

Abstract

A new subseries of substituted piperidines as p53-HDM2 inhibitors exemplified by 21 has been developed from the initial lead 1. Research focused on optimization of a crucial HDM2 Trp23–ligand interaction led to the identification of 2-(trifluoromethyl)thiophene as the preferred moiety. Further investigation of the Leu26 pocket resulted in potent, novel substituted piperidine inhibitors of the HDM2-p53 interaction that demonstrated tumor regression in several human cancer xenograft models in mice. The structure of HDM2 in complex with inhibitors 3, 10, and 21 is described.
Keywords: HDM2, p53, protein−protein interaction, cancer
Cancer is a disease of uncontrolled cell growth of various tissues and organs in the body. According to the American Cancer Society, about 1.7 million new cancer cases are expected to be diagnosed in 2015, with an estimated half million deaths in the US alone. Even with the use of conventional chemotherapy and newly approved targeted therapies, cancer still represents an unmet medical need.1
The tumor suppressor p53 plays many critical roles in surveying and responding to various stress and damage signals. It regulates cell growth, migration, apoptosis, angiogenesis, metabolism, development , and stromal matrix cellular environment.2,3 The loss of p53 function predisposes cells to a cancerous state.4 HDM2 acts as a major negative regulator of p53 activity by repressing p53 transcriptional activity through its binding to p53 and targeting p53 for degradation in the proteasome through its ubiquitin E3 ligase activity. An inhibitor capable of blocking this HDM2-p53 interaction would lead to accumulation of p53 and activation of transcriptional and nontranscriptional activities of p53, leading to cell cycle arrest and apoptosis of tumor cells. In more than 50% of human cancers, p53 is inactivated due to overexpression of HDM2 protein,5−7 making the development of small molecule inhibitors of HDM2-p53 a very attractive approach to cancer therapy.8−11
Recently, multiple teams have reported the discovery of small molecules interfering with the HDM2-p53 protein–protein interaction (PPI).12−19 We previously disclosed the discovery of gem-disubstituted piperidines20−221 with HDM2 binding activity. Detailed SAR and computational studies highlighted the pivotal role of an aliphatic side chain on the piperidine ring conformation leading to potent small molecule inhibitor 2 of the HDM2-p53 PPI (Figure 1).23 Moreover, we demonstrated that this side chain at position 2 of the piperidine could tolerate polar functional groups providing the opportunity to improve physical properties. Similarly, transformation of the methoxy-ethylene side chain to hydroxyl-ethylene led to compound 3, which showed good permeability and an excellent pharmacokinetic profile in mouse.24,25 More importantly, the structure of HDM2 in complex with 3 was solved using X-ray diffraction data (Figure 2).
Figure 1.

Figure 2.
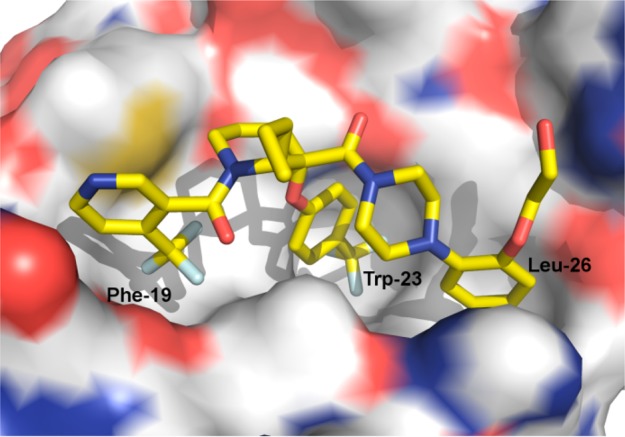
Structure of HDM2 in complex with 3. The inhibitor is shown as stick and the protein as surface, colored according to atom type. The Phe-19, Trp-23, and Leu-26 subsites are indicated.
The compound binds along the region of the HDM2 surface that has been shown to interact with p53, thus effectively disrupting this PPI. The affinity of compound 3 is mostly due to the hydrophobic interactions between the compound and three surface pockets that have been shown to bind p53, residues Phe19, Trp23, and Leu26.26−28 On the basis of the crystal structure, the trifluormethylphenyl group binds in the Trp23 pocket. Although the installation of novel moieties that would bind in the deep Trp 23 pocket would have to take place early in the synthesis, it was envisioned that research optimization focused on this interaction could lead to enhanced binding potency. Thus, bioisosteric replacements of the phenyl group were studied.29 Using the general chemistry outlined in Schemes 1 and 2, a series of analogues of lead 3 were prepared to examine the effect of substitution on binding interactions in the Trp 23 pocket. As described earlier,20,23,24 the team capitalized on the Bargellini reaction of protected 3-piperidinone 26a and 26b with various alcohol moieties to assemble in one-pot the gem-disubstituted piperidine core of type 27a and 27b.20 Efficient acid chloride coupling procedures were developed to install 4-(trifluoromethyl)-nicotinic acid 28 as well as 1-(2-alkoxyphenyl)piperazine hydrochloride of type 32 into final targets. Another synthetic approach relied on standard HATU coupling procedures, installing first 1-(2-alkoxyphenyl)piperazine moieties 32 then 4-(trifluoromethyl)-nicotinic acid 28 to deliver compounds of type 34.
