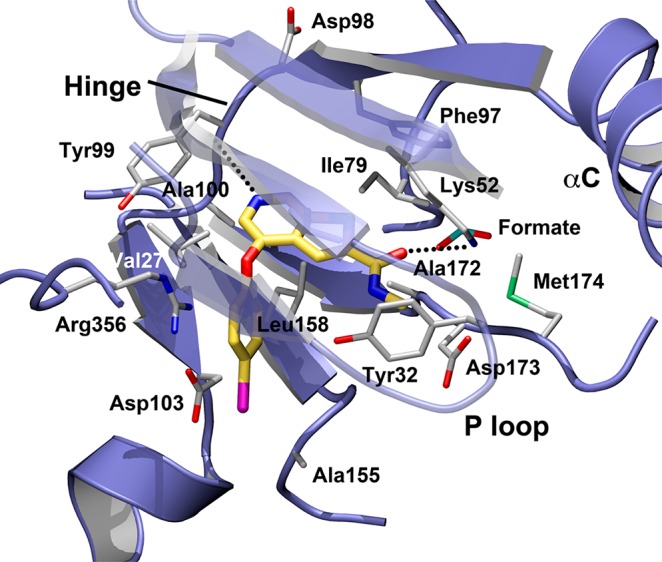
Structure of compound 22. Hydrogen bonding interactions with Ala100 and Lys52 are indicated with dashed lines.
Our first analogues were targeted at probing the region in the vicinity of the hinge binding nitrogen, which accepts a hydrogen bond from the NH of Ala100. We noted that there was space available to make a contact with the main chain of Asp98 from the 7-position of our heterocycle, and we made a series of analogues to access this region (Table 4). Interestingly, hydrophobic analogues 29–31 retained much of their CDK8 binding activity, with 29 losing less than 3-fold, and the others less than 2-fold. Compound 32 was found to bind 4-fold more potently than its parent compound 13, which confirmed our modeling of the compound and indicates that the NH2 can readily make a hydrogen bond to the backbone carbonyl of Asp98.
Table 4. Seven-Substituted Thieno[2,3-c]pyridinesa.
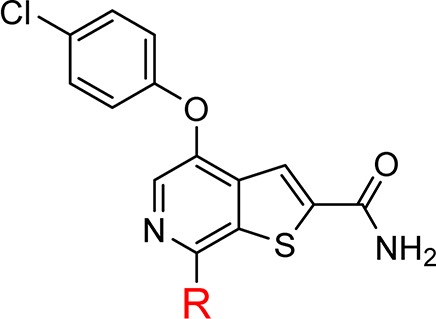
| R | CDK8 IC50 (nM) | PPB % bound (H/R/M) | kinetic solubility (μM) | |
|---|---|---|---|---|
| 13 | H | 6.2 | 98.0/97.0/98.0 | 6.3 |
| 29 | F | 16 | n/d | <1 |
| 30 | Cl | 11 | n/d | <1 |
| 31 | Me | 8 | 99.3/99.2/98.8 | 10 |
| 32 | NH2 | 1.5 | 98.13/97.7/95.9 | 5.5 |
PPB figures in human, rat, and mouse plasma (H/R/M).
Having generated extremely potent binders of CDK8, it was important to examine the selectivity of these compounds relative to other kinases. We focused especially on ensuring that there was minimal activity against CDKs 1 and 4, as these would be expected to have antiproliferative effects via disruption of the cell cycle, confounding our ability to assess the effects of inhibiting CDK8. We therefore submitted compounds 13 and 32 for profiling against a panel of 209 kinases. We were pleased to find that both compounds exhibit excellent selectivity for CDK8 over every other kinase examined (Figure 3) even when tested at concentrations 160- and 670-fold greater than the measured IC50, respectively.
Figure 3.
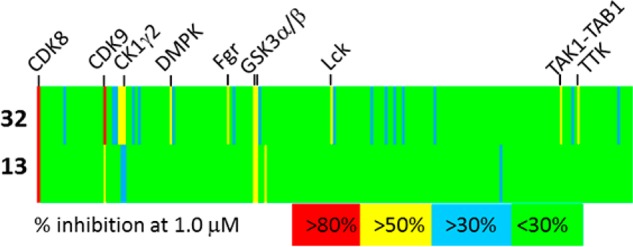
Kinase selectivity for compounds 13 and 32 measured at a concentration of 1 μM in a panel of 209 kinases. Each vertical line represents a single kinase and is colored by the percent inhibition seen. Selected potently inhibited kinases are highlighted. Complete data is in the Supporting Information.
