

Abstract
The optimization, based on computational, thermodynamic, and crystallographic data, of a series of small-molecule ligands of the Phe43 cavity of the envelope glycoprotein gp120 of human immunodeficiency virus (HIV) has been achieved. Importantly, biological evaluation revealed that the small-molecule CD4 mimics (4–7) inhibit HIV-1 entry into target cells with both significantly higher potency and neutralization breadth than previous congeners, while maintaining high selectivity for the target virus. Their binding mode was characterized via thermodynamic and crystallographic studies.
Keywords: HIV, gp120, CD4, entry inhibitor, structure-based drug design, thermodynamics, X-ray crystallography, viral inhibition, protein−protein interactions
With an estimated 35 million people currently living with HIV-1 worldwide in conjunction with 2.1 million new infections each year,1 the development of effective new strategies to achieve both prevention of HIV transmission, complementing known pre-exposure prophylactics2,3 and depletion of the “viral reservoir” in HIV-infected patients remain critically important challenges.
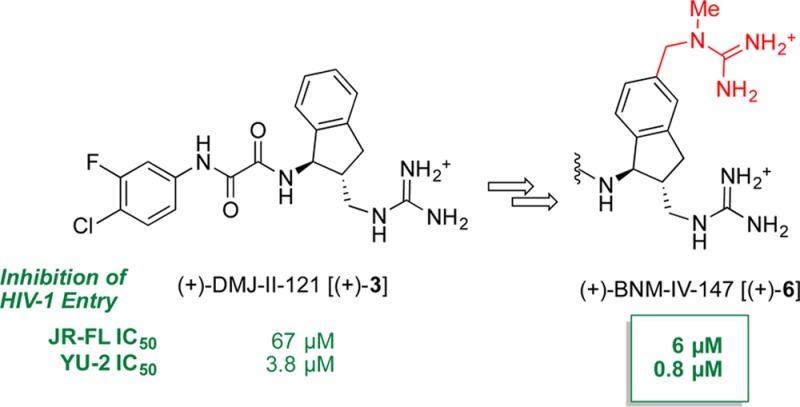
Recently, we reported the structure-based design, synthesis, and evaluation of the small-molecule CD4-mimic DMJ-II-121 (3), a micromolar inhibitor of HIV-1YU-2 entry into human immune cells.4,5 Like the initial congeners reported by Debnath et al. (see 1 and 2, Figure 1),6−83 binds the Phe43 cavity of the viral envelope (Env) glycoprotein gp120, blocking CD4 binding and triggering a conformational change that ultimately results in viral inactivation.
Figure 1.
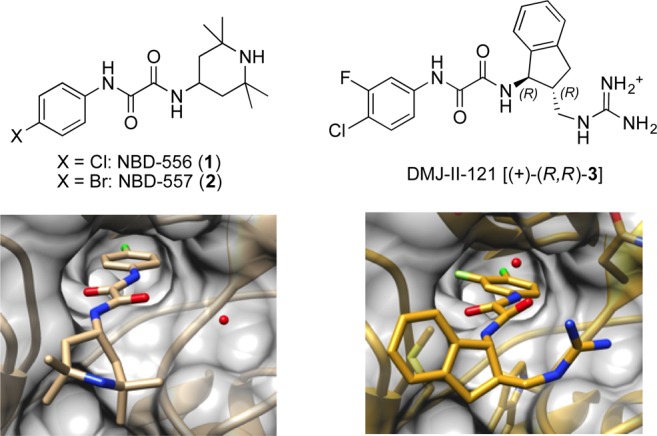
NBD-556 (1), (+)-DMJ-II-121 [(+)-(R,R)-3], and their gp120-bound X-ray structures (PDB accession codes: 3TGS and 4I53, respectively).
A particularly attractive feature of this mechanism of action resides in the opportunity to elicit immune responses specific to epitopes that become exposed on the surface on Env upon the binding of CD4 mimics. Importantly, we have demonstrated that antibodies can indeed recognize gp120 bound to the CD4 small-molecule mimics, both on the surface of the virion9 and, more recently, on the surface of infected cells.10 Thus, in our efforts to enhance the direct inhibitory activity of the small-molecule CD4 mimetics, we have sought to improve our understanding of the underlying molecular mechanisms. Logically, we chose 3 as the starting point for the design of a new generation of more potent small-molecule entry inhibitors.