Scheme 1. Synthesis of 3–25 (General Method Method 1).

Reagents and conditions: (a) R1–OH, NaOH, CHCl3, 0–40 °C, 25–50%; (b) ACN/MeOH (1:1), 2.0 M TMSCHN, 20 °C, 0.5 h, 40–50%; (c) 50% TFA/DCM, 0 °C to rt, 3 h, (100% yield); (d) Chiralcel AD column, 90:10 heptanes/iPrOH; (e) benzene, thionyl chloride, reflux/cat DMF, 100%; (f) 27, DCM/DIPEA/0 °C, 100%; (g) H2, Pd/C, EtOH; (h) KOH, MeOH reflux, 100%; (i) (COCl)2, DMF, DCM 0 °C; (j) Cs2CO3, DMF, R3-Br (50–90% yield); (k) 4.0 N HCl dioxane 100%; (l) 30, THF/H2O, 0 °C to rt, 0.5 h (90–100% yield); (m) final deprotection step.
Scheme 2. Synthesis of 3–25 (General Method 2).

Reagents and conditions: (a) R1–OH, NaOH, CHCl3, 0–40 °C, 25–50%; (b) 32, DMF/DCM (1:1), HATU, DIPEA, 45 °C, 12 h, 40–60%; (c) DCM, TMS-I, 0 °C, 5 h, (100% crude yield); (d) DMF/DCM (1:1), HATU, DIPEA, 45 °C, 12 h, 40–80%; (e) H2, Pd/C, EtOH; (f) final deprotection step and Chiralcel OD column purification.
Of the initial analogues, 2-alkoxy-4-(trifluoromethyl)-thiophene bioisosteric replacement of the trifluoromethyl phenyl group in 3 was especially promising. Compound 27 with R1 = 4-iodo-2-alkoxythiophene was envisioned as a key intermediate to access targets 6 as well as 4, 5, and 7 (Table 1), via a sequence relying on regioselective transmetalation of 2,4-dibromothiophene followed by selective halide displacements. As seen in the poor activity with compound 7, substitution with both iodine and trifluoromethyl at position 4 (compounds 5 and 6 respectively) turned out to be optimal when compared to 4. Modification to a thiazole analogue in 8 resulted in 6-fold loss in potency where pyrazole analogue 9 was the least active of the series. Then the 4-alkoxy-2-(trifluoromethyl)thiophene isomer was prepared. Compound 10 demonstrated significant improvement in both biochemical (FP) and cellular (SJSA-1) inhibition assay not only versus 2-thiophene isomer 6 but original lead 3 as well. The structure of HDM2 in complex with 10 was solved and revealed (Figure 3) that the trifluoromethyl thiophene provides a better complementarity to the pocket than the trifluoromethyl phenyl moiety. The trifluoromethyl thiophene is equidistant from all the side chains that line the binding pocket, while the trifluoromethyl phenyl is asymmetrically bound and in close contact with the side chains of V93 and L54. To capitalize on the additional surface in the Trp23 pocket available for binding, compounds 11–14 were prepared and evaluated for their inhibitory activity. Although the one-carbon homologue of 10 conserved similar biochemical inhibitory potency, 11 did not provide further improvement and suffered 6-fold loss in cellular activity. A small substituent at position 4 of the thiophene ring such as methyl (12) is tolerated, but a bulkier group such as chlorine (13) resulted in reduction of HDM2 binding potency. With 4-fold loss in potency, compound 14 clearly indicated that substitution at position 2 of the thiophene ring was not advantageous.
Table 1. Trp23 Substitution Structure–Activity Relationshipsa.
