

Abstract

By targeting the flap backbone of the BACE1 active site, we discovered 6-dimethylisoxazole-substituted biaryl aminothiazine 18 with 34-fold improved BACE1 inhibitory activity over the lead compound 1. The cocrystal structure of 18 bound to the active site indicated two hydrogen-bond interactions between the dimethylisoxazole and threonine 72 and glutamine 73 of the flap. Incorporation of the dimethylisoxazole substitution onto the related aminothiazine carboxamide series led to pyrazine-carboxamide 26 as a very potent BACE1 inhibitor (IC50 < 1 nM). This compound demonstrated robust brain Aβ reduction in rat dose–response studies. Thus, compound 26 may be useful in testing the amyloid hypothesis of Alzheimer’s disease.
Keywords: BACE1, inhibitor, aminothiazine, Aβ42, amyloid hypothesis, Alzheimer’s disease
Multiple lines of evidence suggest that β-amyloid (Aβ) peptide, particularly the longer 42 amino acid form Aβ42, plays a critical role in the progression of Alzheimer’s disease (AD).1−3 Aβ is derived from the β-amyloid precursor protein (APP) by proteolysis. Human genetic mutations such as the Swedish mutation result in increased β-secretase processing of APP, elevated Aβ levels, and ultimately aggressive early onset AD.4 A more recent study reported a variant of the APP gene (APPA673T) at a site proximal to the BACE1 proteolytic site that confers protection against AD.5 A673T substitution in APP results in reduced production of Aβ peptides secreted from heterologously transfected cells, further supporting the hypothesis that increases in Aβ may underlie the pathology of AD. Cleavage of APP by β-site APP cleaving enzyme-1 (BACE1) results in shedding of the APP ectodomain, and the remaining membrane bound C-terminal fragment, C99, is further processed by γ-secretase to produce Aβ. Thus, inhibition of BACE1 to reduce Aβ production is a promising approach to test the amyloid hypothesis.6−10
In 2009, Eli Lilly reported that 1 (LY2811376) (Figure 1), a small molecule biaryl aminothiazine BACE1 inhibitor, demonstrated sustained brain Aβ reduction in healthy volunteers upon oral administration.11,12 The clinical development of this compound was discontinued due to nonclinical, off-target-related retinal toxicity. As this compound exhibited only modest BACE1 inhibitory activity (IC50: 0.27 μM from our assay, 0.24 μM from the literature12), we sought to increase its potency as an avenue to increase the therapeutic window versus the observed toxicity. To our knowledge, no potent biaryl aminothiazine BACE1 inhibitors have been disclosed. We recently developed a novel approach to improve the activity of the biarylaminothiazines, and we then extended this strategy to the related aminothiazine carboxamide series. These efforts culminated in the discovery of a potent BACE1 inhibitor that elicited robust brain Aβ reduction in rodents. This report describes our progress along these lines.
Figure 1.
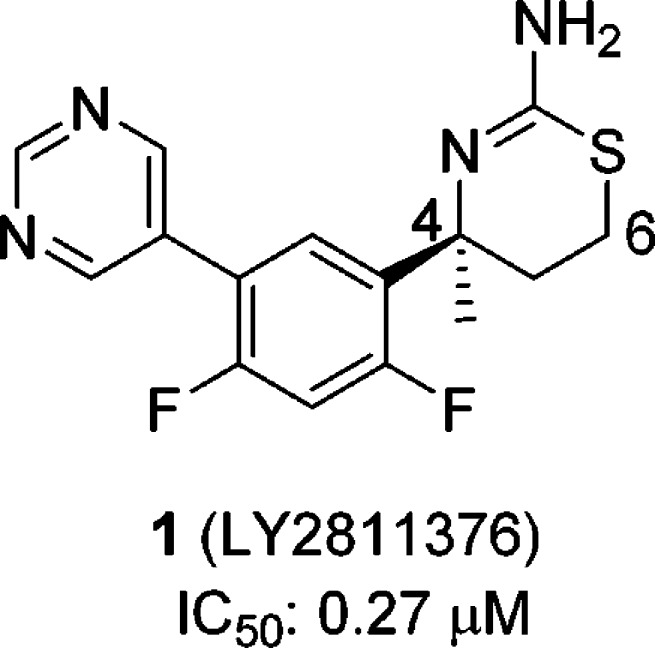
Structure of 1 (LY2811376).
