

Abstract

Cyclophilin D (CypD), a peptidylprolyl isomerase F (PPIase), plays a central role in opening the mitochondrial membrane permeability transition pore leading to cell death. CypD resides in the mitochondrial matrix, associates with the inner mitochondrial membrane, interacts with amyloid beta to exacerbate mitochondrial and neuronal stress and has been linked to Alzheimer’s disease (AD). We report the biological activity of a small-molecule CypD inhibitor (C-9), which binds strongly to CypD and attenuates mitochondrial and cellular perturbation insulted by Aβ and calcium stress. Binding affinities for C-9 were determined using in vitro surface plasmon resonance. This compound antagonized calcium-mediated mitochondrial swelling, abolished Aβ-induced mitochondrial dysfunction as shown by increased cytochrome c oxidase activity and adenosine-5′-triphosphate levels, and inhibited CypD PPIase enzymatic activity by real-time fluorescence capture assay using Hamamatsu FDSS 7000. Compound C-9 seems a good candidate for further investigation as an AD drug.
Keywords: CypD, small molecules, mitochondrial dysfunction, mPTP, amyloid β
Alzheimer’s disease (AD) is a chronic neurodegenerative disease that primarily affects the elderly and for which there are currently only symptomatic treatments. Early pathological features of the AD brain include mitochondrial and synaptic dysfunction, which is considered to be related to progressive amyloid-beta (Aβ) oligomer accumulation in synaptic mitochondria.1,2 This affects membrane potential, membrane permeability transition pore (mPTP) formation, respiration, energy metabolism, oxidative stress, mitochondrial dynamics, and calcium homeostasis.3−11 Since mitochondrial dysfunction is believed to play a role in Aβ-induced toxicity, a compound that restores this dysfunction might have potential therapeutic benefit.12,13 Thus, Aβ oligomer inhibitors or procedures for blocking Aβ oligomer production appear to offer promising approaches for prevention and treatment of AD. One approach being taken is to reduce Aβ oligomer accumulation by blocking the clipping action of secretases with inhibitors.14,15 Another approach is to develop “passive vaccines” that will remove Aβ oligomer directly. Both gamma-secretase inhibitors and beta-secretase inhibitors have been in advanced clinical studies.16,17 Concerns about side effects have also prevented any drugs from entering clinical trials. Given that AD is a multifaceted disease with a poorly understood molecular biology, multitargeted approaches will probably be the most effective method for the treatment.
Cyclophilin D (CypD), a peptidyl prolyl isomerase F, resides in the mitochondrial matrix, associates with the inner mitochondrial membrane, and plays a central role in opening the mitochondrial mPTP leading to cell death.18,19 Neurons in AD-affected regions of the brain have significantly elevated levels of CypD. Our earlier studies have shown that a Aβ-CypD complex is present in the cortical mitochondria of AD brain and Tg mAPP mice. Our surface plasmon resonance (SPR) studies have also shown that recombinant CypD protein binds to Aβ.4
Although the precise role of Aβ in mitochondria is not yet defined, absence of the Aβ binding partner, CypD, has been shown to protect against Aβ-mediated mitochondrial and synaptic dysfunction.4,20−22 These reports indicate that mitochondrial and neuronal stress is increased in transgenic AD mouse models through the interaction of mitochondrial Aβ with the mitochondrial protein CypD, and this points to the utility of CypD as a potential drug target for AD treatment.19 Blocking CypD protects against Aβ- and oxidative stress-induced mitochondrial and synaptic degeneration and also improves mitochondrial and cognitive function. Cyclosporine A (CsA), currently the most specific inhibitor of the mPTP, acts by blocking the peptidyl-prolyl cis–trans isomerase (PPIase) activity of CypD;18,23,24 fortunately, CsA lacks clinical significance in any disorder associated with the central nervous system because it cannot cross the blood–brain barrier (BBB). Several CsA derivatives that lack immunosuppressive effects have been developed, including N-Me-Ala-6-cyclosporin A and N-Me-Val-4-cyclosporin. They are potent inhibitors of CypD PPIase activity and thereby antagonize mPTP opening and apoptosis induction.25,26 Two other potential drugs, Sanglifehrin A and Antamanide, were recently developed for inhibition of the mPTP, but they have severe side effects including nephrotoxicity, neurotoxicity, and hepatotoxicity as well as poor permeability through the BBB.27,28 Small-molecule quinoxaline derivatives, which inhibit mPTP opening,29 have been synthesized by Guo et al.,29 but their effects on the mPTP need to be re-evaluated. Thus, the current CypD inhibitors all have one or more disadvantages such as low solubility, poor ability to cross the BBB, high toxicity, and/or low cell permeability.