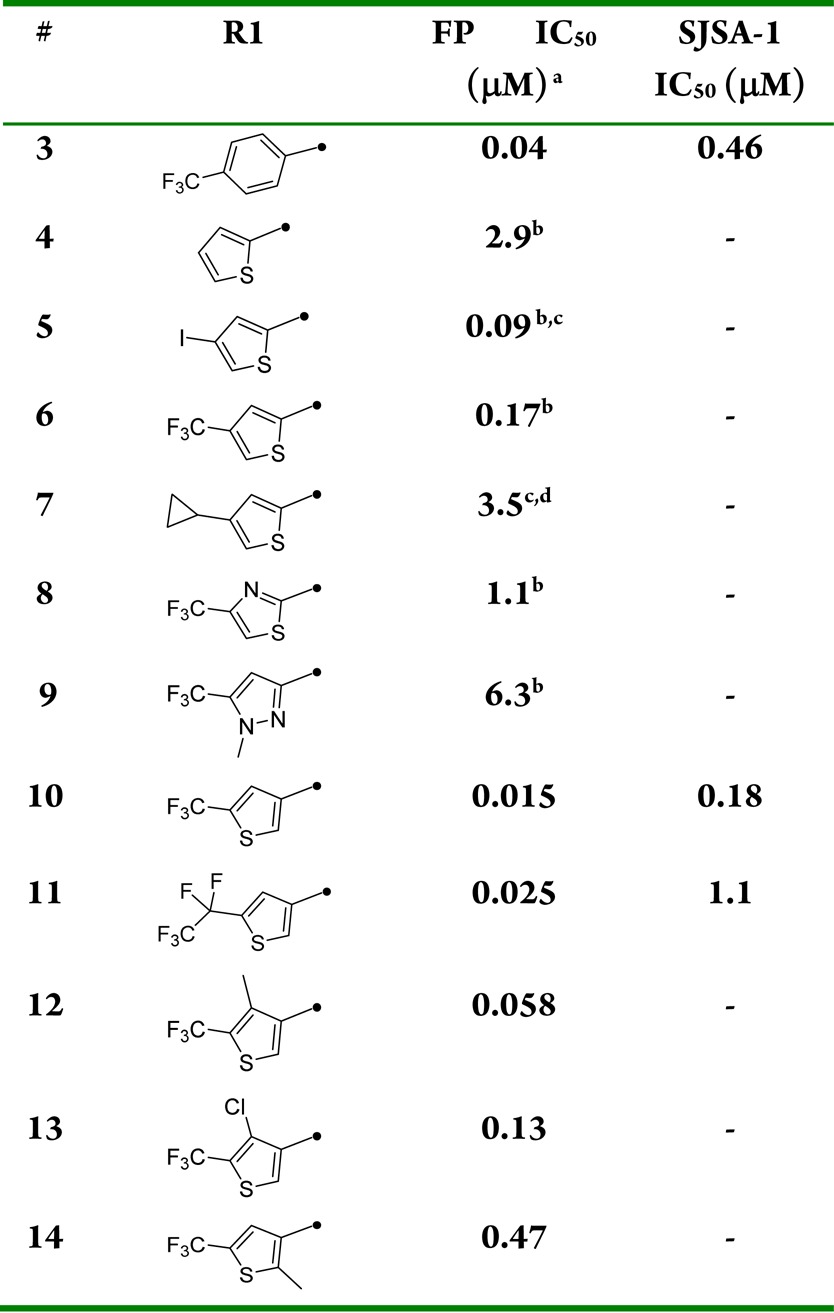
Biochemical p53-HDM2 binding fluorescent polarization (FP) assay and antiproliferative osteosarcoma SJSA-1 assay results for compounds 3–14. Fluorescence polarization (FP) peptide displacement assay value was determined as described in Zhang et al.15 For comparison Nutlin-3a fluorescence polarization (FP) peptide displacement IC50 assay value obtained in house was 0.07 μM. In our antiproliferative assay, Nutlin-3a had an IC50 value of 1.9 μM using osteosarcoma SJSA-1 cells.
Racemic mixture.
Allyl group at position 2 of piperidine.
Phenoxybutanoic acid side chain.
Figure 3.
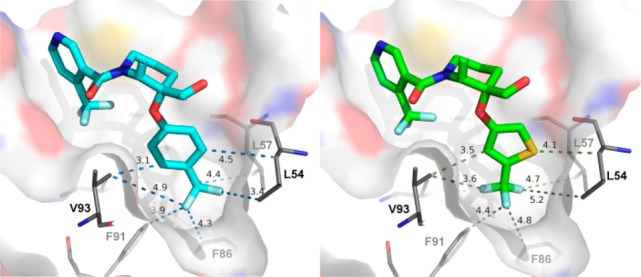
Comparison of 3 and 10 binding on the Trp-23 subside.
