

Abstract
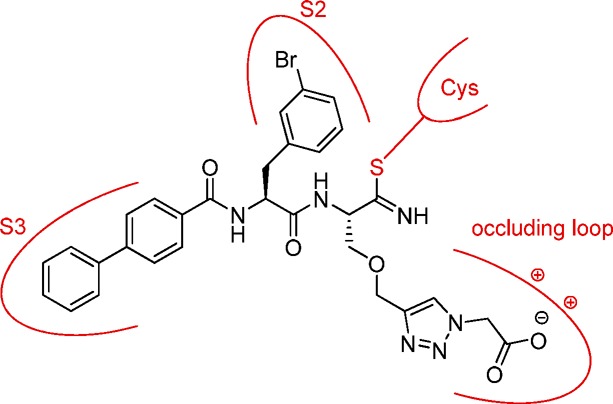
An active site mapping of human cathepsin B with dipeptide nitrile inhibitors was performed for a combinatorial approach by introducing several points of diversity and stepwise optimizing the inhibitor structure. To address the occluding loop of cathepsin B by a carboxylate moiety, click chemistry to generate linker-connected molecules was applied. Inhibitor 17 exhibited Ki values of 41.3 nM, 27.3 nM, or 19.2 nM, depending on the substrate and pH of the assay. Kinetic data were discussed with respect to the conformational selection and induced fit models.
Keywords: Copper-catalyzed azide−alkyne cycloaddition, cysteine proteases, human cathepsin B, nitrile inhibitors
Cathepsin B is a papain-like cysteine protease that is ubiquitously expressed and involved in various physiological processes.1 In nontumor cells, it is localized within endosomes/lysosomes, while in transformed cells it adopts a peripheral distribution in the cytoplasm and is, moreover, associated with the plasma membrane.2,3 At the surface of invasive tumor cells, cathepsin B was found to bind to annexin II or caveolin-1, the structural protein of caveolae. Caveolae serve as a platform for a proteolytic cascade for the degradation of the extracellular matrix (ECM), executed by several proteases, including cathepsin B.2−4
Cathepsin B affects the ECM either directly by extracellular proteolytic degradation of its components or indirectly via activation of other proteases. Partially degraded ECM components can be internalized to intracellular endosomal/lysosomal vesicles where the proteolysis is continued. This ECM breakdown remodels the tumor environment, promotes tumor invasion, and enables angiogenesis and metastasis. Cathepsin B also contributes to angiogenesis by degrading metalloprotease inhibitors and releasing growth factors that are bound to ECM components.2,5,6 The crucial roles of cathepsin B at multiple points of the tumor development have been established in several in vitro and in vivo models and cathepsin B was proposed to be a prognostic marker in patients with various types of cancer.1,2,7
Cathepsin B shows endopeptidase and exopeptidase activity, a unique feature for cysteine cathepsins, arousing from a flexible structural element, referred to as the occluding loop. The occluding loop is a 19–20 amino acid section that blocks the active site cleft at the end of the primed site. This event leaves only space for two amino acid residues of the substrate C-terminal of the scissile bond, that is, in the P1′ and P2′ positions. In this so-called closed conformation, two salt bridges, His110-Asp22 and Arg116-Asp224, hold the occluding loop over the primed subsites of the substrate binding cleft, preventing extended binding of large endopeptidase substrates and conferring an exopeptidase activity to cathepsin B. Mutations of His110 and Asp22 or the deletion of 12 amino acids of the occluding loop to enforce an open conformation reduced the exopeptidase activity of cathepsin B in favor of its endopeptidase activity.8,9 His110 and His111 of the occluding loop contribute to the exopeptidase activity by providing an appropriately spaced acceptor to bind the C-terminal carboxyl group of a peptide substrate.2,3,10,11