Having previously conducted structural optimization on Regions I and II of the parent scaffold, defined respectively by the halogenated aromatic ring and the oxalamide linker (Figure 2), our efforts focused on modification of the substituted indane ring comprising Region III.11 Importantly, the protein:ligand cocrystal structure of 3 permitted definition of potential binding hotspots in proximity to the aromatic portion of the indane.4 Based on the structure and “hotspot” analysis,12 we also demonstrated that a methylene (one carbon) spacer present between the indane skeleton and the guanidinium moiety in 3 proved optimal for viral entry inhibition;4 therefore, no modifications were planned in that section of Region III. Instead, a virtual fragment growing algorithm,13 followed by docking,14,15 was carried out to establish contacts between the potentially new analogues and “hotspot” gp120Gly472, a highly conserved gp120 residue located in the outer domain near the vestibule of the Phe43 cavity, associated with moderate Env restructuring (i.e., allosteric signal) upon CD4 binding.12,16 This computational analysis suggested that addition of a (methylamino)methyl unit at position 6 of the indane skeleton (see 4, Figure 2) might result in hydrogen bonding with the backbone carbonyl of gp120Gly472. Our computational model also predicted binding between the guanidinium terminus and gp120Asp368, an interaction that had been predicted by our prior modeling work, but to date had proven elusive.5
Figure 2.
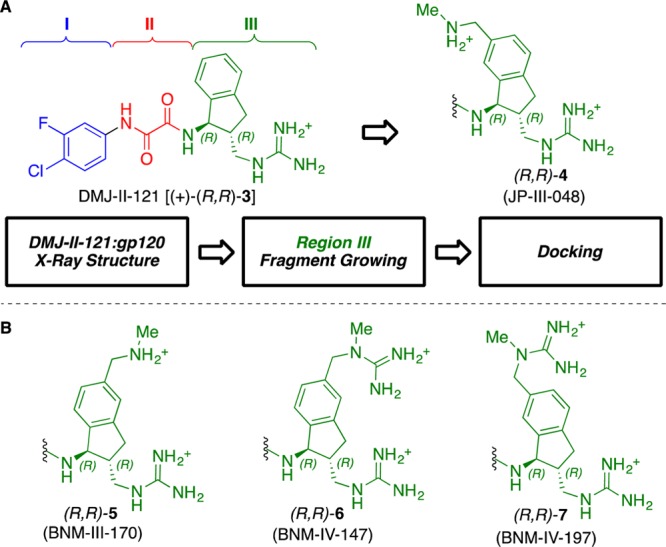
(A) Computational design of 4. (B) Further evolution of inhibitor Region III scaffolds.
To evaluate in rapid fashion the biological impact of this previously unexplored scaffold substitution pattern, the synthesis of the (methylamino)methyl-bearing analogue JP-III-048 (4) was carried out initially in racemic form (see Supporting Information). Pleasingly, a viral infectivity assay employing (±)-4 revealed that the proposed structural modification did not result in significant loss of inhibitory activity relative to 3 (HIV-1YU-2 IC50 6.8 μM). To determine the effect of absolute stereoconfiguration on bioactivity, enantioenriched (+)- and (−)-4 were prepared by separation of the ultimate synthetic intermediate via semipreparative supercritical fluid chromatography (SFC) employing a chiral stationary phase. Global deprotection resulted in (+)- and (−)-4 (in >96% ee). Evaluation of the individual enantiomers revealed that similar to the previous enantiopure congeners, the (R,R)-stereoisomer (+)-4 exhibited 40-fold stronger inhibitory activity than the (S,S)-counterpart (YU-2 IC50 2.1 and 87.8 μM, respectively). This result represents a gain, albeit modest, relative to (+)-3 (YU-2 IC50 3.8 μM). We were, however, pleased to find that the binding affinity of (+)-4 to full-length gp120, measured by isothermal titration calorimetry (ITC), had increased quite significantly [KD 24 vs 110 nM for (+)-3].