Examination of the docked conformation of 1 to the BACE1 active site revealed proximity between threonine 72 and glutamine 73 of the flap and the C-6 position of the thiazine ring. We hypothesized that an appropriately selected substituent at C-6 may interact with the flap residues, thus enhancing binding affinity (Figure 2). Unfortunately, the existing synthesis of the 6-substituted aminothiazines 2 was both lengthy and linear, and therefore we desired to develop a more convergent methodology. Retrosynthetically, aminothiazines 2 could be assembled through a [4 + 2] cycloaddition involving heterodiene 3 and an olefin, and the heterodiene 3 can be visualized from the condensation of an aryl aldehyde or ketone and thiourea (Scheme 1). Indeed, the three-component, one-pot reaction was previously described for the synthesis of 4-desmethyl aminothiazines,13 and with some minor modifications, we were able to obtain a variety of 4-des-methyl 6-substituted aminothiazines (2, R′ = H). However, we were unable to extend this methodology to the synthesis of 4-methyl 6-substituted analogues (2, R′ = Me). At the time this work was carried out (January 2011), the impact of this methyl group was unknown in the literature.14 Therefore, our plan was to identify an active 4-desmethyl, 6-substituted aminothiazine using the [4 + 2] cycloaddition strategy and then to evaluate the impact of the 4-methyl group on potency.
Figure 2.
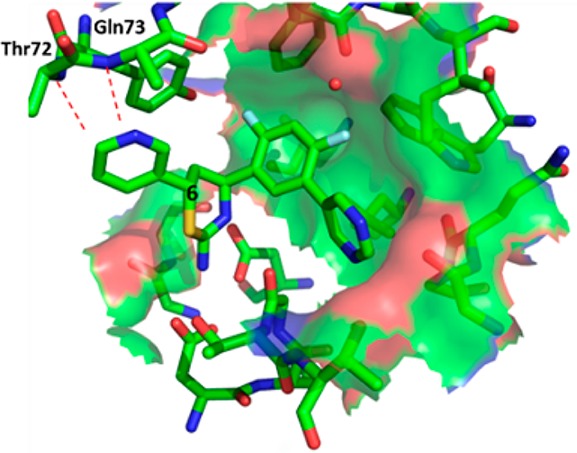
Modeled interaction between the flap backbone of the BACE1 active site and a substituent at the C-6 of aminothiazine.
Scheme 1. Retrosynthesis of 6-Substituted Aminothiazines.

Our work began with the three-component, one-pot reaction of the commercially available 3-(pyrimidin-5-yl)-benzaldehyde (4a) with a variety of substituted styrenes 5 (Scheme 2). In general, these reactions proceeded in moderate yields, and the 4,6-cis products were predominant over the trans isomers. The minor trans isomers were significantly more active than their corresponding cis counterparts (data not shown). The most active compound from this small library was the (4S,6S)-6-(3,4-dimethoxyphenyl)-substituted aminothiazine 7 (IC50: 1.1 μM, Scheme 2). We then introduced the difluoro substituents onto 7 using 2,4-difluoro-5-(pyrimidin-5-yl)-benzaldehyde (4b, Scheme 2) in the three-component reaction, and the resulting analogue 8 exhibited an approximately 4-fold improved BACE1 inhibitory activity (IC50: 0.29 μM). This magnitude of improvement is consistent with that observed in the 6-unsunstituted biaryl aminothiazine series previously reported.12
Scheme 2. Synthesis of 6-Substituted Aminothiazines.

Our next library synthesis was aimed at enhancing potency, reducing lipophilicity (clogP: 3.6 for 8 versus 2.9 for 1), and improving ligand efficiency (LE: 0.29 kcal·mol–1 for 8 versus 0.37 kcal·mol–1 for 1). Molecular modeling of 8 indicated the potential for hydrogen-bond interactions between the two methoxy groups and the flap backbone, and these interactions may be improved by moving the oxygen into the ring, i.e., forming a heterocyclic ring system. Replacement of dimethoxyphenyl with an appropriate heterocycle was also anticipated to reduce lipophilicity and improve ligand efficiency. Thus, three-component reactions of vinyl heterocycles with 3-(pyrimidin-5-yl)-benzaldehyde (4a) were carried out, and this exercise led to 6-(3,5-dimethylisoxazol-4-yl) substituted trans aminothiazine (±)-9 with moderate activity (IC50: 0.23 μM, Figure 3). Again, we introduced the difluoro substituents, and the resulting (4S,6S) enantiomer 10 exhibited activity 5-fold more potent than the lead compound 1. As compared to the dimethoxyphenyl analogue 8, compound 10 also showed reduced lipophilicity (clogP: 2.9 versus 3.6 for 8) and improved ligand efficiency (LE: 0.35 kcal·mol–1 versus 0.29 kcal·mol–1 for 8). As observed in other pairs of enantiomers, the opposite enantiomer of 10 was significantly less active (data not shown). Introduction of a quaternary methyl at C-6 in (±)-11 dramatically reduced activity, while the activity was restored upon removal of the C-3 methyl of the isoxazole in (±)-12 (vide infra).
Figure 3.

Structures of (±)-9, 10, (±)-11, and (±)-12.