Having identified CDK9/cyclin T1 as our most active off-target, we therefore determined the IC50 values for 13 and 32 in a fluorescence polarization activity assay Compound 13 had an IC50 value of 2.2 μM for CDK9/cyclin T1 and therefore a selectivity of 350-fold for CDK8/cyclin C. Compound 32 had an IC50 value of 1.1 μM and a selectivity of 730-fold. The overall profile of these two molecules now met our goal of producing potent, selective CDK8 inhibitors with solubility, plasma protein binding, permeability, and low efflux (Table S2).We therefore evaluated the antiproliferative effects of our compounds in the colorectal carcinoma HCT-116 cell line, which had previously been shown to exhibit sharply reduced proliferation in response to CDK8 protein depletion using targeted shRNA.4 Cells were treated with compounds for 72 h in media containing fetal bovine serum (FBS) at a concentration of either 0.625% or 10%. The lower concentration of FBS was used to mitigate the effects of the low free fraction of some of our compounds. Throughout the course of the experiment, the cells were in growth phase, and yet we did not observe significant growth inhibition (Table 5). Flavopiridol, a potent inhibitor of multiple CDKs17 with known antiproliferative properties18 was used as a positive control. In each case, the activity of the compounds appeared at approximately 1000-fold higher concentration than the IC50 value for the purified enzyme. Concerned that the compounds examined were failing to act on CDK8 in the cellular context, we examined phosphorylation of Ser727 of STAT1, a substrate of CDK819 as a biomarker.
Table 5. Antiproliferative Activity of Compounds 13, 31, and 32 on HCT-116 Cellsa.
| HCT116 EC50 10% FBS (μM) | HCT116 EC50 0.625% FBS (μM) | |
|---|---|---|
| 13 | >10 | 8.8 |
| 31 | >10 | 8.7 |
| 32 | 6.7 | 5.4 |
| flavopiridol | 0.034 | 0.059 |
Compounds 5-8, 11, and 25 were also evaluated, but had EC50 values >10 μM under both high and low serum conditions.
In order to validate STAT1 Ser727 as a substrate in HCT116 cells, CRISPR guide RNAs were created to target CDK8 for genomic deletion.20−23 Three separate clones showed complete ablation of CDK8 protein, while its close homologue CDK1924 and actin expression remained unchanged (Figure 4A). Clone 1 was then used to generate three CDK8/CDK19 double knockouts, for which all three generated clones showed the complete absence of both proteins. Interestingly, cyclin C levels were greatly reduced in the absence of CDK8 alone, and the protein was undetectable in the double knockout, likely due to instability when unable to associate with the other components of the mediator complex, and highlighting the possibility of secondary effects when protein expression is altered using siRNA or CRISPR.
Figure 4.
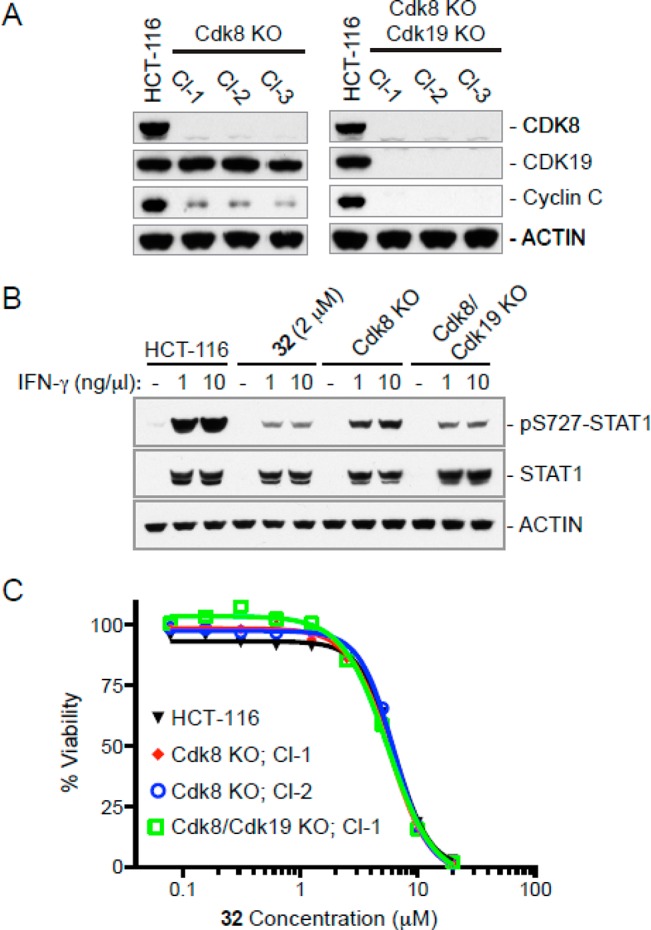
(A) Immunoblot analysis of CDK8 knockout and CDK/CDK19 double knockout cells. Three independent clones of each mutant are shown. (B) Pharmacological inhibition of CDK8 kinase activity reduces STAT1 Ser727 phosphorylation following stimulation with IFNγ. Cells were treated for 24 h in the presence or absence of 2 μM 32. (C) Cell viability in the presence of 32 at 72 h.