In the present study, we first evaluated the CypD inhibitor activity of a novel 4-aminobenzenesulfonamide derivative (C-9) using in vitro SPR to determine whether this synthetic compound binds to human CypD protein. We next examined the biological activity of inhibitor on Aβ-induced mitochondrial dysfunction. Based on the SPR binding studies and biological activity, inhibitor C-9 was selected as a possible drug candidate for AD prevention and treatment.
The synthesized CypD inhibitor C-9(19) (structure shown in Figure 1) exhibited high binding affinity to human CypD along with the improvement of mitochondrial function. In vitro SPR was used to verify the ability of inhibitor C-9 to bind to human recombinant CypD protein. This presumably correlates with its potential CypD inhibitory activity. Figure 2 demonstrates that the synthetic CypD inhibitor C-9 displays a high binding affinity for human CypD. The equilibrium dissociation constant (KD) was determined using a 1:1 Langmuir binding fit model and the association (kon) and dissociation (koff) rate constants were determined using equations 1 and 2.
| 1 |
where R represents the response unit, C is the concentration of the analyte, and
| 2 |
Figure 1.

Structure of the synthesized small molecule CypD inhibitor C-9.
Figure 2.
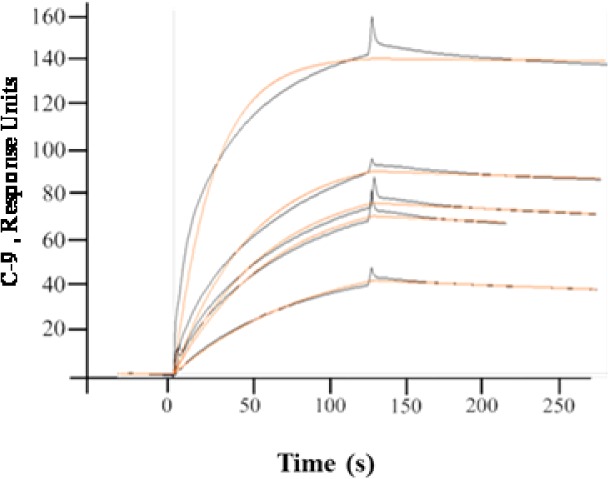
Synthesized CypD inhibitor C-9 binds to immobilized CypD in a dose-dependent manner. Surface plasmon resonance (SPR) analysis of CypD–sulfonamide derivative binding interaction, where the globally fit data (black lines) were overlaid with experimental data (red lines). Human recombinant CypD protein (10 μg/mL) was immobilized directly on the hydrophilic carboxy methylated dextran matrix of the CM5 sensor chip (Biacore-3000) using the standard primary amine coupling reaction according to standard procedures. Compound C-9 (2.5, 5, 10, 15, and 20 μM) was injected at a flow rate of 40 μL/min for the determination of the kinetic and equilibrium constants. All data analyses were carried out using BIA evaluation software, and sensor grams were processed by automatic correction for nonspecific bulk refractive index effects. The dissociation constant (KD) was determined as indicated at 25 °C.
The obtained results were evaluated by χ2 analysis, and all kinetic parameters are listed in Table 1. The SPR results shown in Figure 2 clearly indicate that compound C-9 binds quite strongly to CypD with a KD value of 149 nM (Table 1). This was also verified by the CypD peptidyl prolyl isomerase enzymatic activity.