Low pH values of around 4–5 are connected with the exopeptidase activity of cathepsin B, while the endopeptidase activity increases with a rising pH value. This effect was suggested to result from the protonation/deprotonation state of the residues involved in the stabilizing salt bridges. At low pH, corresponding to the conditions of lysosomal acidic compartments, the closed conformation is preferred and cathepsin B displays a carboxydipeptidase activity.2,3,11 Beyond the localization in the lysosomes and late endosomes, the endopeptidase activity of cathepsin B is thought to predominate at pH values of 6–7.4 and to be typical for the degradation of proteins of the ECM.2,10,11
The introduction of an electrophilic warhead in the position of the scissile peptide bond has been a successful strategy to design peptidomimetic inhibitors. Peptide nitriles form reversible thioimidate adducts resulting from the nucleophilic attack of the active-site cysteine. As a matter of fact, such peptide nitriles can only utilize the noncovalent interactions of the P4–P1 residues with the nonprimed subsites S4–S1. However, as it has been demonstrated with nitrile inhibitors for cathepsin B, a spacer can direct an acidic moiety to the S′ region to allow for an advantageous salt bridging with the histidine residues of the occluding loop.12 Peptide nitriles attracted much attention as synthetic inhibitors for cysteine cathepsins,13−15 in particular for the treatment of osteoporosis, where the cathepsin K inhibitor odanacatib (I, Figure S1 in the Supporting Information) is currently being investigated in clinical phase III.2,3 Further warheads for reversible inhibition of cysteine cathepsins are, for example, peptide aldehydes or ketones, while irreversible inhibition can be achieved with, for example, epoxide derivatives, vinyl sulfones or acyloxymethyl ketones.2,3,13,14,16,17 Other cathepsin B inhibitor types include nitroxoline derivatives,6,18 redox-reactive compounds,19 1,2,4-thiadiazoles,20 aziridinyl peptides,21 and cystatin-derived azapeptides.22
To enhance the selectivity of the broad spectrum cathepsin inhibitor E-64 (II, Figure S1),23 further epoxysuccinyl derivatives, for example, the highly potent and cathepsin B-selective CA-074 and CA-030 (III and IV, Figure S1) have been developed, binding exclusively to the S1′ and S2′ pockets and exploiting interactions with the positively charged histidine residues of the occluding loop.24−27 Noteworthy, such epoxide dipeptides with a free C-terminal proline exhibited stronger cathepsin B inactivation at lower pH values than under neutral conditions.25,28
Herein, we have used the dipeptide nitrile chemotype to map the nonprimed binding region of human cathepsin B. The outcome of these experiments was utilized for a fragment-based approach by combining the appropriate structure with a preselected linker moiety to address the occluding loop of cathepsin B with a terminal carboxylic group. The so designed inhibitors were assembled via click chemistry and investigated with respect to the target’s exo- and endopeptidase activity.
A library of 57 dipeptide nitriles bearing different amino acids in P2, mostly aminoacetonitrile in P1 and different capping groups in P3 position was evaluated at human cathepsin B by performing a photometric assay using pH 6.0 and Cbz-Arg-Arg-pNA as substrate. As for the inhibition of human cathepsins L, S, K, and F by these compounds,29−32 the linear progress curves revealed a “fast-binding” behavior. The obtained IC50 values were converted into the Ki values using the Cheng-Prusoff equation. The kinetic results are depicted in Figure S2 and Table S1 in the Supporting Information; the Ki values span from >500 μM to 0.33 μM.
