

Abstract
Very long chain acyl-coA dehydrogenase deficiency (VLCADD) is an autosomal recessive inborn error of fatty acid oxidation detected by newborn screening (NBS). Follow-up molecular analyses are often required to clarify VLCADD-suggestive NBS results, but to date the outcome of these studies are not well described for the general screen-positive population. In the following study, we report the molecular findings for 693 unrelated patients that sequentially received Sanger sequence analysis of ACADVL as a result of a positive NBS for VLCADD. Highlighting the variable molecular underpinnings of this disorder, we identified 94 different pathogenic ACADVL variants (40 novel), as well as 134 variants of unknown clinical significance (VUSs). Evidence for the pathogenicity of a subset of recurrent VUSs was provided using multiple in silico analyses. Surprisingly, the most frequent finding in our cohort was carrier status, 57% all individuals had a single pathogenic variant or VUS. This result was further supported by follow-up array and/or acylcarnitine analysis that failed to provide evidence of a second pathogenic allele. Notably, exon-targeted array analysis of 131 individuals screen positive for VLCADD failed to identify copy number changes in ACADVL thus suggesting this test has a low yield in the setting of NBS follow-up. While no genotype was common, the c.848T>C (p.V283A) pathogenic variant was clearly the most frequent; at least one copy was found in ∼10% of all individuals with a positive NBS. Clinical and biochemical data for seven unrelated patients homozygous for the p.V283A allele suggests that it results in a mild phenotype that responds well to standard treatment, but hypoglycemia can occur. Collectively, our data illustrate the molecular heterogeneity of VLCADD and provide novel insight into the outcomes of NBS for this disorder.
Keywords: VLCAD, inborn error of metabolism, fatty acid oxidation, newborn screening, ACADVL, VLCADD, NBS, V283A, V243A
1. Introduction
ACADVL encodes for very long chain acyl CoA dehydrogenase (VLCAD), a mitochondrial enzyme that catalyzes the initial rate-limiting step of β-oxidation of long chain fatty acids [1, 2]. Patients with autosomal recessive VLCAD deficiency (VLCADD; OMIM #201475) accumulate high plasma levels of long chain acylcarnitine conjugates, especially the tetradecenoyl (C14:1) acylcarnitine, and can exhibit a wide range of clinical outcomes including (i) a severe neonatal onset disease associated with cardiomyopathy and a high mortality rate, (ii) an infantile onset form usually presenting with non-ketotic hypoglycemia and hepatic dysfunction, and (iii) an adult onset myopathic form characterized by exercise induced muscle weakness/pain and rhadomyolysis [3-5]. ACADVL null alleles are associated with a severe early onset phenotype whereas missense or in frame deletion alleles are often, but not always, associated with a milder later onset form of VLCADD [6].
VLCADD is detected by newborn screening (NBS) laboratories in the United States on the basis of blood spot acylcarnitine levels (typically C14:1 acylcarnitine or a ratio involving this compound)[7, 8]. Large multicenter US NBS studies predicated on standard tandem mass spectrometry based approaches to acylcarnitine analysis have reported positive predictive values for VLCADD of 20-30% [8, 9]. Positive NBS results can occur for many reasons including (i) truly affected early onset forms of the disorder requiring immediate clinical attention, (ii) later onset forms of the disorder that may not manifest symptoms until adulthood, (iii) unaffected carrier status [9, 10], (iv) maternal effect [11], (v) false positives, possibly resulting from fasting, diet, or other factors not related to VLCADD. There have also been multiple reports of confirmed affected individuals that screen positive for VLCADD but then appear asymptomatic by follow-up quantitative plasma acylcarnitine analysis [10, 12-14].
Given this complexity, diagnostic decisions can be challenging and advisory panels have advocated for additional testing to clarify positive NBS results such as enzymatic studies and/or molecular analysis of ACADVL (https://www.acmg.net/StaticContent/ACT/C14.pdf; [15]). Numerous groups have published case reports describing follow-up testing results from clinically interesting screen positive individuals, but there are limited data describing the specific molecular findings in a large unselected cohort of individuals screen positive for VLCADD [5, 9, 11, 16].