Given the difference in activity between (+)- and (−)-4, the original synthetic route was modified to permit facile preparation of the single enantiomers of this new generation of entry inhibitors. To this end, the enantioselective synthesis of (+)-4 began with protection of commercially available 6-bromoindanone (8) as the corresponding dioxolane, followed by lithiation and formylation, to furnish aldehyde 9 (Scheme 1). Reductive amination, protection of the resulting secondary amine as the corresponding Boc carbamate, and selective removal of carbonyl protection then revealed indanone 10, now bearing the desired (methylamino)methyl functionality. Ketone 10 was next carboxylated at the α-position, and the resulting racemic ketoester (±)-11 subjected to dynamic-kinetic resolution employing chiral ruthenium catalyst RuCl(p-cymene)[Ts-DPEN],17 which permitted transfer hydrogenation with excellent stereoselectivity (er 99:1), as demonstrated by chiral phase SFC. This transformation was carried out employing both enantiomers of the ruthenium catalyst, thus permitting access to both enantiomers of β-hydroxyester 12. Each enantiomer was then taken through the following identical sequence. Exhaustive reduction of (+)-12, initially utilizing sodium borohydride in methanol, resulted in partial epimerization (cis/trans ≈ 3:1). Pleasingly, treatment of (+)-12 with the more reactive lithium aluminum hydride in an aprotic solvent (THF) provided the desired diol as a single stereoisomer. Selective silylation of the primary hydroxyl followed by treatment with diphenylphosphorylazide provided the corresponding azide with complete inversion of stereochemistry at the benzylic position. Palladium-catalyzed hydrogenation next furnished amine (+)-13. Following union with oxalic acid 14, fluoride-mediated removal of the silyl group furnished the corresponding primary alcohol, which in turn was subjected to Mitsunobu conditions18 to install the requisite guanidine functionality and reveal, after treatment with trifluoroacetic acid, target inhibitor (+)-(R,R)-4 as the corresponding bis(trifluoroacetate) salt. Gratifyingly, the synthetic route proved highly efficient, furnishing the target compound in 20% yield over 15 steps from commercially available 6-bromoindanone (8, Scheme 1). The alternate enantiomer (−)-(S,S)-4 was synthesized in identical manner from (−)-12.
Scheme 1. Enantioselective Synthesis of (+)-(R,R)-4.

To understand the structural basis of the increased bioactivity, (+)-(R,R)-4 was soaked into preformed crystals of gp120 from HIV Clade C1086,19 and diffraction data was obtained at 2.7 Å Bragg spacings (see Supporting Information). The resulting protein:ligand structure revealed that, compared to (+)-(R,R)-3 entry inhibitor, (+)-(R,R)-4 establishes two additional hydrogen-bonding interactions, one with the backbone carbonyl of gp120Gly431 and another with the carbonyl of gp120Gly472, in agreement with our computational prediction. Disappointingly, however, (+)-(R,R)-4 failed to bind gp120Asp368. Instead, as in the case of (+)-(R,R)-3,4 the guanidinium terminus interacts with the backbone carbonyl of gp120 Met426. Considering (i) the diverse nature of the hotspots located in proximity to the Phe43 cavity (i.e., their varying contributions to the global allosteric signal upon binding of CD4), and (ii) the proximity of the ligand’s guanidinium to the still elusive gp120Asp368, we reasoned that subtle structural modifications resulting in changes in ligand orientation might significantly impact bioactivity. We thus set out to prepare analogues now bearing the (methylamino)methyl moiety at indane position 5 (see 5, Figure 2). The route described above, in this case employing commercially available 5-bromoindanone (see Supporting Information for details) in similar synthetic fashion, readily furnished the target compounds (+)-(R,R)- and (−)-(S,S)-5 (Figure 2).
Although (+)-(R,R)-5 displayed only a modest improvement in activity against HIV-1 monotropic strains YU2 and ADA (clone AD8) (IC50 1.5 and 6.4 μM, respectively), a 5-fold increase in potency relative to congener 3 was observed against strain JR-FL (IC50 14.2 μM). Again, however, (−)-(S,S)-5 displayed a lower level of viral entry inhibition (JR-FL IC50 > 100 μM, Table 1). The neutralization breadths of the more potent CD4 mimics, specifically (+)-(R,R)-4 and (+)-(R,R)-5, were measured against a panel of 52 HIV-1 Env pseudoviruses. Like for previous congeners,4 clade B pseudoviruses were the most sensitive to the new inhibitors (9/9 strains were neutralized), with also significant activity in clades C (11/17) and D (3/3). While both assayed compounds demonstrated similar inhibitory profiles, (+)-(R,R)-5 exhibited 1.3-to-1.8-fold lower geometric mean titer (GMT) inhibitory concentrations than (+)-(R,R)-4 in clades B (GMT IC50 3.5 μM) and C (GMT IC50 17 μM), thus corroborating the trend observed with individual strains (see Supporting Information for further details).