Compound 10 incorporating a 4-alkoxy-2-(trifluoromethyl)thiophene as optimal moiety for binding in the Trp23 pocket, provided a valuable starting point for further optimization. Using the chemistry outlined in Scheme 1, a series of analogues of 10 was prepared to examine the effect of R2 substitution on the phenyl ring that occupies the Leu26 binding pocket (Table 2). The R2 chains bind in a very solvent exposed pocket and are thought to assume different conformations. While we anticipated that potency enhancements could be achieved through modification of the hydrophobic interaction of the hydroxyl-ethylene side chain with the HDM2 protein, our efforts to introduce more lipophilicity as in 15 and 16 proved largely unsuccessful. Due to the positively charged surface near Leu 26, it was anticipated that acidic R2 substituents might improve binding. Thus, straightforward hydroxyl to carboxylic acid functional group manipulation resulted in compound 17 that showed an 8-fold improvement in HDM2 affinity. Unfortunately, in vitro assessment revealed that the acid moiety in 17 was detrimental to permeability (Caco2 permeability = 7 nm/s; AP to BL), a plausible explanation for lack of SJSA-1 cellular activity and poor plasma exposure observed following a single-dose oral mouse PK study (Table 4). Improvement in permeability obtained by masking the acid group with carboxamide 18 (Caco2 permeability = 181 nm/s; AP to BL) resulted in 10-fold improvement in SJSA-1 cellular activity. Similarly, shielding the acid group in compound 19 with steric bulk restored cellular potency. The effect of chain length was also studied with compounds 20–24. A three carbon linker with compound 21 was found to be optimal, having improved potency in both biochemical and antiproliferative assays compared to 10. Substitution along the side chain of 21 was evaluated with compounds 22 and 23 and did not afford additional improvements. The structure of HDM2 in complex with 21, using X-ray diffraction data was solved and showed hydrogen bonding to the nearby side chain of Gln24 (Figure 4). Side chain extension in 24 conserved excellent binding potency for the compound but resulted in weaker cellular activity.
Table 2. Leu 26 Substitution Structure–Activity Relationshipsa.
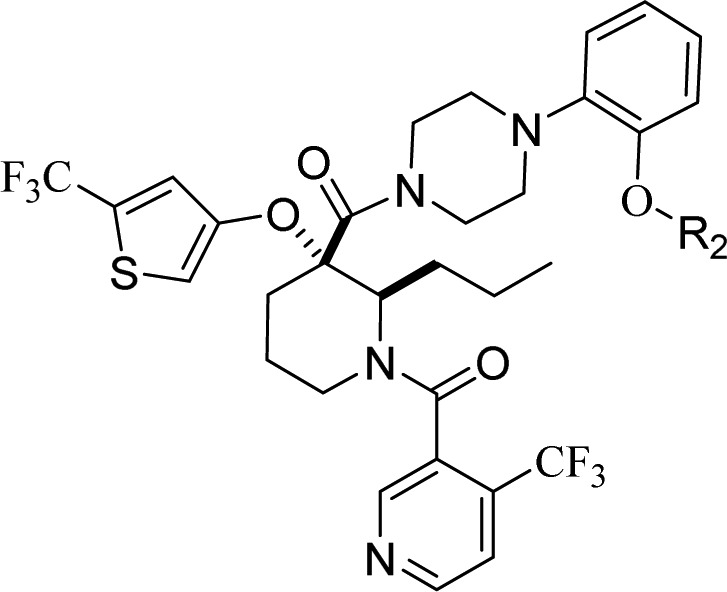
Biochemical p53-HDM2 binding fluorescent polarization (FP) assay, antiproliferative osteosarcoma SJSA-1, and colorectal HCT116 assay results for compounds 10, 15–24.
Table 4. Human Liver Microsomal Stability and Oral Exposure for Compounds 3, 10, 17, 18, and 21a.
| # | 3 | 10 | 17 | 18 | 21 | |
|---|---|---|---|---|---|---|
| Human Clint (mL/min/kg) | 23 | 14 | 13 | 3 | ||
| PO 1 mg/kg DNAUC(0–24h) (μM·h) | Mouse | 1.2 | 2 | 0.02 | 2 | 0.1 |
| Dog | 0.2 | 0.2 | 5.8 | |||
| Monkey | 0.3 | 0.3 | 0.2 |
PO dosing formulation: 20% HPBCD (mouse), 0.4% HPMC (dog and monkey).
Figure 4.