We describe our experiences as a reference laboratory that has completed molecular follow-up analysis for hundreds of VLCADD NBS screen positive individuals from testing facilities across the US. Results from this analysis provide a novel perspective on NBS outcomes that can be used to further advance an evidence-based approach to NBS follow-up for VLCADD.
2. Materials and Methods
2.1 Specimen collection
The 1080 specimens reported in this study represent all patients for whom Sanger sequence analysis of ACADVL was performed at the Baylor College of Medicine (Houston, Texas) from 06/2007 to 12/2014 (Fig. 1). Samples were collected as whole blood in EDTA containing tubes and shipped at ambient temperature to the Baylor College of Medicine. Approximately 50% of samples came from the state of Texas, with nearly all of the remainder collected in 1 of 40 different states; the following were represented by 20 or more unique patients: PA, CA, IA, CT, WI, MA, MO, AL, and OH. Each specimen was submitted with a requisition form intended to gather additional patient information including ethnicity, indication for study, and relevant family history. All procedures were approved by the Baylor College of Medicine Institutional Review Board with a waiver of informed consent.
Fig. 1.
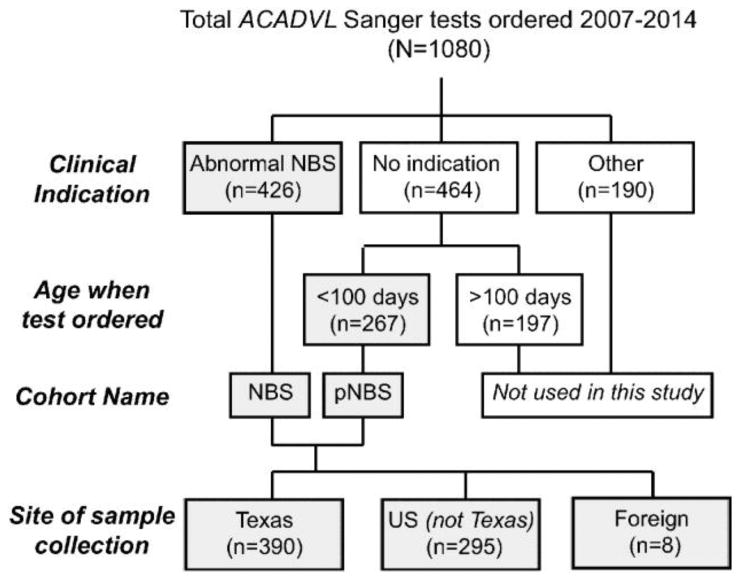
The flowchart describes the samples sent to our laboratory for Sanger sequence analysis of ACADVL over the course of seven years. Grey boxes indicate samples analyzed in this study. pNBS= presumptive newborn screen positive.
2.2 Sanger sequencing
DNA was extracted from EDTA preserved whole blood using a commercially available DNA isolation kit (Gentra Systems Inc., Minneapolis, MN) according to the manufacturer's protocol. The coding regions of the ACADVL gene (NM_000018.2), as well as proximal intronic sequences, were PCR amplified and then sequenced in the forward and reverse directions using automated fluorescent dideoxy sequencing methods. Nucleotide 1 corresponds to the A of the start codon ATG (NM_000018.2). Variants detected in exons and in introns within up to 20 bp of the exon/intron boundaries were studied.
2.3 Array CGH
A custom-designed oligonucleotide 180K exon-targeted CGH array (MitoMet v3) was used to assess for copy number changes involving ACADVL. The average probe density was greater than four probes per exon, with 1-kb spacing in the intronic regions. The targeted region of the aCGH contained probes for the coding exons and 50 bp of the flanking intronic regions. The criteria for a potential CNV call in the aCGH are at least two contiguous probes with a log2 ratio >0.3 for duplication and <−0.3 for deletion in regions of interest. Findings were reported according to human genome build hg19.
2.4 Biochemical analyses
Plasma acylcarnitine analysis described in Fig. 4 was completed as described previously [17]. NBS, enzymatic testing, and quantitative acylcarnitine analysis described in Table 3 and Fig. 5 were completed by outside CLIA certified clinical testing laboratories on a fee for service basis. Enzymatic testing was completed using peripheral leukocytes or fibroblasts.
Fig. 4. Follow-up quantitative plasma acylcarnitine data for patients with a positive NBS suggestive of VLCADD.