From this set of data, first structure–activity relationships were drawn. Regarding the P2 position, several compounds with a Boc capping group at P3 and glycine nitrile at P1 position as common features were compared. Five of these inhibitors, 1–5, are listed in Table 1. Unnatural amino acid residues as 2-naphthylmethyl, meta-bromobenzyl or meta-phenylbenzyl caused a pronounced cathepsin B inhibition (1–3 versus 4 and 5). In comparison with the meta-connected derivatives 2 and 3, a remarkable decrease in potency was observed for the corresponding para-connected isomers 68 and 70 (see Table S1 for Ki values). Furthermore, in P3 position, a more voluminous residue than the Boc capping group turned out to be advantageous for increasing the inhibitory potency. This can be exemplarily seen by comparing 5 and 6 (Table 1) where the Cbz capping group caused a 3-fold lower Ki value. A similar effect was observed when replacing the Boc group in 4 by Cbz in Cbz-phenylalanylglycine nitrile (53, Table S1). However, compounds with a para-phenylbenzoyl group in P3 position possessed a stronger inhibitory capability toward cathepsin B (4 versus 7 and 6 versus 8, Table 1). Compound 99 (Table S1) which shares the structure of 8 but bears a methionine side chain in P1 position was even more potent than 8 and showed the strongest cathepsin B inhibition of all 57 screened compounds (Ki = 0.33 ± 0.03 μM). This result might indicate that the introduction of a side chain in P1 position does not diminish the activity, an issue that supports our design of compounds with a linker reaching into the primed binding region of cathepsin B.
Table 1. Inhibition of Human Cathepsin B by Dipeptide Nitriles 1–8.

Experiments were performed at pH 6.0 with Cbz-Arg-Arg-pNA. Values without standard errors refer to duplicate experiments with a single inhibitor concentration of 50 μM. IC50 values were calculated by using the equation IC50 = [I]/(v0/v – 1), where v and v0 are the rates in the presence and absence of the inhibitor, respectively, and [I] is the inhibitor concentration. Values with standard errors refer to duplicate measurements with five different inhibitor concentrations. IC50 values were determined by nonlinear regression using the equation v = v0/(1 + [I]/IC50). Standard error of the mean (SEM) values refer to this nonlinear regression. Ki values ± SEM were calculated from IC50 values by applying the equation Ki = IC50/(1 + [S]/Km), where [S] is the substrate concentration.
Value was taken from ref (32).
The screening results were used for the synthesis of three new inhibitors with beneficial structural features, that is, a combination of the para-phenylbenzoyl group at P3 with either a 2-naphthylmethyl, meta-bromobenzyl, or meta-phenylbenzyl moiety at the P2 position. The target compounds 9–11 were synthesized as depicted in Scheme S1 in the Supporting Information. Dipeptides 1 and 2 (for structures see Table 1) were deprotected with methanesulfonic acid and subsequently coupled with biphenylcarboxylic acid leading to 9 and 10. Compound 10 was further transformed in a Suzuki reaction with phenylboronic acid to generate compound 11 (for structures 9–11 and kinetic data with human cathepsins B, S, L, and K see Table 2). Derivatives 9 and 10 were found to be potent cathepsin B inhibitors with Ki values of less than 130 nM. By replacing the tert-butyloxy with the biphenyl-4-yl residue, a 27- or 43-fold increase in potency was achieved (1 and 2, Table 1 versus 9 and 10, Table 2). Compound 11 did not show the expected increase in potency toward cathepsin B (11, Table 2 versus 3, Table 1), however, it turned out to be a strong and selective inhibitor of cathepsin K. Compounds 9 and 10 were dual cathepsin B/S inhibitors with selectivity over cathepsins L and K.
Table 2. Inhibition of Human Cathepsins by Dipeptide Nitriles 9–19.


So far, we optimized the P2 and P3 substructures of the envisaged cathepsin B inhibitor. In the next step, the unique structural feature of cathepsin B, the occluding loop, should additionally be taken into consideration. The occluding loop when inserted into the active site cleft leads to a relatively blocked S′ region and dictates the carboxydipeptidase activity of the protease. The two histidine residues can interact with a carboxylate moiety of inhibitors as has been exemplified, for example, with CA-074, CA-030 and peptidomimetic compounds containing a linker-connected benzoic acid moiety.12,20,24−28 These results, together with the aforementioned observation on the potency of the P1 methionine inhibitor 99 (Table S1) justifies our next step in the course of the structural optimization. Model dipeptide nitriles were designed with a Cbz capping group, phenylalanine in P2 position and various linkers to address the carboxylate recognition site. For this purpose, a click-chemistry approach was followed using a CuII salt and sodium ascorbate as reducing agent.33 The preparation of the model compounds 12 and 13 is illustrated in Scheme S2 (for structures see Table 2). Boc-protected serine was O-alkylated to introduce an alkyne moiety. It was converted to the primary carboxamide, deprotected, coupled with Cbz-phenylalanine and transformed to the nitrile. Azidoacetic acid and 3-azidopropanoic acid were prepared from appropriate bromocarboxylic acids and used for the click reaction to form the final products 12 and 13.