Table 1. Small-Molecule CD4Mimics: Inhibition of Viral Entry and Protein–Ligand Affinity.
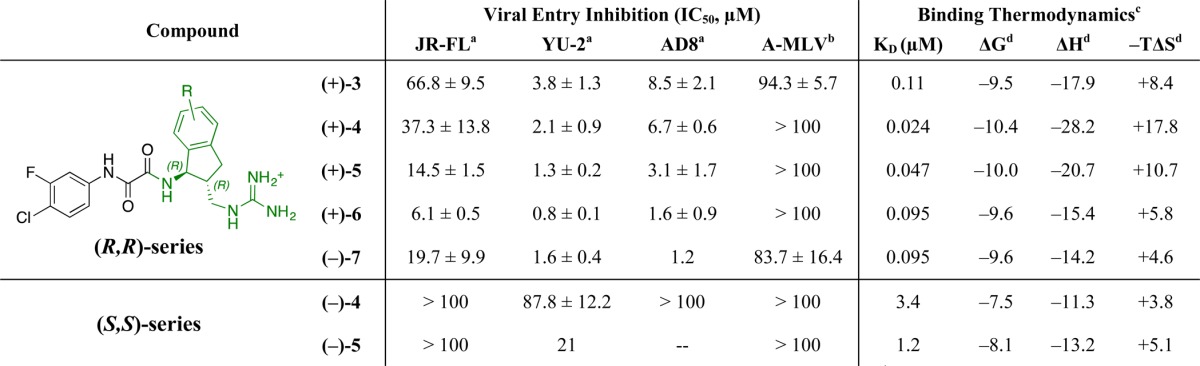
The IC50 was determined in Cf2Th-CD4/CCR5 cells infected with HIV-1 virus of the strains indicated.
The IC50 in cells infected with HIV-1 pseudotyped with the envelope glycoproteins of amphotropic murine leukemia virus (A-MLV).
As deemed by ITC employing YU-2 gp120.
Energies in kcal/mol.
However, when we turned to ITC to evaluate the binding affinities of these new analogues, we discovered that, although more active against viral entry, (+)-(R,R)-5 binds monomeric gp120 with a lower affinity than (+)-(R,R)-4 (KD 47 vs 24 nM). Interestingly, compared to the immediate predecessor, (+)-(R,R)-5 elicits a lowered entropic penalty (−TΔS = +10.7 kcal/mol vs – TΔS = +17.8 kcal/mol, Table 1). The difference in thermodynamic signatures between (+)-(R,R)-4 and (+)-(R,R)-5 upon binding gp120 suggests that subtle structural differences (e.g., in this case, position isomerism) could be responsible for inducing significantly dissimilar binding modes, a hypothesis we again sought to evaluate crystallographically.
The resulting structure of gp120-bound (+)-(R,R)-5, obtained employing the same technique as described above (see Supporting Information), revealed a generally similar binding pose to that of (+)-(R,R)-4. Orientations for the indane rings differ slightly such that the substituent projects into much of the same space despite being moved from position 5 to position 6. Nevertheless, the hydrogen bond from the (methyamino) methyl group to the carbonyl of gp120Gly472 is now lost [dN–O = 3.65 Å in 5 vs 3.08 Å in 4]. This change may explain the decreased enthalpic contribution to the binding energy (Table 1). The observations derived from the structure of (+)-(R,R)-5:gp120 with perhaps more immediate practical implications was the proximity of the ligand’s (methylamino)methyl side-chain to gp120Asp474, a CD4 binding hotspot associated with a purely enthalpic signal.12 To exploit this feature we directly converted the secondary amine in (+)-(R,R)-5 to a guanidinium moiety, which would in effect extend the reach of the side-chain and in turn potentially establish a strong interaction with the nearby carboxylate of gp120Asp474. Pleasingly, selective incorporation of the new guanidine functionality could be readily achieved in one step by subjecting (+)-(R,R)-5 to 1H-pyrazol-1-carboxamidine hydrochloride, to furnish (+)-(R,R)-6 (see Supporting Information). To our delight, (+)-(R,R)-6 inhibited infection of HIV-1 at lower concentrations than all previous analogues in all tested strains (Figure 3; YU-2IC50 0.8 μM; JR-FLIC50 6.2 μM; AD8IC50 3.3 μM; Table 1). Interestingly, however, the binding affinity of (+)-(R,R)-6 for monomeric gp120 again appeared to decrease relative to the less potent congeners (+)-(R,R)-4 and (+)-(R,R)-5. In turn, the diguanidinium congener (−)-(R,R)-7, prepared in a similar fashion from (+)-(R,R)-4, exhibiting a thermodynamic profile nearly identical to that of (+)-(R,R)-6, proved to be approximately half as potent (see Table 1). Crystal structures were also determined for gp120 complexes with (+)-(R,R)-6 and with (−)-(R,R)-7. The binding poses for these (methylguanidino) methyl derivatives are almost the same as those for their (methylamino) methyl congeners, (+)-(R,R)-4 and 5 (Figure 4); however, the added guanidine moieties point away from possible hydrogen-bonding partners, including gp120Asp474. For all four complexes, there are hydrogen bonds from the guanidinium group at indane position 1 to the carbonyl groups of gp120Met426 and gp120Gly431. For (+)-(R,R)-6 and (−)-(R,R)-7, distances to the carbonyl oxygen of gp120Glu429 suggest additional hydrogen bonding (dN–O = 3.05 Å for 6 and 3.21 Å for 7 as compared to 3.3–3.4 Å for 4 and 5).