Table 1. CypD PPIase Enzymatic Activity and SPR Results.
| inhibitor | IC50 (μM) | Ki (μM) | KD (μM) |
|---|---|---|---|
| compound C-9 | 1.49 ± 0.20 | 1.33 ± 0.13 | 0.149 |
| CsA | 0.00360 | 0.00258 |
The ability of inhibitor C-9 to inhibit CypD PPIase enzymatic activity was determined by using a newly developed real time fluorescence method that is described in the SI. The results are presented in Figures 3 and 4 and Table 1. The IC50 value for C-9 is 1.49 ± 0.20 μM. Since inhibition of CypD PPIase activity was essential for potentiation of Aβ cytotoxicity, Aβ-induced cytotoxicity can presumably be decreased if the Aβ-CypD interaction is blocked with small-molecule inhibitors like C-9.
Figure 3.
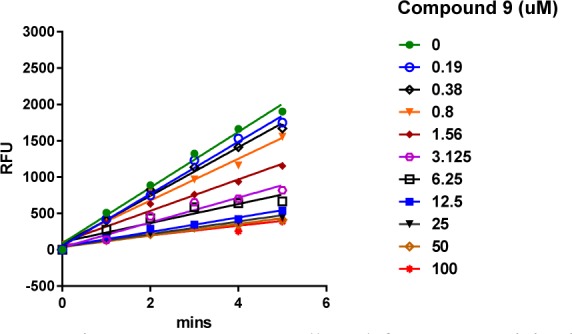
Reaction progress curves collected for CypD activity in DMSO alone or at various compound C-9 concentrations ranging from 0.19 to 100 μM. Linearity was maintained up to 5 min of incubation at 25 °C.
Figure 4.

IC50 of compound C-9 and CsA inhibiting PPIase activity of CypD with real-time kinetics assay and calculated using nonlinear regression analysis in Graphpad Prism 5.0.
Mitochondrial Swelling in Response to Calcium
It is known that CypD-deficient cortical mitochondria are resistant to Aβ- and calcium (Ca2+)-induced mitochondrial swelling and mitochondrial permeability transition. These deficient mitochondria also exhibit increased calcium buffering capacity and generate fewer mitochondrial reactive oxygen species. In order to determine the relationship between Ca2+ and mitochondrial swelling, cortical mitochondria were isolated from mice and subjected to a swelling assay as previously described.4 Mitochondria treated with Ca2+ experienced excessive swelling. However, if compound C-9 was added to the mitochondria, swelling was diminished in a dose-dependent manner (Figure 5). The fact that brain mitochondrial swelling was unaffected by the inhibitors alone implies a direct relationship between Ca2+ and the inhibitor (Figure 5). The addition of CsA as a positive control to the reaction mixture significantly blocked Ca2+-induced mitochondrial swelling.
Figure 5.
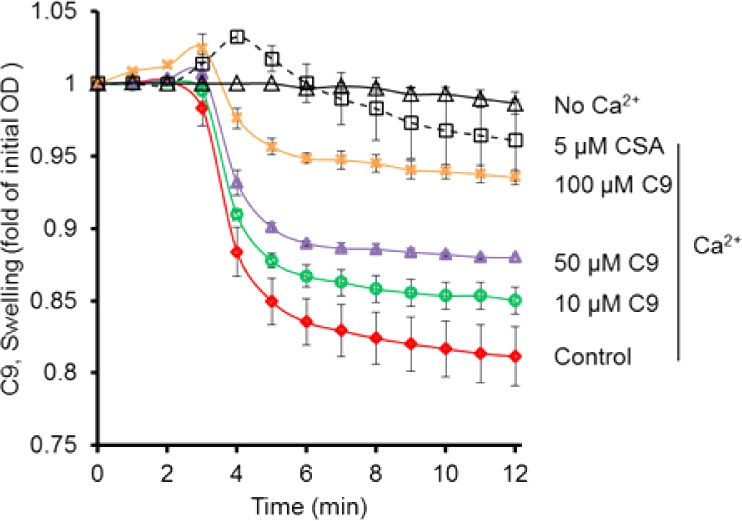
Effect of CypD inhibitor C-9 on calcium-induced mitochondrial swelling (red line) compared to vehicle-treated mitochondria (black line). The isolated cortical mitochondria (100 μg) were incubated with and without CypD inhibitor C-9 on ice for 5 min. Calcium (200 μM) was added to the reaction buffer to trigger mitochondrial swelling. The addition of the indicated concentrations of compound C-9 (0 to 100 μM) or CsA attenuated calcium-induced mitochondrial swelling.