Two further model compounds were designed in which the central part of the linker, OCH2–triazole, was shifted toward the carboxylate recognition site. Essentially the same approach as to 12 and 13 was followed starting from Boc-homoserine (Scheme S3, Supporting Information) to receive compounds 14 and 15 with an extended linker length (for structures see Table 2). With the four new compounds (12–15) in hand, the biochemical evaluation was carried out to ascertain the optimal linker structure for the interaction of the carboxylate with the occluding loop of cathepsin B (Table 2). Although 12–15 were poor inhibitors of human cathepsin B, compounds 13–15 showed a particular weak inhibition and thus the structure of 12 was considered for our further design. The high Ki values from 24 to 196 μM determined for cathepsin B inhibition by 12–15 were highly unexpected, the more so as the other cathepsins were inhibited to a reasonable extent. Presumably, the not optimized amino acid residues in P2 and P3 position prevented a pronounced cathepsin B inhibition by 12–15. Nonetheless, by maintaining the linker present in 12 and combining the results of our fragment-based approach, a potent cathepsin B inhibitor was finally obtained.
The synthesis of the optimized cathepsin B inhibitor 17 and the corresponding methyl ester 16 (for structures see Table 2) is shown in Scheme S4 in the Supporting Information. Furthermore, two analogs of 17 with the same P3 and P2 moieties were designed in which the length of the linker between the P1 α-carbon and the carboxylate was reduced. The corresponding final compounds 18 and 19 are listed in Table 2. Their synthesis was performed starting from the clickable amino acid Boc-propargylglycine (Scheme S5, Supporting Information).
The dipeptide nitrile 16 exhibited a general and unexpectedly strong inhibition profile toward the four investigated human cathepsins (Table 2). The extended neutral P1 residue of 16 was tolerated by the cathepsins and even provided additional affinity for cathepsins B, L and K, as can be seen from the data of inhibitor 10 and those of 16. Next, we compared the ester 16 with the free acid 17. The latter was 2-, 15-, and 5-fold less active at cathepsin S, L and K, respectively. Similar Ki values were determined for 16 and 17 at cathepsin B. Thus, we concluded that additional benefit was not gained by the free carboxylate function and that cathepsin B is mainly present in the open conformation under the assay conditions used. Unexpectedly, 18 and in particular 19, although optimized with respect to their P3 and P2 moieties, were only weak inhibitors when examined at pH 6.0.
It has been reported that a pH value of 6.0 and the use of “small” substrates such as Cbz-Arg-Arg-pNA (or Cbz-Arg-Arg-AMC) caused a preferred endopeptidase activity which is exerted by cathepsin B in the open conformation.6,8,10,18 However, as the occluding loop is considered to be a highly flexible segment, the closed conformation might be formed effortlessly and might be stabilized by inhibitors that can favorably interact, for example, with the histidine residues of the occluding loop.12,24−27 Moreover, the pNA (or AMC) substrate only occupies a small part of the primed substrate binding region and is therefore not expected to prevent the formation of the closed conformation. Accordingly, Cbz-Arg-Arg-AMC or Cbz-Phe-Arg-AMC were also mentioned not to be appropriate model substrates to exclusively study the endoproteolysis of cathepsin B.8,10 Mutations in cathepsin B which influence the formation of the closed conformation affected the cleavage of true endopeptidase substrates, for example, Abz-Ala-Phe-Arg-Ser-Ala-Ala-Gln-EDDnp, cleaved after arginine, but had little effect on the hydrolysis of Cbz-Phe-Arg-AMC.8 Moreover, peptidic inhibitors with a linker-connected benzoic acid moiety targeting the occluding loop have been reported to be highly active under assay conditions (pH 5.8, Cbz-Arg-Arg-AMC)12 similar to those that we have used (pH 6.0, Cbz-Arg-Arg-pNA).