To better understand the binding conformation of the dimethylisoxazole group, we obtained a cocrystal structure of thiazine 10 bound in the BACE1 active site (Figure 4).15 As expected, the aminothiazine functionality formed intricate hydrogen-bond donor–acceptor interactions with aspartates 32 and 228 at the BACE1 active site; the C-2 hydrogen (see 10 in Figure 3) attached to the difluorophenyl ring formed a potential C–H hydrogen-bond with the backbone carbonyl of glycine 230; and the pyrimidine ring was projected into the opening of S3 pocket where it interacted with the carbonyl of serine 229 via a bridging water molecule. The two heteroatoms of the isoxazole ring served as hydrogen-bond acceptors to the amide NH groups of the threonine 72 and glutamine 73 located on the flap backbone.16 The C-6 isoxazole ring was arranged nearly perpendicularly to the thiazine ring, and introduction of a quaternary methyl at C-6 as in compound 11 would cause severe steric repulsion between the two methyl groups (Figure 5). As a result, the orthogonal arrangement would be unfavorable, thus resulting in diminished activity ((±)-11 IC50: 3.7 μM). However, when the C-3 methyl of the isoxazole was removed, as in compound 12, activity was restored. Indeed, the active enantiomer of compound (±)-12 was estimated to have an IC50 value of 90 nM (only one enantiomer was active based on SAR), comparable to that obtained with compound 10 (IC50: 70 nM, Figure 3).
Figure 4.
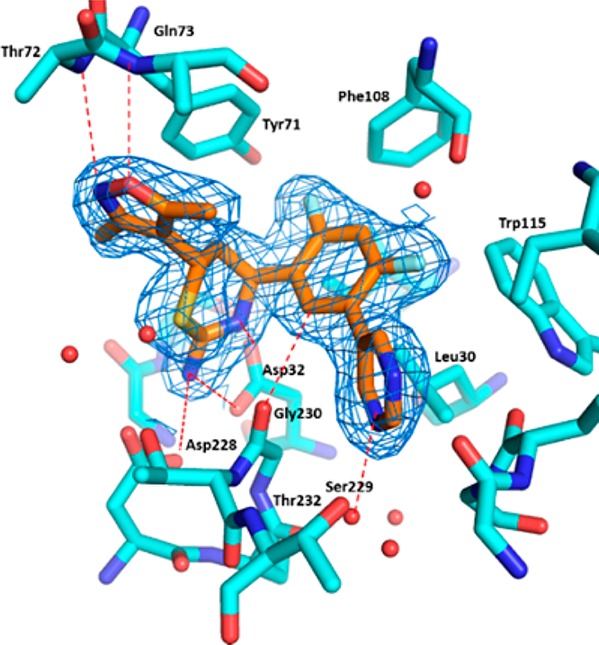
Cocrystal structure of 10 bound with BACE1 active site, with electron density contoured around 10 in blue mesh.15
Figure 5.
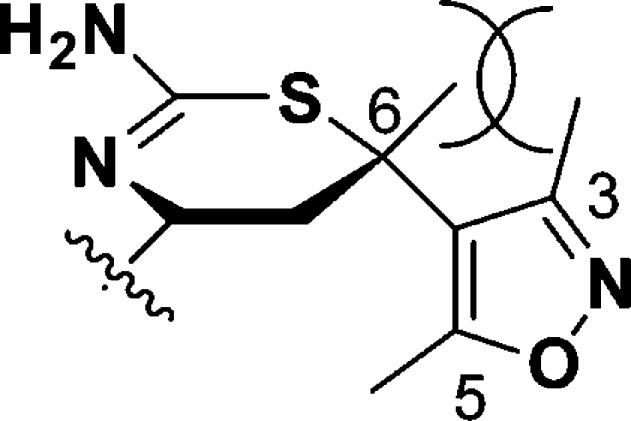
Steric interaction of the C-6 methyl of the thiazine ring with the C-3 methyl of isoxazole in 11.
With the C-4 des-methyl thiazine 10 identified as a moderately potent BACE1 inhibitor, our next step was to prepare the C-4 methyl-substituted aminothiazine 18 (Scheme 3). Reaction of amino alcohol 13, readily available using methodology recently developed in our laboratory,17 with benzoyl isothiocyanate provided the benzoyl thiourea derivative 14. Treatment of 14 with hydrochloric acid at 100 °C brought about ring closure and subsequent hydrolysis of the benzoyl group to give the 4,6-trans-aminothiazine 15, which was converted to its NHBoc derivative 16. The relative and absolute stereochemistry of 16 were confirmed by single crystal X-ray crystallography (Scheme 3).18 Sukuki coupling of 16 with pyrimidin-5-yl-boronic acid and subsequent N-Boc deprotection furnished 18.
Scheme 3. Synthesis of Aminothiazine 18.