HCT-116 cells along with their respective CDK8 knockout and CDK8/CDK19 double knockout clones were then probed with an antibody specific for phosphorylated Ser727 on STAT1. As had been previously reported in bone marrow derived macrophages19 and peripheral blood mononuclear cells,25 stimulation of HCT-116 cells with interferon-γ resulted in increased expression of STAT1 and a large increase in the amount of phosphorylated Ser727 (Figure 4B). This phosphorylation was reduced in the CDK8 knockout, and further reduced in the CDK8/CDK19 double knockout clones stimulated in the same manner, while the total amount of STAT1 expressed remained relatively constant. Encouragingly, cells treated with 2 μM compound 32 showed levels of phosphorylated Ser727 nearly identical to that of the double knockout cell line, clearly demonstrating that this molecule is able to downregulate signaling in cultured cells.
Having demonstrated that 32 was in fact inhibiting CDK8 function in HCT-116 cells, we used the CDK8 and CDK8/19 knockout cell lines created to demonstrate that the observed antiproliferative effects of 32 were independent of their inhibition of the targeted protein. Wild type HCT-116 cells, along with the CDK8 and CDK8/19 deletion clones derived from them, were incubated with varying concentrations of 32. As can be seen in Figure 4C, each of these cell lines showed identical growth inhibition curves, indicating that growth inhibition in these cells is an off-target effect.
During the preparation of this letter, additional selective CDK8 inhibitors have been reported, both of which have been shown to sharply reduce the presence in cells of STAT1-pS727 analogously to 32. Cortistatin A is reported26 to have mixed antiproliferative effects on cell lines with sensitivity driven by the set of genes upregulated upon treatment, with HCT-116 cells among the insensitive lines. The compound CCT251545, identified through its inhibition of the WNT pathway in cells, binds to and inhibits CDK8 and CDK1927 and inhibits proliferation of SW620 colon cancer cells after 96 h in culture.
Our development of potent, highly specific CDK8 inhibitors and the lack of CDK8-dependent antiproliferative activity for 32 on the HCT-116 colon cancer cell line has therefore served to substantiate the emerging, complex picture of the role of CDK8 in cancer. STAT1 pSer727 has been potently downregulated by inhibition of CDK8/19 by different compounds and appears to be a useful biomarker of inhibition. The impact of CDK8 kinase inhibition in proliferation is cell line dependent, and possibly condition dependent, and appears to diverge from the scaffolding functions of CDK8 as evidenced by the differing antiproliferative effects of CDK8 knockdown and kinase inhibition in HCT-116 cells. Further investigation of these differences is clearly important to understanding the roles of CDK8.
Acknowledgments
We thank members of the DMPK and purification groups within Genentech Small Molecule Drug Discovery for purification and analytical support. The crystallographic experiments were performed on the X06SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00278.
Description of the preparation of 1–32 along with kinase selectivity and experimental procedures used in the determination of the cocrystal structure of 22 with CDK8 (PDB: 5CEI), and the preparation and characterization of our genetically altered HCT-116 cell line (PDF)
The authors declare the following competing financial interest(s): M.F.T.K., P.B., E.M.B., K.B., K.R.C., R.F., J.R.K., M.L.M., L.S., S.S., J.W., and M.H.B. are employees of Genentech, Inc; K.M. and E.V.S. are employees of Proteros Biostructures, GmbH.