In order to address the effect of pH on the inhibitory activity of our compounds 17–19, we repeated the measurements at pH 4.5. The kinetic data of cathepsin B inhibition by 17−19 and, for reasons of comparison, 10 and 16 are given in Table 2. When performing the assay at pH 4.5 and maintaining the “small” substrate Cbz-Arg-Arg-pNA, a weak reduction of potency was observed for 10, whereas the inhibition of 16 was attenuated to a stronger extent (26.0 nM at pH 6 versus 136 nM at pH 4.5). On the contrary, inhibitors 17−19 exhibited lower Ki values under acidic assay conditions (e.g., 17: 41.3 nM at pH 6 versus 27.3 nM at pH 4.5). These data support the interaction of the carboxylate moiety of 17–19 with the protonated histidine residues of the occluding loop. Next, we examined the inhibition profile of the five peptide nitriles when an exopeptidase substrate of cathepsin B was used. These measurements were performed with Abz-Gly-Ile-Val-Arg-Ala-Lys(Dnp)-OH at pH 4.5. The “quenched” fluorescent substrate is cleaved after arginine and provides the free terminal carboxylate moiety for salt bonds to the occluding loop. Whereas 10 and 16 exhibited a similar Ki value as before, the inhibition of cathepsin B by 17–19 was further enhanced (e.g., 17: Ki = 19.2 nM). Nevertheless, the unfavorable reduction of the linker length could not be overcome, since 18 (Ki = 132 nM) was 7-fold and 19 (Ki = 368 nM) was 19-fold less active than 17.
Our data indicate that the acids, for example 17, but not the corresponding ester 16, can gain affinity when the equilibrium of cathepsin B is shifted to the closed conformation, where the exopeptidase character predominates. The enhanced affinity is assumed to result from the favorable ionic contacts between the carboxylate of 17 and the occluding loop’s histidines. Accordingly, the ester 16, unable to exert such ionic contacts, lost affinity when shifting the assay conditions to pH 4.5.
The similar Ki values obtained at pH 4.5 for 10, 16 and 17 either with the “small” or the exopeptidase substrate led to the conclusion that the choice of the substrate did not strongly affect the equilibrium between the open and closed form of cathepsin B. Under acidic conditions, inhibitor 17 is expected to bind to this closed conformation as defined by the conformational selection model.34 However, a still pronounced inhibition by 17 (but not by 18 or 19) was obtained at pH 6 when the salt bridges that bind the loop to the enzyme body are thought to be disrupted and the equilibrium to be shifted to the open conformation. Under these conditions, an induced fit model might be applicable. Hence, inhibitor 17 binds to the open form of cathepsin B and causes a conformational readjustment.34 To reach an optimal complementarity of the enzyme’s binding sites and the ligand’s substructures, the occluding loop adopts the closed conformation, thus providing additional ionic contacts to the inhibitor.
It was a striking feature of our kinetic data, that the optimization of the inhibitor structure with respect to the P3 and P2 moieties made a major impact on activity. The interaction of these moieties with their corresponding subsites, S3 and S2, is thought to be the primary binding event and can occur irrespective of the open or closed conformation of the enzyme. However, an optimized P3–P2 structure could not compensate the inappropriate linker structure as present in 18 and particularly in 19. Of course, the crucial step to elongate the residence time of the inhibitor-enzyme complex is the formation of the covalent thioimidate through the attack of the active site cysteine at the nitrile warhead. Noteworthy, when using the “small” substrate Cbz-Arg-Arg-pNA in the assay, it can possibly bind to cathepsin B in both the closed and open conformation with similar affinity, as it could also be deduced from similar Km values.