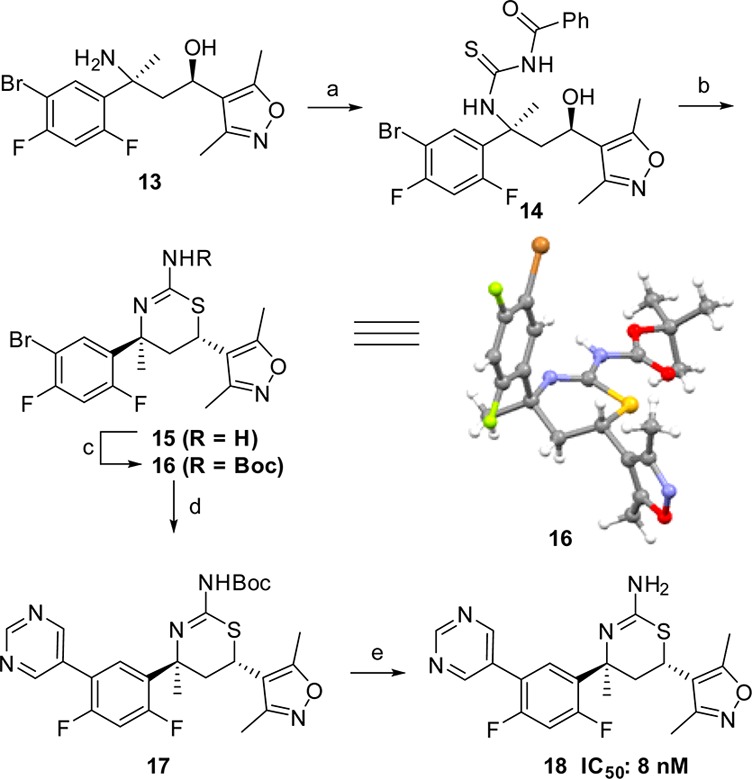
Reagents and conditions: (a) benzoyl isothiocyanate, CH2Cl2, rt, 94%; (b) 6 N HCl, dioxane, 100 °C, 6 h, 49%; (c) Boc2O, saturated NaHCO3, 1,4-dioxane, water, rt, 75%; (d) pyrimidin-5-yl-boronic acid, PdCl2(PPh3)2, Cs2CO3, DME, EtOH, H2O, 100 °C, 5 min, 60%; (e) TFA, CH2Cl2, rt, 100%. The thermal ellipsoid plot (50% ellipsoids) of 16 is also shown above.
Compound 18 (IC50: 8 nM) was nearly 10-fold more potent than the 4-des-methyl analogue 10. A similar trend was also observed in the iminopyrimidinone BACE1 inhibitors.19 Again, we obtained a cocrystal structure of 18 in the BACE1 active site,20 and Figure 6 shows the overlay of cocrystal structures of both 10 and 18. The biaryl group in both inhibitors is oriented in a pseudoaxial conformation to occupy the contiguous S1–S3 hydrophobic pockets. A similar effect was seen in the related iminopyrimidinone analogue;19 however, we believe the underlying energetics are different. In the iminopyrimidinone case, the pseudoequatorial conformation was preferred by 1.4 kcal/mol with a C-4 hydrogen substituent; addition of the C-4 methyl group led to a loss of this energy barrier to access the preferred pseudoaxial conformation (Figure 7). In the case of compounds 10 and 18, quantum mechanical calculations at the HF/6-31+G* level of theory showed that both compounds highly preferred the bound pseudoaxial conformations, by 10.2 kcal/mol for 10 and 5.3 kcal/mol for 18. We conclude that the 4-methyl group of 18 contributes to activity primarily through a favorable hydrophobic interaction with isoleucine 118, which lines the active site instead of conformational preference as shown in the iminopyrimidinone analogues.
Figure 6.
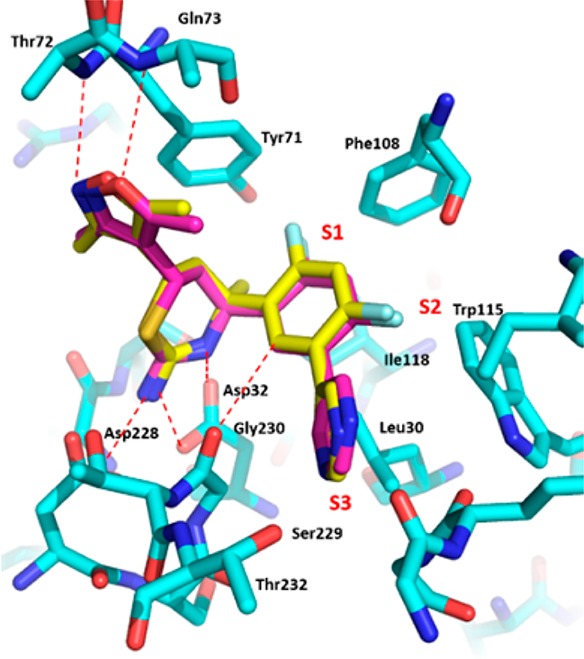
Overlay of cocrystal structures of 10 (violet) and 18 (yellow) bound with BACE1 active site.
Figure 7.

Energy difference between pseudoaxial and pseudoequatorial conformations of 10 and 18.