Figure 3.
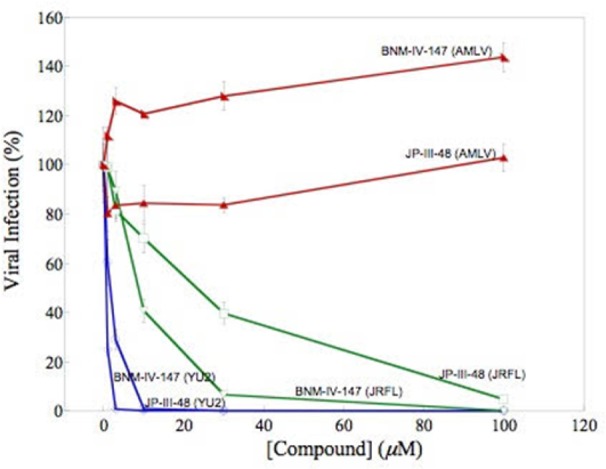
Effect of (+)-(R,R)-4 (JP-III-048) and (+)-(R,R)-6 (BNM-IV-147) on the infection of Cf2Th-CD4-CCR5 cells by recombinant, luciferase-expressing HIV-1 pseudoviruses carrying envelope glycoproteins of the YU2 or JR-FL strains or that of the amphotropic murine leukemia virus (AMLV, negative control). Virus infection is expressed as the percentage of infection (measured by luciferase activity in the target cells) observed in the presence of increasing concentrations of compound relative to the level of infection observed in the absence of compound.
Figure 4.
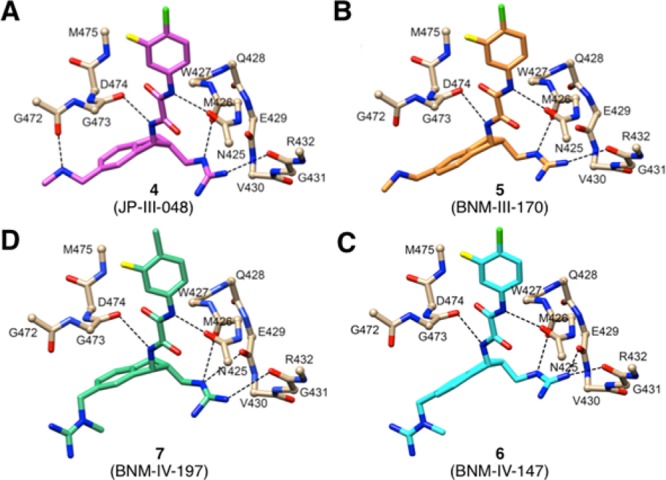
Comparison of gp120-bound poses of 4 (A), 5 (B), 6 (C), and 7 (D), all (R,R)-series. Orientations are the same based on superpositioning of gp120 Cα positions. Hydrogen-bond interactions are shown as dashed lines.