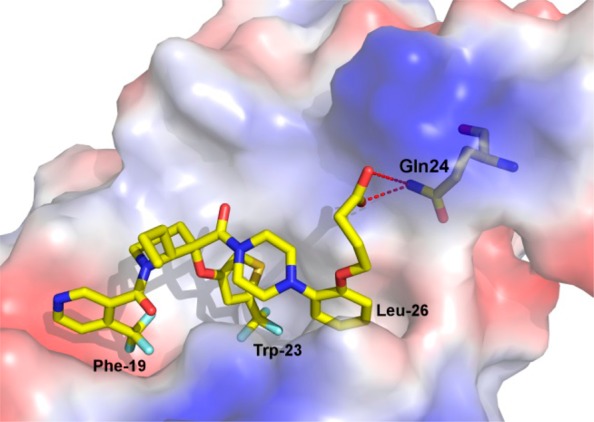
Structure of HDM2 in complex with 21. The inhibitor is shown as stick and the protein as surface, colored according to atom type. The Phe-19, Trp-23, and Leu-26 subsites are indicated as well as Gln 24 residue.
Inhibitors 10, 18, and 21 demonstrated selective inhibition of cell growth in cancer cells expressing wild-type p53 (SJSA-1 and HCT116), but not cancer cells harboring mutant p53 (SKUT-1) and p53-null cells (H1299) (Table 3). Furthermore, activation of p53 transcription evaluated by P53RE-Luc (measured in HCT116) and apoptotic response in Casp3/7 (measured in SJSA-1) were used to confirm on-target activity. Compound 21 exhibited excellent selectivity with mechanism-based on-target activity showing more than 500-fold selectivity for wild-type p53 tumor cells versus mutant (SKUT-1) and null cells (H1299).
Table 3. Leu 26 Substitution Selectivity and on Target Activity Assay Results for Compounds 10, 18, and 21.
| SKUT-1 IC50 (μM) | H1299 IC50 (μM) | P53RE-Lu EC50 (μM) | Casp 3/7 IC50 (μM) | |
|---|---|---|---|---|
| 10 | >17 | >17 | 0.4 | 0.3 |
| 18 | >10 | >17 | 0.4 | 0.3 |
| 21 | >50 | >50 | 0.1 | 0.09 |
Compared to original lead 3, compounds 10 and 18 showed similar in vitro profiles, while 21 clearly differentiated both in terms of solubility30 and human Clint (Table 4). Single-dose mouse, dog, and monkey PK studies were used to assess exposure of compounds 10, 18, and 21 following oral administration (Table 4). Oral exposure of 21 in rodent and monkey was low but showed superiority when dosed in dog versus all the other leads.31 Compound 21 was evaluated for inhibition of a panel of CYP’s (3A4, 2D6, 2C8, 2C9, 1A2; IC50s > 50 μM). While 21 was not a substrate for cytochrome P450 3A4, it was found to have a potential for time-dependent inhibition that could require close monitoring when part of a combination regimen with other cancer therapies. Plasma protein binding (% free fraction) was similar across human, monkey, dog, and mouse (4%, 5%, 2%, and 3%, respectively) with excellent permeability (Caco-2 = 180 nm/s). Human half-life projection of 21 based on observed preclinical PK by applying allometric scaling is estimated to be 6 h, consistent with an oral BID therapeutic. Compound 21 showed no cardiovascular liabilities with 20% hERG (VC) inhibition at 6 μM, translating in 80-fold window for a human dose as high as 1000 mg BID.
Since compound 21 demonstrated the best cellular antiproliferative potency and selectivity, activity for additional cancer cell lines was evaluated and confirmed cell growth inhibition in a broad panel of human tumor cell lines expressing wild-type p53 (Figure 5). Thus, several cell lines were implanted in nude mice for study in cancer xenograft models. Efficacy of 21 both as a single agent (in SJSA-1 osteosarcoma) as well as in combination with cytotoxics in A549 nonsmall cell lung (NSCLC) and in A2780 ovarian cancer xenograft model were evaluated. Single agent efficacy of 21 in an SJSA-1 osteosarcoma model following oral administration of 21 at either intermittent schedule (oral daily dosing with cycle of 3 days on, 4 days off) or daily continuous at 100 and 200 mpk resulted in tumor regression in the SJSA-1 osteosarcoma xenograft model (Figure 6) without any observable toxicity (no body weight loss was observed in any treatment group compare to vehicle control group). Single agent efficacy of 21 in an A549 (NSCLC) model following oral administration of 21 daily at 200 mg/kg resulted in 75% tumor growth inhibition (Figure 7). Intermittent dosing of 21 (daily for 3 days followed by 4 days off schedule) in combination with cytotoxic paclitaxel (20 mg/kg, intraperitoneal, weekly) in the A549 NSCLC model resulted in increased antitumor response compared to either single agent used alone. To further exemplify activity of 21, wild-type p53 mice bearing A2780 tumor xenograft were treated with 50 mg/kg 21 (oral, twice daily) or 40 mg/kg docetaxel (ip, weekly, 2×) or the combination of the two agents (Figure 8). Continuous daily dosing of 21 at 50 mpk BID resulted in 41% tumor growth inhibition while combination with cytotoxic Docetaxel resulted in increased antitumor response (81% tumor growth inhibition) in the A2780 ovarian cancer xenograft model compared to either single agent used alone.