Despite its potent activity, compound 18 demonstrated only modest reduction in brain Aβ peptides in rodents relative to the vehicle treated controls even at 30 mg/kg po, a result of moderate brain penetration. The exposure was limited by poor metabolic stability across species: only 23% (human), 49% (rat), and 48% (mouse) remained after a 10 min incubation with liver microsomes at 0.5 μM. The major metabolites resulted from oxidation of the pyrimidine ring, a pathway difficult to address because BACE1 inhibitory activity was sensitive to substitution of this ring. In the end, we were unable to improve metabolic stability while maintaining potent BACE1 inhibitory activity.
While our work to increase the potency of biaryl aminothiazines was being undertaken, workers at Eli Lilly also disclosed the discovery of furo[3,4-d][1,3]thiazin-2-amine picolinamide 19 (LY2886721) (Figure 8), a potent BACE1 inhibitor that reached phase II clinical trials in mild cognitive impairment.21,22 This new disclosure from Eli Lilly prompted us to replace the pyrimidyl moiety of 18 with a picolinamide. To test this approach, we prepared the 4-des-methyl aminothiazine 20 using the three-component, one-pot reaction of 5-chloro-N-(4-fluoro-3-formylphenyl)picolinamide, 3,5-dimethyl-4-vinylisoxazole and thiourea (scheme not shown). Compound 20 displayed good BACE1 inhibitory activity (IC50: 5 nM). As the C-4 quaternary methyl of the thiazine ring and the C-4 fluoro substituent of the phenyl ring generally contributed to the activity of the biaryl aminothiazine BACE1 inhibitors, we prepared picolinamide 22 from bromide 16 in four steps: palladium-catalyzed azidation, reduction of azide to aniline 21, amidation of the aniline, and removal of the N-Boc group (Scheme 4). Interestingly, compound 22 was 6-fold less active than the 4-desmethyl analogue 20. The docking conformation of 22 (Figure 9) in the BACE1 active site indicated that the NH of the amide formed a hydrogen-bond with the backbone carbonyl of Gly230. The electronic repulsion between the 2-fluorine substituent on the phenyl and the amide carbonyl resulted in a nonoptimal hydrogen-bond configuration for the backbone carbonyl of Gly230. Relocation of fluoro substituent from C-4 to C-5 of the phenyl was expected to minimize this electronic repulsion. Indeed, the 5,6-difluorophenyl analogue 23 was 44-fold more potent than 22 with an IC50 value of less than 1 nM. The superior activity of the 5,6-difluorophenyl picolinamides over their 4,6-difluorophenyl counterparts appeared to be general, but the magnitude of the enhancement depended on the substituent(s) of the picolinic acid moiety. The 5,6-difluorophenyl picolinamides displayed either comparable or slightly improved activity over the 5-fluorophenyl analogues (data not shown). As we had expected, fluoro substitution at C-2 of the phenyl ring was not well tolerated (25, IC50: 160 nM) as this eradicated a potential C–H hydrogen-bond between the C-2 hydrogen of the phenyl and the carbonyl of Gly230 in the BACE1 backbone, as shown in the cocrystal structure of biaryl aminothiazine 10 (Figure 4). Overall, given a specific picolinamide, the activity of the analogues can be arranged in the order of 5,6-difluorophenyl (23) ≥ 5-fluorophenyl (24) > 4,6-difluorophenyl (22) > 2-fluorophenyl (25).
Figure 8.

Structures of 19 (LY2886721) and 20.
Scheme 4. Synthesis of Aminothiazine 24.

Reagents and conditions: (a) NaN3, CuSO4·5H2O, l-ascorbic acid sodium salt, trans-1,2-(bismethylamino)cyclohexane, 80 °C; (b) Pd/C (10%), MeOH, H2 balloon, rt, 37% for two steps; (c) 5-chloropicolinic acid, HATU, Hunig’s base, CH2Cl2, rt, 80%; (d) TFA, 61%
Figure 9.
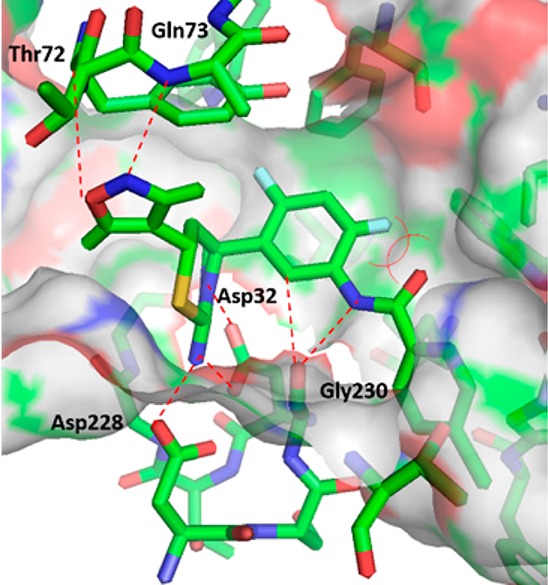
Docking conformation of 22 bound in the BACE1 active site.