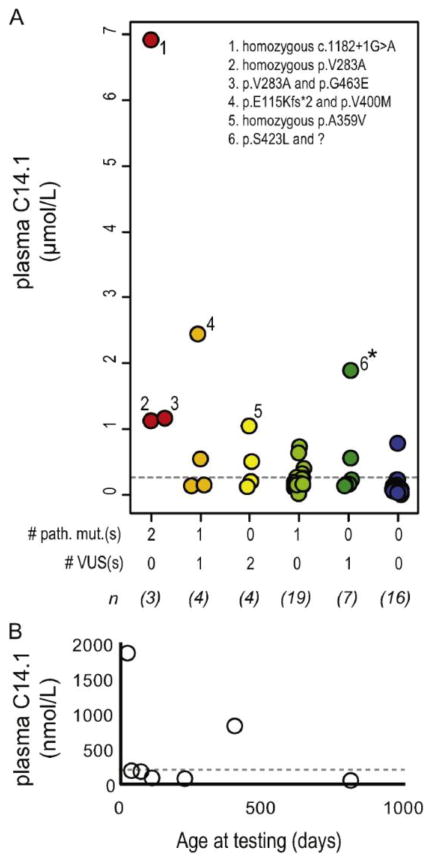
(A) Plasma C14:1 levels from the first quantitative plasma acylcarnitine analysis following a positive NBS are plotted in relation to the patient's molecular finding. For example, red dots indicate C14.1 values for patients harboring two pathogenic variants and blue dots indicate C14.1 values for patients with no variants in ACADVL. The number of unique patients in each genotypic class is shown (n) and specific genotypes are listed for a few notable cases- indicated by numbers. The asterisk indicates findings for case PAT0165 that is further explored in Fig. 2B. (B) Plasma C14:1 levels are shown for seven different follow-up analyses completed over the course of 3 years for PAT0165. Grey dotted lines indicate the upper limit of normal (95th percentile for unaffected individuals tested in our laboratory).
Table 3. Description of clinical outcomes for homozygous p.V283A patients.
| ID | NBS C14:1 (μmol/L)¶ | NBS normal range | Presenting feature | Age at last consult (years) | Neonatal symptoms | Echocardiogram result (age at test date) | Treatment § | Enzyme testing (normal range)¥ |
|---|---|---|---|---|---|---|---|---|
| PAT0472 | 1.53 | <0.6 | NBS | 1.5 | hypoglycemia and hyperbilirubinemia | normal (21d and 322d) | M, D | deficient |
| PAT0306 | 1.03 | <0.65 | NBS | 1 | prematurity, respiratory distress, hyperbilirubinemia | normal | M, D | 0.8 (3.9-9.6) |
| PAT0044 | abnormal | NBS | 3 | none | normal (26d) | M, D | deficient | |
| PAT0692 | 1.27 | <0.18 | NBS | 0.15 | none | normal | M, C | - |
| PAT0613 | 0.74 | <0.52 | NBS | 1 | hypoglycemia | normal | M, C | - |
| PAT0135 | 2.89 | <0.79 | NBS | 5.5 | none | normal (8d) small ASD (31d) | M, C, D | - |
| PAT0131 | 1.18 | <0.79 | NBS | 4.5 | none | and normal (1, 2, 3, 4y) | M, C, D | - |
Qualitative NBS values were not available for all patients. Results are from NBS bloodspot cards collected in first 3 days of life.
M = supplementation with MCT oil of MCT containing formula, D = dietary fat restriction, C = supplemental carnitine
values are in nmol/min/mg protein (completed on peripheral leukocytes). “Deficient” is used for a qualitative enzyme testing results indicating affected status (completed on fibroblasts).
Fig. 5. Quantitative plasma acylcarnitine analysis in patients homozygous for the p.V283A allele.