In conclusion, we have employed a library of 57 dipeptide nitriles for a mapping of the active site of human cathepsin B. A para-phenylbenzoyl group in P3 and 3-bromophenylalanyl in the P2 position as favorable moieties were considered to be combined in a fragment-based approach with an optimized linker structure connecting the α-carbon of the P1 position with a free carboxylate group. This structure was successfully assembled through click chemistry, leading to the selective and potent covalent cathepsin B inhibitor 17. Despite the assumption that 17 addressed the recognition site of the occluding loop, potency differences at varying assay conditions and thus at a changed equilibrium between the open and closed conformations of the target were rather small. The complex interplay of the target, substrate, and inhibitor in the light of the conformational selection and induced fit models represents a subject for future investigations.
Glossary
Abbreviations
- Abz
2-aminobenzoyl
- AMC
7-amido-4-methylcoumarin
- Dnp
2,4-dinitrophenyl
- EDDnp
N-(2,4-dinitrophenyl)ethylenediamine
Biography
Michael Gütschow studied Biochemistry and received his Ph.D. in Pharmaceutical Chemistry from the University of Leipzig, Germany. After postdoctoral training at the Georgia Institute of Technology, School of Chemistry and Biochemistry, he returned to the University of Leipzig and was appointed as a professor for Pharmaceutical Chemistry at the University of Bonn in 2001. His research interests include (i) synthesis of bioactive heterocycles, (ii) peptides and peptidomimetic drugs, (iii) development of inhibitors and activity-based probes of proteases and esterases, and (iv) biochemistry of enzyme−drug interactions. He has published approximately 190 publications on these scientific topics.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00474.
Table of kinetic evaluation of dipeptide nitrile inhibitors at human cathepsin B; structures of cathepsin B inhibitors; progress curves and IC50 determination for cathepsin B inhibition by 17; syntheses of dipeptide nitriles 9–11 and 12–19; general methods and materials; synthetic procedures and characterization of all compounds; enzyme inhibition assays; 1H NMR and 13C NMR spectra. (PDF)
Author Contributions
The manuscript was written through contributions of all authors, who have given approval to the final version of the manuscript.
J.S. was supported by the Gender Equality Center of the Bonn-Rhein-Sieg University of Applied Sciences.
The authors declare no competing financial interest.
Supplementary Material
References
- Fonović M.; Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 2560–2570. 10.1016/j.bbagen.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Kos J.; Mitrović A.; Mirković B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med. Chem. 2014, 6, 1355–1371. 10.4155/fmc.14.73. [DOI] [PubMed] [Google Scholar]
- Löser R.; Pietzsch J. Cysteine cathepsins: their role in tumor progression and recent trends in the development of imaging probes. Front. Chem. 2015, 3, 37. 10.3389/fchem.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane B. F.; Yan S.; Podgorski I.; Linebaugh B. E.; Cher M. L.; Mai J.; Cavallo-Medved D.; Sameni M.; Dosescu J.; Moin K. Cathepsin B and tumor proteolysis: contribution of the tumor microenvironment. Semin. Cancer Biol. 2005, 15, 149–157. 10.1016/j.semcancer.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Premzl A.; Zavasnik-Bergant V.; Turk V.; Kos J. Intracellular and extracellular cathepsin B facilitate invasion of MCF-10A neoT cells through reconstituted extracellular matrix in vitro. Exp. Cell Res. 2003, 283, 206–214. 10.1016/S0014-4827(02)00055-1. [DOI] [PubMed] [Google Scholar]