The fluoro substituents exerted a profound impact not only on BACE1 inhibitory activity of the aminothiazine picolinamides, but also on their metabolic stability (Figure 10). For example, C-2 fluoro substitution (25) was detrimental to both activity and metabolic stability. Our structure–metabolic stability relationship studies revealed the following order of metabolic stability: 5,6-difluorophenyl (23), 4,6-difluorophenyl (22) > 2-fluorophenyl (25), 5-fluorophenyl (24). Taken together, we concluded that the 5,6-difluorophenyl picolinamides provided the optimal balance between BACE1 inhibitory activity and metabolic stability. Accordingly, we focused on 5,6-difluorophenyl based carboxamides. Optimization of the amide side chain in the S3 pocket culminated in the discovery of pyrazine-carboxamide 26 (Scheme 5) as a potent BACE1 inhibitor (IC50: 0.9 nM) with a good metabolic stability profile. Compound 26 was prepared from aniline 27, which was available following the exact sequence described for compound 21 (Scheme 4).
Figure 10.

Metstab (H/R/M): % percent compound that remained after a 10 min incubation with (H)uman, (R)at, or (M)ouse liver microsomes at 0.5 μM.
Scheme 5. Synthesis of Aminothiazine 26.

Reagents and conditions: (a) 5-methoxypyrazine-2-carboxylic acid, HATU, Hunig’s base, CH2Cl2; (b) TFA, CH2Cl2, 63% for two steps.
Compound 26 demonstrated robust brain Aβ reduction in rodents. A single oral administration of 26 at 10 mg/kg in mice resulted in a reduction of brain Aβ42 peptide relative to vehicle-treated control by 85% (data not shown). In rat studies, this compound demonstrated a dose-dependent reduction of brain Aβ40, Aβ42, and total Aβ (Aβ1-x) when dosed orally (Figure 11). The exposure of 26 was also measured in rat plasma and brain samples (Table 1) to allow quantification of the PK/PD relationship (Figure 12). Analysis of the rat brain Aβ42 reduction versus the total plasma concentration curve (Figure 12) resulted in the estimation of a brain Aβ42 EC50 of 260 nM total plasma concentration or 13 nM free drug plasma level (using rat plasma protein binding data for 28, 95%), which is 14-fold the cellular IC50 value of the compound. In terms of brain exposure required for efficacy, 260 nM total plasma exposure was achieved close to the 3 mg/kg drug dose (Table 1), which gave 360 nM plasma and 160 nM total brain exposure. Therefore, the total brain exposure at the ED50 was near 160 nM. Comparing this value to the cell IC50 (1 nM) implied significant brain drug binding for compound 26.
Figure 11.
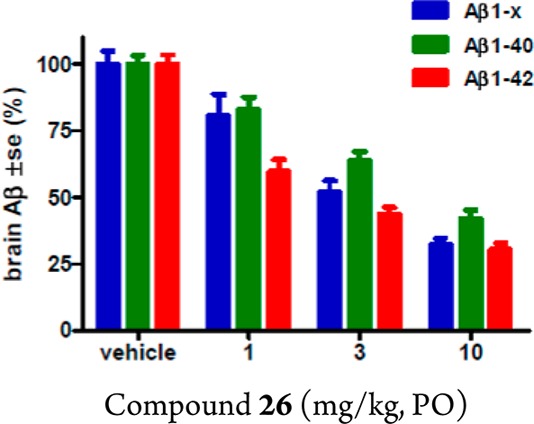
Dose-dependent inhibition of Aβ40, Aβ42, and total Aβ (Aβ1–x) in rat brain by 28 at 5 h after oral administration (n = 5 rats).
Table 1. Brain, Plasma Concentration, and Brain/Plasma Ratio of 26a.
| dose (mg/kg) | plasma conc (nM) (SD) | brain conc (nM) (SD) | brain/plasma |
|---|---|---|---|
| 1 | 150 (65) | 67 (46) | 0.44 (0.14) |
| 3 | 360 (75) | 160 (30) | 0.46 (0.10) |
| 10 | 1500 (320) | 420 (190) | 0.30 (0.10) |
Compound dosed PO to female rats in PEG-400/EtOH/Tween 80 (84:15:1); plasma and brain collected 5 h postdose (5 rats/dose) for determination of exposure and brain Aβ levels. Data are mean values (n = 5) with SD in parentheses.
Figure 12.
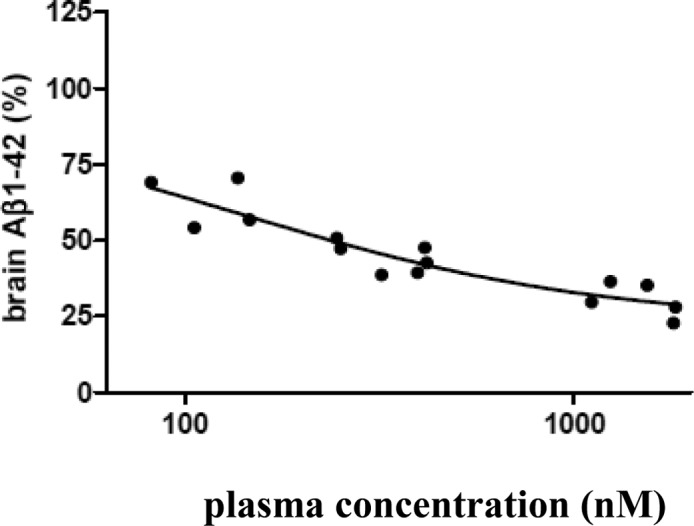
Brain Aβ42 reduction and plasma concentration curve of 26.