Cytochrome c Oxidase (CcO)
The effect of inhibitor C-9 on Aβ-induced mitochondrial respiratory function was assessed by determining cytochrome c oxidase (CcO) activity. CcO is a vital enzyme associated with the mitochondrial electron transport chain. SK-N-SH cells were treated with 5 μM oligomer Aβ1–42 in the presence of 10 and 20 μM C-9 and 1 μM CsA for 48 h, after which samples were tested for CcO enzymatic activity levels. Aβ1–42 treatment significantly decreased CcO activity, whereas the addition of CypD inhibitor C-9 rescued CcO activity in Aβ-treated cells as compared with vehicle-treated cells (Figure 6). Treating cells with inhibitor alone had no effect on CcO activity in the absence of Aβ1–42, thereby demonstrating that the CypD inhibitor does not disrupt normal cell function. These results provide evidence that inhibition of CypD rescues CcO activity in Aβ-affected cells without harming healthy cells, suggesting that they are desirable candidates for continued study in AD research and treatment.
Figure 6.

Effect of CypD inhibitor C-9 and CsA on Aβ-induced reduction in CcO activity. SK-N-SH cells were treated with 5 μM oligomer Aβ plus: vehicle without inhibitor, C-9 at the indicated concentrations (10 and 20 μM), CsA (1 μM), and Aβ alone (5 μM), respectively. After 48 h, CcO activity was determined in cell lysates. *P < 0.01: Aβ vs 10 μM C-9 + Aβ, 20 μM C-9 + Aβ, and CsA + Aβ, respectively.
Levels of Adenosine-5′-Triphosphate (ATP)
To determine the effect of the CypD inhibitor C-9 on Aβ-induced energy metabolism impairment, we analyzed cellular ATP levels. SK-N-SH cells were exposed to 5 μM of Aβ1–42 in the presence of 10 and 20 μM compound C-9 and 1 μM CsA, respectively. After 48 h, we measured the ATP concentrations in the cell lysates. As shown in Figure 7, Aβ1–42 treatment significantly decreased ATP levels, whereas the addition of CypD inhibitor C-9 restored ATP levels in a dose-dependent manner. ATP levels in the cells exposed to CypD inhibitor C-9 were comparable to those of vehicle-treated cells without Aβ1–42, suggesting no toxic effect of the inhibitors on mitochondrial energy metabolism and ATP production. These data suggest that the synthesized CypD inhibitor C-9 prevents mitochondrial dysfunction induced by Aβ (Figure 7).
Figure 7.

Effect of CypD inhibitor C-9 and CsA on Aβ-induced decline of ATP levels. SK-N-SH cells were treated with 5 μM oligomer Aβ plus: vehicle without inhibitor, C-9 at the indicated concentrations (10 and 20 μM), CsA (1 μM), and Aβ alone (5 μM), respectively. After 48 h, ATP levels were determined in cell lysates. *P < 0.01: Aβ vs 10 μM C-9 + Aβ, 20 μM C-9 + Aβ, and CsA + Aβ, respectively.
Cell Viability
Having observed excellent reversal effects of the CypD inhibitor C-9 on Aβ-affected processes, including diminished mitochondrial swelling and enhanced CcO activity and ATP production, we next sought to assess the overall cell health of SK-N-SH cells treated with compound C-9. A wide dosing range was utilized to examine the effect of the CypD inhibitor on cell viability using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay (0, 5, 10, 25, 50, and 100 μM of inhibitor diluted in cell culture medium). As shown in Figure 8, cells treated with C-9 inhibitor exhibited similar reduction of MTT to the cells without C-9 inhibitor treatment. Further, the increasing dosage compound C-9 had no significant effect on SK-N-SH cell viability, indicating that inhibitor C-9 produces no severe toxic effects at either low or high doses in healthy cells.