- Sosič I.; Mirković B.; Arenz K.; Stefane B.; Kos J.; Gobec S. Development of new cathepsin B inhibitors: combining bioisosteric replacements and structure-based design to explore the structure-activity relationships of nitroxoline derivatives. J. Med. Chem. 2013, 56, 521–533. 10.1021/jm301544x. [DOI] [PubMed] [Google Scholar]
- Reiser J.; Adair B.; Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Invest. 2010, 120, 3421–3431. 10.1172/JCI42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägler D. K.; Storer A. C.; Portaro F. C.; Carmona E.; Juliano L.; Ménard R. Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry 1997, 36, 12608–12615. 10.1021/bi971264+. [DOI] [PubMed] [Google Scholar]
- Illy C.; Quraishi O.; Wang J.; Purisima E.; Vernet T.; Mort J. S. Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 1997, 272, 1197–1202. 10.1074/jbc.272.2.1197. [DOI] [PubMed] [Google Scholar]
- Schenker P.; Alfarano P.; Kolb P.; Caflisch A.; Baici A. A double-headed cathepsin B inhibitor devoid of warhead. Protein Sci. 2008, 17, 2145–2155. 10.1110/ps.037341.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil D.; Zucic D.; Turk D.; Engh R. A.; Mayr I.; Huber R.; Popovic T.; Turk V.; Towatari T.; Katunuma N.; Bode W. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan P. D.; Clark K. L.; Tommasi R. A.; Cowen S. D.; McQuire L. W.; Farley D. L.; van Duzer J. H.; Goldberg R. L.; Zhou H.; Du Z.; Fitt J. J.; Coppa D. E.; Fang Z.; Macchia W.; Zhu L.; Capparelli M. P.; Goldstein R.; Wigg A. M.; Doughty J. R.; Bohacek R. S.; Knap A. K. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J. Med. Chem. 2001, 44, 4524–4534. 10.1021/jm010206q. [DOI] [PubMed] [Google Scholar]
- Gobec S.; Frlan R. Inhibitors of cathepsin B. Curr. Med. Chem. 2006, 13, 2309–2327. 10.2174/092986706777935122. [DOI] [PubMed] [Google Scholar]
- Vicik R.; Busemann M.; Baumann K.; Schirmeister T. Inhibitors of cysteine proteases. Curr. Top. Med. Chem. 2006, 6, 331–353. 10.2174/156802606776287081. [DOI] [PubMed] [Google Scholar]
- Borišek J.; Vizovišek M.; Sosnowski P.; Turk B.; Turk D.; Mohar B.; Novič M. Development of N-(functionalized benzoyl)-homocycloleucyl-glycinonitriles as potent cathepsin K inhibitors. J. Med. Chem. 2015, 58, 6928–6937. 10.1021/acs.jmedchem.5b00746. [DOI] [PubMed] [Google Scholar]
- Santos M. M.; Moreira R. Michael acceptors as cysteine protease inhibitors. Mini-Rev. Med. Chem. 2007, 7, 1040–1050. 10.2174/138955707782110105. [DOI] [PubMed] [Google Scholar]
- Ettari R.; Pinto A.; Tamborini L.; Angelo I. C.; Grasso S.; Zappalà M.; Capodicasa N.; Yzeiraj L.; Gruber E.; Aminake M. N.; Pradel G.; Schirmeister T.; De Micheli C.; Conti P. Synthesis and biological evaluation of papain-family cathepsin L-like cysteine protease inhibitors containing a 1,4-benzodiazepine scaffold as antiprotozoal agents. ChemMedChem 2014, 9, 1817–1825. 10.1002/cmdc.201402079. [DOI] [PubMed] [Google Scholar]
- Mirković B.; Renko M.; Turk S.; Sosič I.; Jevnikar Z.; Obermajer N.; Turk D.; Gobec S.; Kos J. Novel mechanism of cathepsin B inhibition by antibiotic nitroxoline and related compounds. ChemMedChem 2011, 6, 1351–1356. 10.1002/cmdc.201100098. [DOI] [PubMed] [Google Scholar]