Compound 26 showed an IC50 of 0.64 μM in the hERG whole-cell patch clamp assay, which provided a 49-fold separation versus the brain Aβ42 EC50 of 13 nM free drug level in plasma. More studies would be required to determine if hERG inhibition is a potential safety issue for this compound.
The pyrazine-carboxamide 26 contains an amide bond, which could potentially be hydrolyzed in vivo to release the free aniline 28 (Figure 13). Indeed, in the mouse Aβ reduction studies, aniline 28 was detected in the plasma 3 h after oral administration. Free anilines per se are not inherently mutagenic, but further metabolism generates species that could be mutagenic. To address this concern, we submitted 28 to the standard Ames test, in which it was determined to be nonmutagenic. Compound 28 was obtained from 27 (Scheme 5) via removal of the N-Boc group. The absolute configuration of 28 was confirmed by way of single-crystal X-ray diffraction analysis (Figure 13).23
Figure 13.
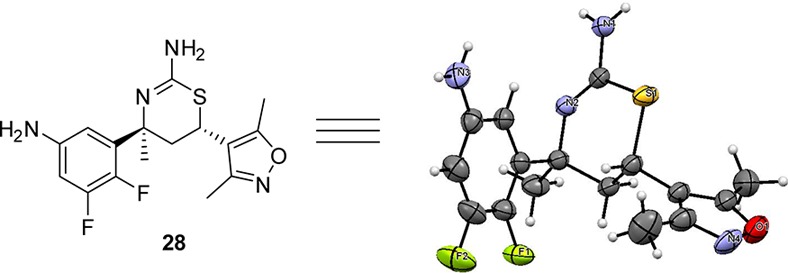
Thermal ellipsoid plot (50% ellipsoids) of aniline 28.
The cytochrome P450 inhibitory potential of compound 26 was also determined using recombinant CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 in order to assess the potential likelihood of drug–drug interactions. Low levels of inhibition were observed at all major human P450 isozymes (IC50 > 20 μM) with the highest level of inhibition against CYP2C8 (IC50: 5 μM).
In summary, we recognized certain features in 1 and used them to develop a general approach to enhance its potency by targeting the flap backbone in the active site of BACE1. This approach led to dimethoxyphenyl at C-6 of the thiazine ring as an initial binder, which incorporated weak hydrogen-bond acceptors to the flap backbone. The interactions with the flap were optimized with the dimethylisoxazole heterocycle. The quaternary methyl substituent at C-4 was introduced, and the resulting aminothiazine 18 was 34-fold more potent than 1. We then replaced the pyrimidyl moiety with a carboxamide, and our SAR studies showed that the 5,6-difluorophenyl moiety in the S1 pocket was optimal in terms of activity and metabolic stability. Optimization of the amide side chain in the S3 pocket culminated in the discovery of the pyrazine-carboxamide 26 as a potent BACE1 inhibitor with good metabolic stability. This compound demonstrated robust brain Aβ lowering after oral dosing in rat dose–response studies. Taken together, we believe that compound 26 can be considered for further preclinical evaluations for testing the amyloid cascade hypothesis.
Acknowledgments
We thank Dr. Nicholas Meanwell for critically reviewing this manuscript. We also thank Rudy Krause, Alan Xu Lin, and Tracey Hall for various biological evaluations, and A. Arumugam, K. Mahammed, Murali Botlagunta, P. N. Arunachalam, Arun Kumar Gupta, and Richard Rampulla for scaling up intermediate 15.
Glossary
Abbreviations Used
- TFA
trifluoroacetic acid
- TEA
triethylamine
- DMF
N,N-dimethylformamide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- rt
room temperature
- Metstab
metabolic stability
- CYP
cytochrome P450 enzymes
- po
oral administration
- PK/PD
pharmacokinetic/pharmacodynamic
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00432.