Figure 8.

Effect of CypD inhibitor C-9 on cell survival and toxicity. SK-N-SH cells were incubated with vehicle (0) and CypD inhibitor C-9 at 37 °C for 48 h, before being subjected to an MTT reduction assay for examination of cell survival and toxicity. Various concentrations of the CypD inhibitor were used to assess cell toxicity (0, 5, 10, 25, 50, and 100 μM).
We next examined the effect of the CypD inhibitor on Aβ-induced neuronal cell death, commonly observed in AD. As demonstrated in Figure 9, the treatment of SK-N-SH cells with Aβ1–42 alone resulted in a significant decrease in cell viability as assessed by an MTT reduction assay. The addition of various dosages of inhibitor C-9 to the cells in the presence of Aβ1–42 increased cell viability. Compound C-9 exhibited a significant effect on cell health at both 10 and 20 μM concentrations (Figure 9). These results, in addition to the rescued mitochondrial function data, indicate that this CypD inhibitor can reduce the harmful effects of Aβ on neuronal cell health without producing toxic side effects in unaffected cells. Thus, these results support the notion that inhibition of CypD with specific inhibitors can have potentially beneficial effects on neuronal and mitochondrial function.
Figure 9.

Effect of CypD inhibitor C-9 on Aβ-induced cell death. SK-N-SH cells were treated with 5 μM oligomer Aβ plus: vehicle without inhibitor, C-9 at the indicated concentrations (10 and 20 μM), CsA (1 μM), and Aβ alone (5 μM), respectively, at 37 °C for 48 h. Following the incubation, cells were subjected to an MTT reduction assay for examination of cell survival. #P < 0.05: Aβ vs 10 μM C-9 + Aβ, respectively. *P < 0.01: Aβ vs 20 μM C-9 + Aβ and CsA + Aβ, respectively.
As a final check, we also performed extended Lipinski rule of five computational studies30 (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp) to test for drug-like properties of compound C-9 and CsA. The results shown in Table 2 demonstrate that, while compound C-9 satisfies all five of the Lipinski rules, CsA satisfies just one (LogP = 3.2). To summarize these results for C-9: its molecular weight is less than 500 Da, there are less than five hydrogen-bond donor atoms and less than 10 hydrogen-bond acceptors, LogP is less than 5 and the molar refractivity is in the range of 40–130. Compound C-9 should therefore have excellent drug-like properties. However, the results for CsA show it only satisfies one of the Lipinski rules, and therefore, CsA should be a much less desirable drug. Furthermore, CsA has a molecular weight of 1201 Da, five hydrogen-bond donor atoms and 23 hydrogen-bond acceptors, LogP is 3.2, and its molar refractivity is 328.44. In vivo BBB penetration is also a crucial pharmacokinetic property because central nervous system (CNS) active compounds must pass across it, and CNS-inactive compounds ideally should not pass across it in order to avoid detrimental CNS side effects. Compound C-9 was predicted to have moderate to low absorption into the CNS. The (C.brain/C.blood) value of 0.0752 was calculated using the preADMET online server (http://preadmet.bmdrc.org/).31,32 Even though this value might indicate a lower likelihood of crossing the BBB, this should be checked experimentally. If C-9 does not cross the BBB, it could still serve as a useful starting point for developing additional active CypD inhibitors. All of this implies that the drug-like properties of C-9 may be superior to those of CsA.
Table 2. Lipinski Rule of Five Computational Studies for C-9 and Cyclosporin A.