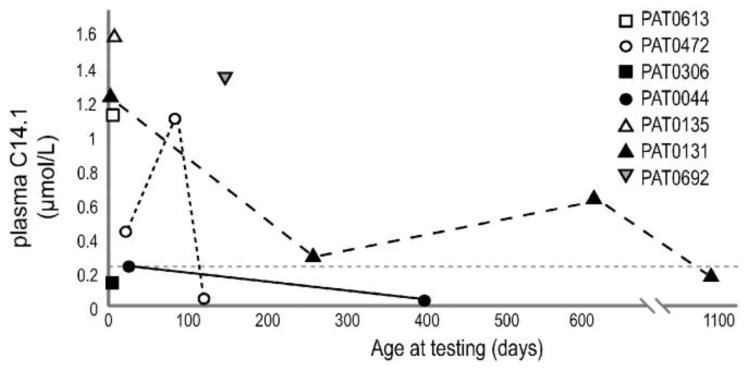
All available plasma C14:1 test results are shown in relation to the patient's age at sampling. Presumably many of these tests were completed when the patient was receiving treatment for VLCADD. The dotted grey line is representative of the normal range of plasma C14:1 levels in unaffected individuals.
2.5 Data analysis
Classification of variants was completed by a team of Fellow of the American College of Medical Genetics credentialed molecular geneticists following ACMG standards and guidelines [18]. Novel pathogenic variants were classified as base pair changes that resulted in a premature stop codon or that occurred within the canonical splice donor or acceptor region of ACADVL but that were not listed in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php). Variant enrichment analysis was completed using a Fisher's exact test and the allele frequencies reported in the ExAC Database. Expected allele numbers were calculated by multiplying the ExAC allele frequency by the number of alleles in our cohort of NBS and likely NBS patients (n=1386). To determine if a variant had been previously detected, we searched the Baylor College of Medicine exome database (accessed 11/14/2014), the ExAC database (accessed 2/17/2015), and the exome variant server (accessed 2/17/2015) with each database comprised of exome data from ∼5000, 60,542, and ∼6500 individuals, respectively, (Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) and Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/). Normal reference ranges for quantitative acylcarnitine analysis were calculated using data from all patients receiving testing in our biochemical genetics laboratory over the course of 10 years. Samples used for reference range calculations were collected within the first 100 days of life from patients that did not have a biochemical genetics diagnosis (n=2438). The upper limit of normal was defined as the 95th percentile of this reference population.
2.6 In silico predictions
Variant pathogenicity predictions were completed using PolyPhen-2 (version2.2.2) and SIFT (Ensembl 63) [19, 20]. The structure of human VLCAD was examined using a previously generated 1.45 Å resolution crystal structure (PDB# 2UXW). PyMOL (DeLano Scientific, CA) was used to visualize the protein structure and model amino acid changes in the three-dimensional structure.
3. Results
3.1 Recurrent variants in ACADVL in patients screen positive for VLCADD
Over the course of 7 years, we completed Sanger sequence analysis of all ACADVL exonic and proximal intronic sequence for 1080 individuals (Fig. 1). Testing was sent for a variety of clinical concerns, but the most common was an abnormal newborn screening result (n=426); the second most common indication listed was hypoglycemia (n=27). An additional 267 cases were collected from patients within the first 100 days of life, but no clinical information was provided; for the remainder of this manuscript, we refer to these as “presumptive NBS” or “pNBS” cases.
Within the group of patients receiving sequencing for NBS or pNBS related findings, we identified 94 pathogenic variants, as well as 134 variants of unknown clinical significance (VUSs) (Fig. 2 and Supplementary Tables S1 and S2). Forty of the pathogenic variants that we detected had not been previously reported in patients with VLCADD (Supplementary Table S2). Although private variants were common, there were a number of pathogenic variants identified in multiple unrelated patients. Chief among these was the c.848T>C (p.V283A) change (sometimes refered to as c.848T>C (p.V243A) depending on the reference isoform) which was found at an allele frequency of 6.6% and comprising 24.3% of all pathogenic alleles identified in our cohort. This represents a significant enrichment for p.V283A when compared to the general population minor allele frequency (MAF) reported in the ExAC database for this variant (MAF = 0.143%; p-value= 2.2 × 10-16).
Fig. 2. Pathogenic variants detected in patients soliciting molecular analysis after a postive NBS suggestive of VLCADD.