Synthetic methods and characterization data for compounds 6–28 and methods for in vitro, in vivo, and pharmacokinetic assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Koh J.-Y.; Yang L.-L.; Cotman C. W. Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990, 533, 315–320. 10.1016/0006-8993(90)91355-K. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Vassar R.; Kuhn P. H.; Haass C.; Kennedy M. E.; Rajendran L.; Wong P. C.; Lichtenthaler S. F. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 2014, 130, 4–28. 10.1111/jnc.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg G. D.; Montine T. J. The Genetics and Neuropathology of Alzheimer’s Disease. Acta Neuropathol. 2012, 124, 305–323. 10.1007/s00401-012-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T.; Atwal J. K.; Steinberg S.; Snaedal J.; Jonsson P. V.; Bjornsson S.; Stefansson H.; Sulem P.; Gudbjartsson D.; Maloney J.; Hoyte K.; Gustafson A.; Liu Y.; Lu Y.; Bhangale T.; Graham R. R.; Huttenlocher J.; Bjornsdottir G.; Andreassen O. A.; Jonsson E. G.; Palotie A.; Behrens T. W.; Magnusson O. T.; Kong A.; Thorsteinsdottir U.; Watts R. J.; Stefansson K. A. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- larson M. E.; Lesne S. E. Soluble Aβ oligomer production and toxicity. J. Neurochem. 2012, 120 (suppl. 1), 125–139. 10.1111/j.1471-4159.2011.07478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B.; Vassar R.; Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamford A.; Strickland C. Inhibitors of BACE for treating Alzheimer’s disease: a fragment-based drug discovery story. Curr. Opin. Chem. Biol. 2013, 17, 320–8. 10.1016/j.cbpa.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Yan R.; Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319. 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlrich D.; Prokopcova H.; Gijsen H. J. M. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2033–45. 10.1016/j.bmcl.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Audia J. E.; Mergott D. J.; Dustin J.; Sheehan S. M.; Watson B. M.. Aminodihydrothiazine derivatives as BACE inhibitors for the treatment of Alzheimer’s diseases. WO-2009/134617.
- May P. C.; Dean R. A.; Lowe S. L.; Martenyi F.; Sheehan S. M.; Boggs L. N.; Monk S. A.; Mathes B. M.; Mergott D. J.; Watson B. M.; Stout S. L.; Timm D. E.; LaBell E. S.; Gonzales C. R.; Nakano M.; Jhee S. S.; Yen M.; Ereshefsky L.; Lindstrom T. D.; Calligaro D. O.; Cocke P. J.; Hall D. G.; Friedrich S.; Citron M.; Audia J. E. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011, 31, 16507–16516. 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Huang S.; Wan J.; Yan L.; Pan Y.; Wu A. Two novel diastereoselective three-component reactions of alkenes or 3,4-dihydro-(2H)-pyran with urea/thiourea-aldehyde mixtures: [4 + 2] cycloaddition vsbBiginelli-type reaction. Org. Lett. 2006, 8, 2599–2602. 10.1021/ol060874b. [DOI] [PubMed] [Google Scholar]
- After this work was near completion, Lilly disclosed LY2811376 in ref (12) by the end of 2011. This paper suggests that the purpose of the quaternary methyl group is to exacerbate the desired topology and enhance chemical stability, but no details were provided.
- Structure coordinates have been deposited with the RSCB Protein Data Bank (PDB ID 5ENM).
- For an example of applying hydrogen-bond interactions involving 3,5-dimethylisoxazole in drug discovery, see:Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Panagis Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–70. 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guernon J.; Marcin L.; Higgins M.; Yang F.; Shi J.; Snyder L.; Thompson L. A.; Wu Y.-J. Synthesis of β-disubstituted β-amino isoxazolyl ketones by addition of ketimines with isoxazolyl methyl ketone enolates. Tetrahedron Lett. 2014, 55, 2134–2137. 10.1016/j.tetlet.2014.02.053. [DOI] [Google Scholar]
- The crystal structure of 16 has been deposited in the Cambridge Crystallographic Data Centre (CCDC #: 1436245).
- Edwards P. D.; Albert J. S.; Sylvester M.; Aharony D.; Andisik D.; Callaghan O.; Campbell J. B.; Carr R. A.; Chessari G.; Congreve M.; Frederickson M.; Folmer R.H. A.; Geschwindner S.; Koether G.; Kolmodin K.; Krumrine J.; Mauger R. C.; Murray C. W.; Olsson L.; Patel S.; Spear N.; Tian G. Application of fragment-based lead generation to the discovery of novel, cyclic amidine β-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J. Med. Chem. 2007, 50, 5912–5925. 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]
- Structure coordinates have been deposited with the RSCB Protein Data Bank (PDB ID 5ENK).
- May P. C.; Willis B. A.; Lowe S. L.; Dean R. A.; Monk S. A.; Cocke P. J.; Audia J. E.; Boggs L. N.; Borders A. R.; Brier R. A.; Calligaro D. O.; Day T. A.; Ereshefsky L.; Erickson J. A.; Gevorkyan H.; Gonzales C. R.; James D. E.; Jhee S. S.; Komjathy S. F.; Li L.; Lindstrom T. D.; Mathes B. M.; Martènyi F.; Sheehan S. M.; Stout S. L.; Timm D. E.; Vaught G. M.; Watson B. M.; Winneroski L. L.; Yang Z.; Mergott D. J. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1200. 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audia J. E.; Mergott D. J.; Shi C. E.; Vaught G. M.; Watson B. M.; Winneroski L. L. Jr.. BACE inhibitors. WO-2011/005738.
- The crystal structure of 29 has been deposited in the Cambridge Crystallographic Data Centre (CCDC #: 1436246).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.