| Lipinski properties | C-9 | cyclosporin A |
|---|---|---|
| mass (Daltons) | 470.574 | 1201 |
| hydrogen bond donor | 3 | 5 |
| hydrogen bond acceptors | 8 | 23 |
| LogP | 2.9 | 3.2 |
| molar refractivity | 122.88 | 328.44 |
In summary, we previously identified 20 pyrimidine and sulfonamide small molecules that serve as CypD inhibitors, and we have now further characterized one of these, which has the best ligand binding affinities as shown by in vitro SPR binding studies. This CypD inhibitor, compound C-9, rescued mitochondrial function by reducing CypD peptidyl prolyl isomerase enzymatic activity and antagonizing calcium-mediated-induced mitochondrial swelling. Importantly, compound C-9 also reversed Aβ-induced mitochondrial dysfunction as shown by increased CcO and ATP levels, suggesting protective effects on mitochondrial function for this molecule. Furthermore, compound C-9 significantly protected from Aβ-induced neuronal cell death without deleterious effects on normal neuronal and mitochondiral function. These results suggest that small molecule CypD inhibitors like compound C-9 may have considerable potential as drugs for the prevention and treatment of neurodegenerative disease including AD if they can cross the BBB and have beneficial pharmacokinetic and metabolic properties with no off-target liabilities.
We performed statistical analysis of the data using one-way analysis of variance (ANOVA) in the Statview statistics software (SAS Institute, Version 5.0.1) with Fisher post hoc testing. P < 0.05 was considered significant. All data are expressed as the mean ± SE.
Glossary
ABBREVIATIONS
- AD
Alzheimer’s disease
- CypD
cyclophilin D
- PPIase
peptidylprolyl isomerase F
- Aβ
amyloid-beta
- mPTP
membrane permeability transition pore
- SPR
surface plasmon resonance
- CcO
cytochrome c oxidase
- ATP
levels of adenosine-5′-triphosphate
- sA
cyclosporine A
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- (BBB)
blood–brain barrier
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00451.
Detailed experimental methods (PDF)
Author Contributions
∥ These authors contributed equally to this work.
This study was supported by grant awards from the National Institute of Health (R37AG037319 R01GM095355, R01AG044793, and R01NS065482). Research in this publication was also supported by a donation to The University of Kansas Cancer Center from the Hall Family Foundation and by the Lead Development and Optimization Shared Resource supported by the National Cancer Institute Cancer Center Support Grant P30 CA168524.
The authors declare no competing financial interest.
Supplementary Material
References
- Lin M. T.; Beal M. F. Alzheimer’s APP Mangles Mitochondria. Nat. Med. 2006, 12, 1241–1243. 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- Du H.; Guo L.; Yan S. Q.; Sosunov A. A.; McKhann G. M.; Yan S. S. Early Deficits in Synaptic Mitochondria in an Alzheimer’s Disease Mouse Model. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18670–18675. 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C.; Wang N.; Yao J.; Sosunov A.; Chen X.; Lustbader J. W.; Xu H. W.; Stern D.; McKhann G.; Yan S. D. Mitochondrial Abeta: A Potential Focal Point for Neuronal Metabolic Dysfunction in Alzheimer’s Disease. FASEB J. 2005, 19, 2040–2041. 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Du H.; Guo L.; Fang F.; Chen D.; Sosunov A. A.; McKhann G. M.; Yan Y.; Wang C.; Zhang H.; Molkentin J. D.; Gunn-Moore F. J.; Vonsattel J. P.; Arancio O.; Chen J. X.; Yan S. D. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation And Ameliorates Learning And Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K.; Fang F.; Zhang W. S.; Yan S. Q.; Fukuzaki E.; Du H.; Sosunov A.; McKhann G.; Funatsu Y.; Nakamichi N.; Nagai T.; Mizoguchi H.; Ibi D.; Hori O.; Ogawa S.; Stern D. M.; Yamada K.; Yan S. S. RAGE-Mediated Signaling Contributes to Intraneuronal Transport of Amyloid-Beta and Neuronal Dysfunction. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20021–20026. 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V. K.; Carlson E. A.; Yan S. S. Mitochondrial Permeability Transition Pore is A Potential Drug Target for Neurodegeneration. Biochim. Biophys. Acta, Mol. Basis Dis. 2014, 1842, 1267–1272. 10.1016/j.bbadis.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X. Q.; Huang S. B.; Wu L.; Wang Y. F.; Hu G.; Li G. Y.; Zhang H. J.; Yu H. Y.; Swerdlow R. H.; Chen J. X.; Yan S. S. Inhibition of ERK-DLP1 Signaling and Mitochondrial Division Alleviates Mitochondrial Dysfunction in Alzheimer’s Disease Cybrid Cell. Biochim. Biophys. Acta, Mol. Basis Dis. 2014, 1842, 220–231. 10.1016/j.bbadis.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H.; Guo L.; Yan S. S. Synaptic Mitochondrial Pathology in Alzheimer’s Disease. Antioxid. Redox Signaling 2012, 16, 1467–1475. 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X. Q.; Wu L.; Huang S. B.; Zhong C. J.; Shi H. L.; Li G. Y.; Yu H. Y.; Swerdlow R. H.; Chen J. X.; Yan S. S. Oxidative Stress-Mediated Activation of Extracellular Signal-Regulated Kinase Contributes to Mild Cognitive Impairitent-Related Mitochondrial Dysfunction. Free Radical Biol. Med. 2014, 75, 230–240. 10.1016/j.freeradbiomed.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H.; Guo L.; Wu X.; Sosunov A. A.; McKhann G. M.; Chen J. X.; Yan S. S. Cyclophilin D Deficiency Rescues Abeta-Impaired PKA/CREB Signaling and Alleviates Synaptic Degeneration. Biochim. Biophys. Acta, Mol. Basis Dis. 2014, 1842, 2517–2527. 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.; Du H.; Yan S.; Wu X.; McKhann G. M.; Chen J. X.; Yan S. S. Cyclophilin D Deficiency Rescues Axonal Mitochondrial Transport in Alzheimer’s Neurons. PLoS One 2013, 8, e54914. 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva D. F.; Esteves A. R.; Arduino D. M.; Oliveira C. R.; Cardoso S. M. Amyloid-beta-induced Mitochondrial Dysfunction Impairs the Autophagic Lysosomal Pathway in a Tubulin Dependent Pathway. J. Alzheimers Dis. 2011, 26, 565–581. [DOI] [PubMed] [Google Scholar]
- Chen J. X.; Yan S. D. Amyloid-beta-Induced Mitochondrial Dysfunction. J. Alzheimers Dis. 2007, 12, 177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A.; Morizane A.; Nakajima Y.; Miyamoto S.; Takahashi J. gamma-Secretase Inhibitors Prevent Overgrowth of Transplanted Neural Progenitors Derived from Human-Induced Pluripotent Stem Cells. Stem Cells Dev. 2013, 22, 374–382. 10.1089/scd.2012.0198. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Rao K. V.; Yadav N. D.; Anderson D. D.; Gavande N.; Huang X. P.; Terzyan S.; Tang J. Structure-Based Design of Highly Selective beta-Secretase Inhibitors: Synthesis, Biological Evaluation, and Protein-Ligand X-ray Crystal Structure. J. Med. Chem. 2012, 55, 9195–9207. 10.1021/jm3008823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert J. S. Progress in the Development Of Beta-Secretase Inhibitors for Alzheimer’s Disease. Prog. Med. Chem. 2009, 48, 133–161. 10.1016/S0079-6468(09)04804-8. [DOI] [PubMed] [Google Scholar]
- Panza F.; Frisardi V.; Solfrizzi V.; Imbimbo B. P.; Logroscino G.; Santamato A.; Greco A.; Seripa D.; Pilotto A. Interacting with Gamma-Secretase for Treating Alzheimer’s Disease: from Inhibition to Modulation. Curr. Med. Chem. 2011, 18, 5430–5447. 10.2174/092986711798194351. [DOI] [PubMed] [Google Scholar]
- Galat A.; Metcalfe S. M. Peptidylproline cis/trans isomerases. Prog. Biophys. Mol. Biol. 1995, 63, 67–118. 10.1016/0079-6107(94)00009-X. [DOI] [PubMed] [Google Scholar]