Although the disparity between potency and protein affinity still remains an unresolved paradox, a possible explanation lies in that thermodynamic measurements employ monomeric gp120, which only comprises part of the fully functional Env (a trimer of gp41-gp120). Indeed, interactions of the more potent inhibitors and the trimeric functional Env might also entail interactions between the Env protomers themselves that in vitro assays employing monomeric gp120 would not reveal. Logically, nonetheless, thermodynamic data are in agreement with crystallographic data, also obtained on monomeric glycoprotein.
In summary, the design of a more potent and more broadly active small molecule CD4-mimetic inhibitor of HIV-1 infection has been achieved exploiting structural and thermodynamic data. In particular, (+)-(R,R)-6 exhibited 1 order of magnitude improvement in inhibitory potency, over our last generation of compounds, against the neutralization-resistant HIV-1 strain JR-FL. Virus neutralization strategies employing these new constructs, in addition to direct viral entry inhibition, are currently being explored and will be reported in due course. These strategies rely on the unique ability of the CD4-mimetic inhibitors to expose, upon Env binding, epitopes recognized by antibodies present in HIV-infected individuals.9,10 That we have already demonstrated the capability of the earlier congeners of (+)-(R,R)-6, namely, 3 and 4, to enhance the efficacy of immune responses both against the HIV virus itself7 and against infected cells10 is very encouraging.
Acknowledgments
We thank Irwin Chaiken and all the members of the P01 Consortium Structure-Based Antagonism of HIV-1 Envelope Function in Cell Entry. Dr. Rakesh Kohli, and Drs. George Furst and Jun Gu (University of Pennsylvania) are also acknowledged for their assistance obtaining mass and NMR spectra, respectively.
Glossary
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- SIV
simian immunodeficiency virus
- sCD4
soluble CD4
- ITC
isothermal titration calorimetry
- A-MLV
amphotropic murine leukemia virus
- GMT
geometric mean titer
- THF
tetrahydrofuran
- Boc
tert-butoxycarbonyl
- HMDS
hexamethyldisilazide
- DPEN
diphenylethylenediamine
- TBS
tert-butyldimethylsilyl
- DPPA
diphenylphosphorylazide
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- TBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- DIPEA
diisopropylethyl amine
- TBAF
tributylammonium fluoride
- DEAD
diethylazodicarboxylate
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00471.
Synthesis, experimental methods and crystallographic data (PDF)
Accession Codes
Accession Codes Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: 5F4L, 5F4P, 5F4R, and 5F4U.
Author Present Address
J.P.: LG Chem, Daejeon, South Korea.
Author Present Address
J.R.C.: Seattle Genetics, Bothell, Washington 98021, United States.
Author Present Address
J.M.L.: Teva Pharmaceuticals USA Inc., Horsham, Pennsylvania 19044, United States.
Author Present Address
D.J.W.: Max Planck Florida Institute for Neuroscience, Jupiter, Florida 33458, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by a Program Project (NIH GM 56550) to J.L., E.F., W.A.H., A.B.S., and J.S.
The authors declare no competing financial interest.
Supplementary Material
References
- United Nations. UNAIDS Fact Sheet 2014.
- Karim Q.; Karim S.; Frohlich J. Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of HIV Infection in Women. Science 2010, 329, 1168–1174. 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celum C.; Baeten J. Tenofovir-Based Pre-Exposure Prophylaxis for HIV Prevention: Evidence and Evolving Questions. Curr. Opin. Infect. Dis. 2012, 25, 51–57. 10.1097/QCO.0b013e32834ef5ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J. M.; Le-Khac M.; Jones D. M.; Courter J. R.; Park J.; Schön A.; Princiotto A. M.; Wu X.; Mascola J. R.; Freire E.; Sodroski J.; Madani N.; Hendrickson W. A.; Smith A. B. III Structure-Based Design and Synthesis of an HIV-1 Entry Inhibitor Exploiting X-Ray and Thermodynamic Characterization. ACS Med. Chem. Lett. 2013, 4, 338–343. 10.1021/ml300407y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courter J. R.; Madani N.; Sodroski J.; Schön A.; Freire E.; Kwong P. D.; Hendrickson W. A.; Chaiken I. M.; LaLonde J. M.; Smith A. B. III Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist. Acc. Chem. Res. 2014, 47, 1228–1237. 