- Mirković B.; Sosič I.; Gobec S.; Kos J. Redox-based inactivation of cysteine cathepsins by compounds containing the 4-aminophenol moiety. PLoS One 2011, 6, e27197. 10.1371/journal.pone.0027197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Toung R.; Wodzinska J.; Li W.; Lowrie J.; Kukreja R.; Desilets D.; Karimian K.; Tam T. F. 1,2,4-thiadiazole: a novel Cathepsin B inhibitor. Bioorg. Med. Chem. 2003, 11, 5529–5537. 10.1016/j.bmc.2003.09.040. [DOI] [PubMed] [Google Scholar]
- Schirmeister T.; Peric M. Aziridinyl peptides as inhibitors of cysteine proteases: effect of a free carboxylic acid function on inhibition. Bioorg. Med. Chem. 2000, 8, 1281–1291. 10.1016/S0968-0896(00)00058-4. [DOI] [PubMed] [Google Scholar]
- Wieczerzak E.; Drabik P.; Łankiewicz L.; Ołdziej S.; Grzonka Z.; Abrahamson M.; Grubb A.; Brömme D. Azapeptides structurally based upon inhibitory sites of cystatins as potent and selective inhibitors of cysteine proteases. J. Med. Chem. 2002, 45, 4202–4211. 10.1021/jm020850k. [DOI] [PubMed] [Google Scholar]
- Barrett A. J.; Kembhavi A. A.; Brown M. A.; Kirschke H.; Knight C. G.; Tamai M.; Hanada K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982, 201, 189–198. 10.1042/bj2010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M.; Miyashita S.; Yokoo C.; Tamai M.; Hanada K.; Hatayama K.; Towatari T.; Nikawa T.; Katunuma N. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett. 1991, 280, 307–310. 10.1016/0014-5793(91)80318-W. [DOI] [PubMed] [Google Scholar]
- Schaschke N.; Assfalg-Machleidt I.; Machleidt W.; Turk D.; Moroder L. E-64 analogues as inhibitors of cathepsin B. On the role of the absolute configuration of the epoxysuccinyl group. Bioorg. Med. Chem. 1997, 5, 1789–1797. 10.1016/S0968-0896(97)00105-3. [DOI] [PubMed] [Google Scholar]
- Schaschke N.; Assfalg-Machleidt I.; Machleidt W.; Moroder L. Substrate/propeptide-derived endo-epoxysuccinyl peptides as highly potent and selective cathepsin B inhibitors. FEBS Lett. 1998, 421, 80–82. 10.1016/S0014-5793(97)01538-X. [DOI] [PubMed] [Google Scholar]
- Turk D.; Podobnik M.; Popovic T.; Katunuma N.; Bode W.; Huber R.; Turk V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. 10.1021/bi00014a037. [DOI] [PubMed] [Google Scholar]
- Cathers B. E.; Barrett C.; Palmer J. T.; Rydzewski R. M. pH Dependence of inhibitors targeting the occluding loop of cathepsin B. Bioorg. Chem. 2002, 30, 264–275. 10.1016/S0045-2068(02)00009-3. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Lülsdorff M.; Gütschow M. Facing the gem-dialkyl effect in enzyme inhibitor design: preparation of homocycloleucine-based azadipeptide nitriles. Chem. - Eur. J. 2011, 17, 11419–11423. 10.1002/chem.201101350. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Furtmann N.; Kläs J.; Gütschow M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. 10.1021/jm101272p. [DOI] [PubMed] [Google Scholar]
- Löser R.; Schilling K.; Dimmig E.; Gütschow M. Interaction of papain-like cysteine proteases with dipeptide-derived nitriles. J. Med. Chem. 2005, 48, 7688–7707. 10.1021/jm050686b. [DOI] [PubMed] [Google Scholar]
- Schmitz J.; Furtmann N.; Ponert M.; Frizler M.; Löser R.; Bartz U.; Bajorath J.; Gütschow M. Active site mapping of human cathepsin F with dipeptide nitrile inhibitors. ChemMedChem 2015, 10, 1365–1377. 10.1002/cmdc.201500151. [DOI] [PubMed] [Google Scholar]
- Pedersen D. S.; Abell A. 1,2,3-Triazoles in peptidomimetic chemistry. Eur. J. Org. Chem. 2011, 2011, 2399–2411. 10.1002/ejoc.201100157. [DOI] [Google Scholar]
- Copeland R. A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–501. 10.4155/fmc.11.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.