Figure 5.
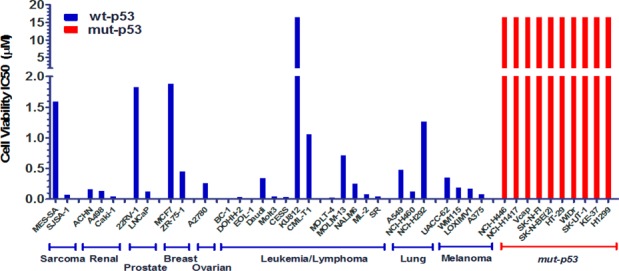
Cancer cell lines were treated with 21 for 72 h, and IC50 of cell viability was determined by CellTiter GLO. p53 gene mutation (coding region) status was determined by direct sequencing and cells with mutated p53 shown in red on the right.
Figure 6.
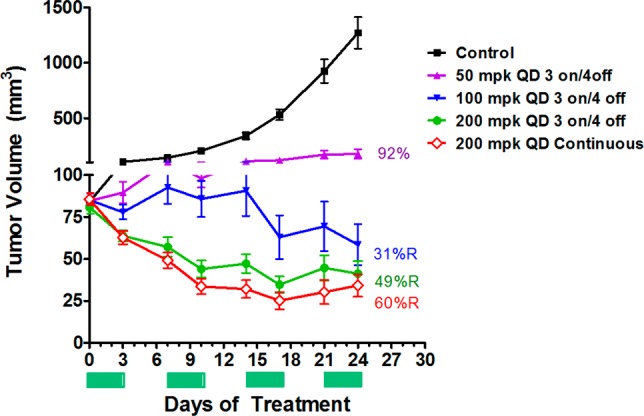
Efficacy of 21 on SJSA-1 osteosarcoma xenograft in Mice. Oral in vivo efficacy profile of 21 on nude mice implanted with SJSA1 osteosarcoma. N = 5 per group. PO dosing formulation 20% HPBCD.
Figure 7.
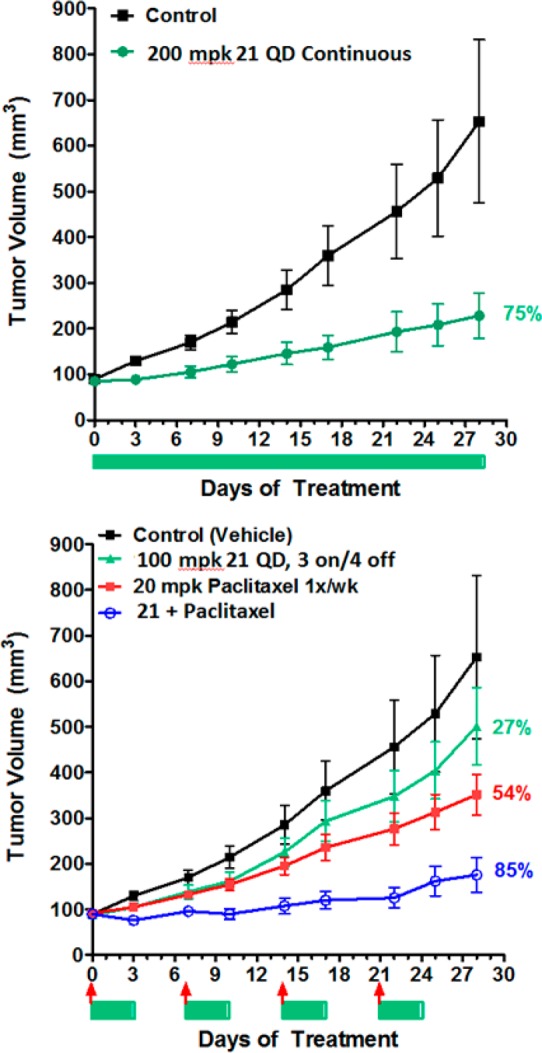
Efficacy of 21 as single agent and in combination with Cytotoxic in A549 non-small cell lung cancer (NSCLC) xenograft model. Oral in vivo efficacy profile of 21 on nude mice implanted with A549 NSCLC. N = 10 per group. Bars and arrows below the graph indicates treatment days of 21 (bar) and Paclitaxel (arrow).
Figure 8.