Supplementary Material
References
- Conaway R. C.; Sato S.; Tomomori-Sato C.; Yao T.; Conaway J. W. The Mammalian Mediator Complex and Its Role in Transcriptional Regulation. Trends Biochem. Sci. 2005, 30, 250–255. 10.1016/j.tibs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Nemet J.; Jelicic B.; Rubelj I.; Sopta M. The Two Faces of Cdk8, a Positive/Negative Regulator of Transcription. Biochimie 2014, 97, 22–27. 10.1016/j.biochi.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Li N.; Fassl A.; Chick J.; Inuzuka H.; Li X.; Mansour M. R.; Liu L.; Wang H.; King B.; Shaik S.; et al. Cyclin C Is a Haploinsufficient Tumour Suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Bass A. J.; Kim S. Y.; Dunn I. F.; Silver S. J.; Guney I.; Freed E.; Ligon A. H.; Vena N.; Ogino S.; et al. CDK8 Is a Colorectal Cancer Oncogene That Regulates Beta-Catenin Activity. Nature 2008, 455, 547–551. 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J.-O.; Han S. I.; Lim S.-C. Role of CDK8 and β-Catenin in Colorectal Adenocarcinoma. Oncol. Rep. 2010, 24, 285–291. 10.3892/or_00000858. [DOI] [PubMed] [Google Scholar]
- Adler A. S.; McCleland M. L.; Truong T.; Lau S.; Modrusan Z.; Soukup T. M.; Roose-Girma M.; Blackwood E. M.; Firestein R. CDK8Maintains Tumor De-Differentiation and Embryonic Stem Cell Pluripotency. Cancer Res. 2012, 72, 2129–2139. 10.1158/0008-5472.CAN-11-3886. [DOI] [PubMed] [Google Scholar]
- Firestein R.; Shima K.; Nosho K.; Irahara N.; Baba Y.; Bojarski E.; Giovannucci E. L.; Hahn W. C.; Fuchs C. S.; Ogino S. CDK8 Expression in 470 Colorectal Cancers in Relation to B-Catenin Activation, Other Molecular Alterations and Patient Survival. Int. J. Cancer 2010, 126, 2863–2873. 10.1002/ijc.24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A.; Goldberg M. S.; Cumberland L. K.; Ratnakumar K.; Segura M. F.; Emanuel P. O.; Menendez S.; Vardabasso C.; Leroy G.; Vidal C. I.; Polsky D.; Osman I.; Garcia B. A.; Hernando E.; Bernstein E. The Histone Variant macroH2A Suppresses Melanoma Progression Through Regulation of CDK8. Nature 2010, 468, 1105–1109. 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen M.; Spaeth J. M.; Keskitalo S.; Park M. J.; Kivioja T.; Clark A. D.; Mäkinen N.; Gao F.; Palin K.; Nurkkala H.; et al. Uterine Leiomyoma-Linked MED12 Mutations Disrupt Mediator-Associated CDK Activity. Cell Rep. 2014, 7, 654–660. 10.1016/j.celrep.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland M. L.; Soukup T. M.; Liu S. D.; Esensten J. H.; de Sousa e Melo F.; Yaylaoglu M.; Warming S.; Roose-Girma M.; Firestein R. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J. Pathol. 2015, 237, 508–519. 10.1002/path.4596. [DOI] [PubMed] [Google Scholar]
- Schneider E. V.; Böttcher J.; Blaesse M.; Neumann L.; Huber R.; Maskos K. The Structure of CDK8/CycC Implicates Specificity in the CDK/Cyclin Family and Reveals Interaction with a Deep Pocket Binder. J. Mol. Biol. 2011, 412, 251–266. 10.1016/j.jmb.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Porter D. C.; Farmaki E.; Altilia S.; Schools G. P.; West D. K.; Chen M.; Chang B. D.; Puzyrev A. T.; Lim C.; Rokow-Kittell R. Cyclin-Dependent Kinase 8 Mediates Chemotherapy-Induced Tumor-Promoting Paracrine Activities. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13799–13804. 10.1073/pnas.1206906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George D.; Friedman M.; Allen H.; Argiriadi M.; Barberis C.; Clabbers A.; Cusack K.; Dixon R.; Fix-Stenzel S.; Gordon T.; Janssen B.; Jia Y.; Moskey M.; Quinn C.; Salmeron J.-A.; Wishart N.; Woller K.; Yu Z. Discovery of Thieno[2,3-C]Pyridines as Potent COT Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4952–4955. 10.1016/j.bmcl.2008.08.037. [DOI] [PubMed] [Google Scholar]