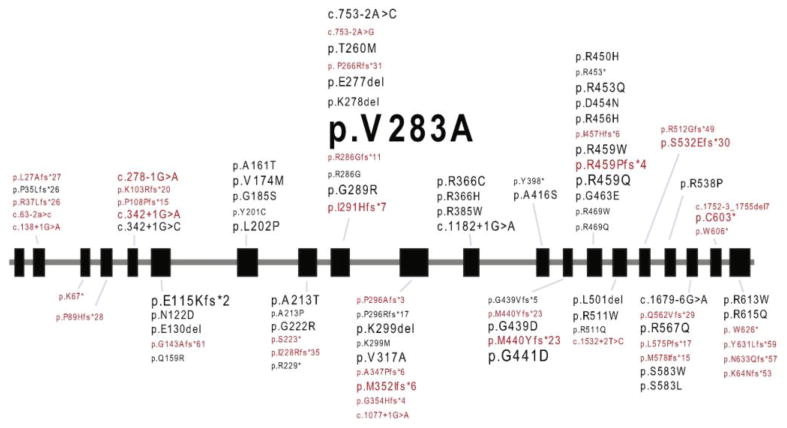
All pathogenic variants detected in our NBS or pNBS cohort are listed. The font size is proportional to the allele frequency. Novel pathogenic variants are shown in red. Black boxes indicate exons and grey lines indicate introns of ACADVL.
Recurrent VUSs were also detected. Particularly interesting were the VUSs that were significantly enriched within the NBS and pNBS population as compared to the unaffected population (Table 1). For example, the previously unclassified variant, c.1076C>T (p.A359V) was found in a heterozygous or homozygous state in 4 different patients out of 684 total in our cohort but was found in a heterozygous state (never homozygous) in only 3 individuals out of 60,542 reported in the ExAC database (p-value = 1.02×10-8). These findings remained significant when recalculated using the highest population allele frequency noted in ExAC, the African population (p-value = 3.95×10-4). Consistent with the pathogenicity of this VUS, C14:1 elevations were detected in an individual homozygous for p.A359V during follow-up quantitative plasma acylcarnitine analysis (patient #5 in Fig. 4A). An additional three VUSs were found in 4 or more unrelated individuals in our cohort but were not present in our institution's exome database, the ExAC database, or the Exome Variant Server. The pathogenicity of two recurrent VUSs was not supported by this analysis, thus raising the possibility that c.1066A>G (p.I356V) and c.1600G>A (p.E534K) may be benign variants common in certain populations.
Table 1. VUSs enriched in patients with an abnormal NBS suggestive of VLCADD.
| VUS | Allele count* | Expected count¶ | P-value¥ | Previously reported | SIFT / Polyphen-2$ | Structural location§ | Summary of results |
|---|---|---|---|---|---|---|---|
| c.1066A>G (p.I356V) | 11 | 1.3-13.9 | 2.38×10-7 to 0.56 | no | D/D | alpha-helical bundle | unclear |
| c.1273G>A (p.A425T) | 8 | 0.05-0.5 | 1.24×10-13 to 1.16×10-5 | (Chien et al 2013) | T/D | alpha-helical bundle | likely pathogenic |
| c.1001T>G (p.M334R) | 6 | 0.03-0.06 | 1.71×10-10 to 5.75×10-9 | no | D/D | near active site | likely pathogenic |
| c.538G>A (p.A180T) | 5 | 0.02-0.12 | 3.79×10-9 to 7.65×10-5 | no | D/D | near active site | likely pathogenic |
| c.640T>G (p.F214V) | 5 | NA | NA | no | D/D | near active site | likely pathogenic |
| c.1076C>T (p.A359V) | 5 | 0.03-0.27 | 1.02×10-8 to 3.95×10-4 | no | D/D | alpha-helical bundle | likely pathogenic |
| c.1019G>T (p.G340V) | 5 | NA | NA | no | D/D | near active site | likely pathogenic |
| c.889_891delGAG (p.E297del) | 4 | NA | NA | no | NA | dimeric interface | likely pathogenic |
| c.1600G>A (p.E534K) | 4 | 3.6-40.0 | 0.79 to 2.14×10-12 | (Mathur et al 1999) | T/T | surface | likely benign |
| c.1103A>C (p.Q368P) | 4 | 0.01-0.02 | 8.06×10-8 to 8.45×10-7 | no | D/D | near active site | likely pathogenic |
The allele count equals the number of unique patients with the variant, all patients were heterozygous for the listed variants. The exception is p.A359V which was found homozygous in one individual.
Expected counts were calculated using the allele frequencies in the ExAC database: (low value) uses overall allele frequency and (high value) uses highest population allele frequency. NA indicates that the allele was not present in the ExAC database.