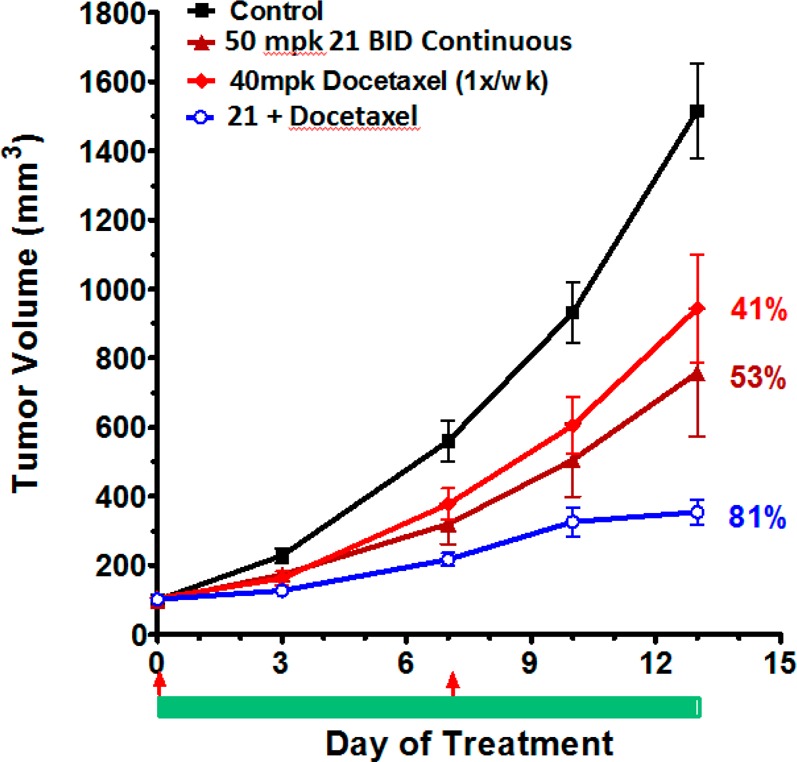
Efficacy of 21 as single agent and in combination with Cytotoxic in A2780 ovarian cancer xenograft model. Oral in vivo efficacy profile of 21 on nude mice implanted in A2780 ovarian cancer xenograft model. N = 10 per group. Bar and arrows below the graph indicates treatment days of 21 (bar) or Docetaxel (arrow).
In summary, the structure of HDM2 in complex with a novel piperidine-based inhibitor was described. Investigation of bioisosteric replacements of moieties that binds into the Trp23 pocket led to a new subseries of HDM2-p53 inhibitors with improved potency as exemplified by 10. Exploration of various functional groups binding in the Leu 26 pocket led to the discovery of 21 as a potent, selective, and orally active HDM2-p53 inhibitor that demonstrated tumor growth inhibition in a variety of cancer cell lines expressing wild-type p53 in vivo.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00472.
General synthesis and characterization data for key intermediates and final compounds 3, 4, 6, 8, 10, 14, 15, 17–19, and 21–25 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- American Cancer Society, Inc. Surveillance Research, 2013. [Google Scholar]
- Mandinova A.; Lee S. W. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci. Transl. Med. 2011, 3, 64rv1. 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raycroft L.; Wu H. Y.; Lozano G. Transcriptional activation by wild-type but not transforming mutants of the p53 antioncogene. Science 1990, 249, 1049–1051. 10.1126/science.2144364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainaut P.; Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. 10.1016/S0065-230X(08)60785-X. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Lane D. P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Bond G. L.; Hu W.; Levine A. J. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr. Cancer Drug Targets 2005, 5, 3–8. 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- Wade M.; Wang Y. V.; Wahl G. M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky M. V.; Prives C. Unleashing the power of p53: lessons from mice and men. Genes Dev. 2006, 20, 125–131. 10.1101/gad.1397506. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Lain S.; Verma C. S.; Fersht A. R.; Lane D. P. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Wade M.; Li Y. C.; Wahl G. M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Aguilar A.; Denzil Bernard D.; Wang S. Small-Molecule Inhibitors of the MDM2–p53 Protein–Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem. 2015, 58, 1038–1052. 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovie Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Hu C.-Q.; Hu Y.-Z. Small molecule inhibitors of the p53-MDM2. Curr. Med. Chem. 2008, 15, 1720–1730. 10.2174/092986708784872375. [DOI] [PubMed] [Google Scholar]
- Patel S.; Player M. R. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin. Invest. Drugs 2008, 17, 1865–1882. 10.1517/13543780802493366. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Yu S.; Sun W.; Liu L.; Lu J.; McEachern D.; Shargary S.; Bernard D.; Li X.; Zhao T. Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. J. Med. Chem. 2013, 56, 5553–5561. 10.1021/jm4005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lopez de Turiso F.; Sun D.; Rew Y.; Bartberger M. D.; Beck H. P.; Canon J.; Chen A.; Chow D.; Correll T. L.; Huang X.; et al. Rational design and binding mode duality of MDM2-p53 inhibitors. J. Med. Chem. 2013, 56, 4053–4070. 10.1021/jm400293z. [DOI] [PubMed] [Google Scholar]