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ma L.; Jiang S.; Lu H.; Liu S.; He Y.; Strick N.; Neamati N.; Debnath A. K. Identification of N-Phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a New Class of HIV-1 Entry Inhibitors that Prevent gp120 Binding to CD4. Virology 2005, 339, 213–225. 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- For recent work by Debnath et al. in the area of HIV entry inhibitors see:Curreli F.; Kwon Y.; Zhang H.; Scacalossi D.; Belov D. S.; Tikhonov A. A.; Andreev I. A.; Altieri A.; Kurkin A. V.; Kwong P. D.; Debnath A. K. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. 10.1021/acs.jmedchem.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Haque K.; Xie L.; Qiu Q.; Xu J.; Yong W.; Tong X.; Debnath A. K. Synthesis, Antiviral Activity and Resistance of a Novel Small Molecule HIV-1 Entry Inhibitor. Bioorg. Med. Chem. 2015, 23, 7618. 10.1016/j.bmc.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N.; Princiotto A. M.; Schön A.; LaLonde J.; Feng Y.; Freire E.; Park J.; Courter J. R.; Jones D. M.; Robinson J.; Liao H.-X.; Moody M. A.; Permar S.; Haynes B.; Smith A. B.; Wyatt R.; Sodroski J. CD4-Mimetic Small Molecules Sensitize Human Immunodeficiency Virus to Vaccine-Elicited Antibodies. J. Virol. 2014, 88, 6542–6555. 10.1128/JVI.00540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Veillette M.; Brassard N.; Iyer S. S.; Roger M.; Martin L.; Pazgier M.; Schön A.; Freire E.; Routy J.-P.; Smith A. B. III; Park J.; Jones D. M.; Courter J. R.; Melillo B. N.; Kaufmann D. E.; Hahn B. H.; Permar S. R.; Haynes B. F.; Madani N.; Sodroski J. G.; Finzi A. CD4Mimetics Sensitize HIV-1-Infected Cells to ADCC. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E2687–E2694. 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For efforts by Tamamura et al. toward the optimization of CD4-mimetic HIV entry inhibitors, in particular via modification of the so-called Region III, see:Mizuguchi T.; Harada S.; Miura T.; Ohashi N.; Narumi T.; Mori H.; Irahara Y.; Yamada Y.; Nomura W.; Matsushita S.; Yoshimura K.; Tamamura H. A Minimally Cytotoxic CD4Mimic as an HIV Entry Inhibitor. Bioorg. Med. Chem. Lett. 2016, 26, 397–400. and refs cited therein 10.1016/j.bmcl.2015.11.103. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Schön A.; Freire E. Optimization of CD4/gp120 Inhibitors by Thermodynamic-Guided Alanine-Scanning Mutagenesis. Chem. Biol. Drug Des. 2013, 81, 72–78. 10.1111/cbdd.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2012. [Google Scholar]
- Jones G.; Willet P.; Glen R. C.; Leach A. R.; Taylor R. Development And Validation of A Genetic Algorithm For Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved Protein-Ligand Docking Using GOLD. Proteins: Struct., Funct., Genet. 2003, 52, 609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Kwong P. D.; Wyatt R.; Robinson J.; Sweet R. W.; Sodroski J.; Hendrickson W. A. Structure of an HIV gp120 Envelope Glycoprotein in Complex with the CD4 Receptor and a Neutralizing Human Antibody. Nature 1998, 393, 648–659. 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros A.; Magriz A.; Dietrich H.; Lassaletta J. M.; Fernández R. Stereoselective Synthesis of syn β-Hydroxy Cycloalkane Carboxylates: Transfer Hydrogenation of Cyclic β-Keto Esters via Dynamic Kinetic Resolution. Tetrahedron 2007, 63, 7532–7537. 10.1016/j.tet.2007.05.058. [DOI] [Google Scholar]
- Odadzic D.; Bramsen J. B.; Smicius R.; Bus C.; Kjems J.; Engels J. W. Synthesis of 2′-O-Modified Adenosine Building Blocks and Application for RNA Interference. Bioorg. Med. Chem. 2008, 16, 518–529. 10.1016/j.bmc.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Kwon Y. D.; Finzi A.; Wu X.; Dogo-Isonagie C.; Lee L. K.; Moore L. R.; Schmidt S. D.; Stuckey J.; Yang Y.; Zhou T.; Zhu J.; Vicic D. A.; Debnath A. K.; Shapiro L.; Bewley C. A.; Mascola J. R.; Sodroski J. G.; Kwong P. D. Unliganded HIV-1 gp120 Core Structures Assume the CD4-Bound Conformation with Regulation by Quaternary Interactions and Variable Loops. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5663–5668. 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.