- Cusack K.; Allen H.; Bischoff A.; Clabbers A.; Dixon R.; Fix-Stenzel S.; Friedman M.; Gaumont Y.; George D.; Gordon T.; Gronsgard P.; Janssen B.; Jia Y.; Moskey M.; Quinn C.; Salmeron A.; Thomas C.; Wallace G.; Wishart N.; Yu Z. Identification of a Selective Thieno[2,3-C]Pyridine Inhibitor of COT Kinase and TNF-Alpha Production. Bioorg. Med. Chem. Lett. 2009, 19, 1722–1725. 10.1016/j.bmcl.2009.01.088. [DOI] [PubMed] [Google Scholar]
- Zhu G.-D.; Gunawardana I. W.; Boyd S. A.; Melcher L. M. A Facile and General Synthesis of 2,4-Di- and 2,4,7-Trisubstituted Thieno[2,3-C]pyridines. J. Heterocycl. Chem. 2008, 45, 91–96. 10.1002/jhet.5570450106. [DOI] [Google Scholar]
- Schneider E. V.; Böttcher J.; Huber R.; Maskos K.; Neumann L. Structure-Kinetic Relationship Study of CDK8/CycC Specific Compounds. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8081–8086. 10.1073/pnas.1305378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; et al. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Jung C.; Motwani M.; Kortmansky J.; Sirotnak F. M.; She Y.; Gonen M.; Haimovitz-Friedman A.; Schwartz G. K. The Cyclin-Dependent Kinase Inhibitor Flavopiridol Potentiates Gamma-Irradiation-Induced Apoptosis in Colon and Gastric Cancer Cells. Clin. Cancer Res. 2003, 9, 6052–6061. [PubMed] [Google Scholar]
- Bancerek J.; Poss Z. C.; Steinparzer I.; Sedlyarov V.; Pfaffenwimmer T.; Mikulic I.; Dölken L.; Strobl B.; Müller M.; Taatjes D. J.; et al. CDK8 Kinase Phosphorylates Transcription Factor STAT1 to Selectively Regulate the Interferon Response. Immunity 2013, 38, 250–262. 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L.; Ran F. A.; Cox D.; Lin S.; Barretto R.; Habib N.; Hsu P. D.; Wu X.; Jiang W.; Marraffini L. A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; East A.; Cheng A.; Lin S.; Ma E.; Doudna J. RNA-Programmed Genome Editing in Human Cells. eLife 2013, 2, e00471. 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Yang L.; Esvelt K. M.; Aach J.; Guell M.; DiCarlo J. E.; Norville J. E.; Church G. M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver M. C.; Bauer D. E.; Dass A.; Yien Y. Y. Characterization of Genomic Deletion Efficiency Mediated by Clustered Regularly Interspaced Palindromic Repeats (CRISPR)/Cas9 Nuclease System in Mammalian Cells. J. Biol. Chem. 2014, 289, 21312–21324. 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui T.; Fukasawa R.; Tanaka A.; Hirose Y.; Ohkuma Y. Identification of target genes for the CDK subunits of the mediator complex. Genes to Cells 2011, 16, 1208–1218. 10.1111/j.1365-2443.2011.01565.x. [DOI] [PubMed] [Google Scholar]
- Lehtonen A.; Matikainen S.; Julkunen I. Interferons Up-Regulate STAT1, STAT2, and IRF Family Transcription Factor Gene Expression in Human Peripheral Blood Mononuclear Cells and Macrophages. J. Immunol. 1997, 159, 794–803. [PubMed] [Google Scholar]
- Pelish H. E.; Liau B. B.; Nitulescu I. I.; Tanpeerachaikul A.; Poss Z. C.; Da Silva D. H.; Caruso B. T.; Arefolov A.; Fadeyi O.; Christie A. L.; Du K.; Banks D.; Schneider E. V.; Jestel A.; Zou G.; Si C.; Ebmeier C. C.; Bronson R. T.; Krivtsov A. V.; Myers A. G.; Kohl N. E.; Kung A. L.; Armstrong S. A.; Lemieux M. E.; Taatjes D. J.; Shair M. D. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T.; Clarke P. A.; Esdar C.; Waalboer D.; Adeniji-Popoola O.; Ortiz-Ruiz M.-J.; Mallinger A.; Samant R. S.; Czodrowski P.; Musil D.; Schwarz D.; Schneider K.; Stubbs M.; Ewan K.; Fraser E.; TePoele R.; Court W.; Box G.; Valenti M.; Brandon A. d.; Gowan S.; Rohdich F.; Raynaud F.; Schneider R.; Poeschke O.; Blaukat A.; Workman P.; Schiemann K.; Eccles S. A.; Wienke D.; Blagg J. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.