- Zak K.; Pecak A.; Rys B.; Wladyka B.; Domling A.; Weber L.; Holak T. A.; Dubin G. Diaryl- and triaryl-pyrrole derivatives: inhibitors of the MDM2–p53 and MDMX–p53 protein–protein interactions. Expert Opin. Ther. Pat. 2013, 23, 425–448. 10.1517/13543776.2013.765405. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Chu X.-J.; Liu J.-J.; Ding Q.; Zhang J.; Bartkovitz D.; Jiang N.; Karnachi P.; So S.-S.; Tovar C.; Filipovic Z. M.; Higgins B.; Glenn K.; Packman K.; Vassilev L.; Graves B. Discovery of potent and orally active p53-MDM2 inhibitors RO5353 and RO2468 for potential clinical development. ACS Med. Chem. Lett. 2014, 5, 124–127. 10.1021/ml400359z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A. Z.; Eksterowicz J.; Bartberger M. D.; Beck H. P.; Canon J.; Chen A.; Chow D.; Duquette J.; Fox B. M.; Fu J.; Huang X.; Houze J. B.; Jin L.; Li Y.; Li Z.; Ling Y.; Lo M.-C.; Long A. M.; McGee L. R.; McIntosh J.; McMinn D. L.; Oliner J. D.; Osgood T.; Rew Y.; Saiki A. Y.; Shaffer P.; Wortman S.; Yakowec P.; Yan X.; Ye Q.; Yu D.; Zhao X.; Zhou J.; Olson S. H.; Medina J. C.; Sun D. Selective and potent morpholinone inhibitors of the MDM2–p53 protein–protein interaction. J. Med. Chem. 2014, 57, 2472–2488. 10.1021/jm401767k. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Lahue B.; Shipps G.; Brookes J.; Wang Y. Substituted piperidines as HDM2 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1026–1030. 10.1016/j.bmcl.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang R.; Ma Y.; Lahue B.; Shipps G.. Method of Using Substituted Piperidines that Increase p53 Activity. U.S. Pat. Appl. Publ. US2008004286, 2008.
- Ma Y.; Lahue B.; Shipps G.; Wang Y.; Bogen S.; Voss M.; Nair L.; Tian Y.; Doll R.; Guo Z.; Strickland C.; Zhang R.; McCoy M.; Pan W.; Siegel E.; Gibeau C.. Preparation of Substituted Piperidines that Increase p53 Activity and the Uses Thereof. U.S. Pat. Appl. Publ. US200800428, 2008.
- Ma Y.; Lahue B. R.; Gibeau C. R.; Shipps G. W. Jr.; Bogen S. L.; Wang Y.; Guo Z.; Guzi T. J. Pivotal role of an aliphatic side chain in the development of an HDM2 inhibitor. ACS Med. Chem. Lett. 2014, 5, 572–575. 10.1021/ml500019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W.; Lahue B. R.; Ma Y.; Nair L. G.; Shipps G. W. Jr.; Wang Y.; Doll R.; Bogen S. L. Core modification of substituted piperidines as novel inhibitors of HDM2–p53 protein–protein interaction. Bioorg. Med. Chem. Lett. 2014, 24, 1983–1986. 10.1016/j.bmcl.2014.02.055. [DOI] [PubMed] [Google Scholar]
- Caco2 permeability = 99 nm/sec (AP to BL) ; Mouse PK (PO/IV, 20/2 mpk) AUC = 8.7 μM·h; F = 92%.
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Chen J.; Marechal V.; Levine A. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. 10.1128/MCB.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottger A.; Bottger V.; Garcia-Echeverria C.; Chene P.; Hochkeppel H. K.; Sampson W.; Ang K.; Howard S. F.; Picksley S. M.; Lane D. P. Molecular characterization of the hdm2–p53 interaction. J. Mol. Biol. 1997, 269, 744–756. 10.1006/jmbi.1997.1078. [DOI] [PubMed] [Google Scholar]
- Meanwell N. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; Lima L. M.; Barreiro E. J. Bioisosterism: a useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 100 μM for 21, 10 μM for 10, 12 μM for 18 (solubility assessed at pH 7.4 in PB).
- Cl values following IV dosing of compound 21: Mouse (2 mpk, 20% HPBCD, 133 mL/min/kg), monkey (1 mpk, 40% HPBCD, 23 mL/min/kg), dog (1 mpk, 40% HPBCD, 4.1 mL/min/kg).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.