- Valasani K. R.; Vangavaragu J. R.; Day V. W.; Yan S. S. Structure Based Design, Synthesis, Pharmacophore Modeling, Virtual Screening, and Molecular Docking Studies for Identification of Novel Cyclophilin D Inhibitors. J. Chem. Inf. Model. 2014, 54, 902–912. 10.1021/ci5000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H.; Guo L.; Zhang W.; Rydzewska M.; Yan S. Cyclophilin D Deficiency Improves Mitochondrial Function and Learning/Memory in Aging Alzheimer Disease Mouse Model. Neurobiol. Aging 2011, 32, 398–406. 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader J. W.; Cirilli M.; Lin C.; Xu H. W.; Takuma K.; Wang N.; Caspersen C.; Chen X.; Pollak S.; Chaney M.; Trinchese F.; Liu S.; Gunn-Moore F.; Lue L. F.; Walker D. G.; Kuppusamy P.; Zewier Z. L.; Arancio O.; Stern D.; Yan S. S.; Wu H. ABAD Directly Links Abeta to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Takuma K.; Yao J.; Huang J.; Xu H.; Chen X.; Luddy J.; Trillat A. C.; Stern D. M.; Arancio O.; Yan S. S. ABAD Enhances Abeta-Induced Cell Stress Via Mitochondrial Dysfunction. FASEB J. 2005, 19, 597–608. [DOI] [PubMed] [Google Scholar]
- Tanveer A.; Virji S.; Andreeva L.; Totty N. F.; Hsuan J. J.; Ward J. M.; Crompton M. Involvement of Cyclophilin D in the Activation of a Mitochondrial Pore by Ca2+ and Oxidant Stress. Eur. J. Biochem. 1996, 238, 166–172. 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- Griffiths E. J.; Halestrap A. P. Further Evidence that Cyclosporin A Protects Mitochondria from Calcium Overload by Inhibiting a Matrix Peptidyl-prolyl cis-trans isomerase. Implications for the Immunosuppressive and Toxic Effects of Cyclosporin. Biochem. J. 1991, 274, 611–614. 10.1042/bj2740611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaspekov L.; Friberg H.; Halestrap A.; Viktorov I.; Wieloch T. Cyclosporin A and its Nonimmunosuppressive Analogue N-Me-Val-4-cyclosporin A Mitigate Glucose/Oxygen Deprivation-Induced Damage to Rat Cultured Hippocampal Neurons. Eur. J. Neurosci 1999, 11, 3194–3198. 10.1046/j.1460-9568.1999.00743.x. [DOI] [PubMed] [Google Scholar]
- Nicolli A.; Basso E.; Petronilli V.; Wenger R. M.; Bernardi P. Interactions of Cyclophilin with the Mitochondrial Inner Membrane and Regulation of the Permeability Transition Pore, and Cyclosporin A-Sensitive Channel. J. Biol. Chem. 1996, 271, 2185–2192. 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- Clarke S. J.; McStay G. P.; Halestrap A. P. Sanglifehrin A Acts as a Potent Inhibitor of the Mitochondrial Permeability Transition and Reperfusion Injury of the Heart by Binding to Cyclophilin-D at a Different site from Cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- Azzolin L.; Antolini N.; Calderan A.; Ruzza P.; Sciacovelli M.; Marin O.; Mammi S.; Bernardi P.; Rasola A. Antamanide, a Derivative of Amanita Phalloides, is a Novel Inhibitor of the Mitochondrial Permeability Transition Pore. PLoS One 2011, 6, e16280. 10.1371/journal.pone.0016280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H. X.; Wang F.; Yu K. Q.; Chen J.; Bai D. L.; Chen K. X.; Shen X.; Jiang H. L. Novel Cyclophilin D Inhibitors Derived from Quinoxaline Exhibit Highly Inhibitory Activity Against Rat Mitochondrial Swelling and Ca2+ Uptake/ Release. Acta Pharmacol. Sin. 2005, 26, 1201–1211. 10.1111/j.1745-7254.2005.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- Irvine J. D.; Takahashi L.; Lockhart K.; Cheong J.; Tolan J. W.; Selick H. E. MDCK (Madin-Darby Canine Kidney) Cells: a Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- Valasani K. R.; Hu G.; Chaney M. O.; Yan S. S. Structure-Based Design and Synthesis of Benzothiazole Phosphonate Analogues with Inhibitors of Human ABAD-Aβ for Treatment of Alzheimer’s Disease. Chem. Biol. Drug Des. 2013, 81, 238–249. 10.1111/cbdd.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.