P-values were calculated using a Fischer's exact test and the same allele frequencies used to generate expected count.
Variant prediction; D=deleterious and T=tolerated
See Fig. 2
To further explore the putative pathogenicity of enriched VUSs, we completed a variety of in silico analyses, which are summarized in Table 1. Particularly informative was a structural modeling analysis using a previously generated 1.45 Å resolution human VLCAD crystal structure (PDB# 2UXW). Five variants (p.M334R, p.A180T, p.F214V, p.G340V, and pQ368P) mapped in close proximity to the internally embedded VLCAD active site wherein the catalytic residue E462 and the cofactor FAD localize (Fig. 3 and Fig. S1A-E) [21]. Other variants mapped outside the catalytic domain but within regions that may interfere with protein stability or dimer formation. For instance, p.A425T, p.I356V, and p.A359V exist in alpha-helical bundles (Fig. 3 and Fig. S1F-H). The variant p.E297del is found within the dimeric interface and appears to be important for anchoring of W249 through an extensive hydrogen network (Fig. 3 and Fig. S1I). Finally, the p.E534K variant was found on the solvent exposed exterior of VLCAD; at this position the substitution of one hydrophilic residue for another (E to K) is predicted to have little impact on function (Fig. 3 and Fig. S1J). This structural result is consistent with our statistical enrichment analysis, which also called into question the pathogenicity of this variant. In summary, the combined enrichment and in silico prediction data was sufficient to reclassify a number of VUSs as likely pathogenic when using the American College of Medical Genetics criteria for variant interpretation; these variants are indicated in Table 1 [22].
Fig. 3. Structural analysis of VLCAD variants.
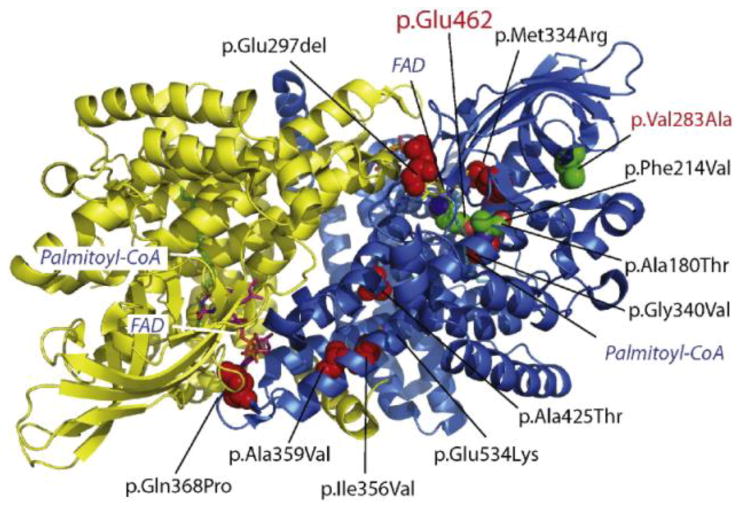
Enriched VUSs, described in Table 1, were plotted on the crystal structure of the VLCAD homodimer (PDB# 2UXW) where the two VLCAD moieties are indicated by yellow and blue ribbon structures. Red spheres indicate the location of VUSs. Stick diagrams indicate the location of the substrate (Palmitoyl-CoA) and the cofactor, flavin adenine dinucleotide (FAD). The location of the catalytic residue, Glu462, and the p.V283A pathogenic variant are shown by green spheres.
3.2 Molecular analysis following an abnormal NBS identifies a high number of carriers
For a total of 426 patients, the test requisition form explicitly stated that ACADVL Sanger sequence analysis was ordered in response to an abnormal NBS; their results are summarized in Table 2. Within this group only 13.0% of individuals had two variants within ACADVL. This includes pathogenic variants or VUSs but not benign polymorphisms. The most common finding was carrier status: 57.3% of all patients in this cohort, with 39.4% and 17.8% having a single pathogenic variant or a VUS, respectively. The remaining 29.8% of patients had no variants detected by Sanger analysis.
Table 2. Molecular findings in patients soliciting ACADVL sequence analysis in response to an abnormal NBS suggestive of VLCADD.
| # Patients | %of Total | |
|---|---|---|
|
|
||
| 2 pathogenic variants | 25 | 5.9 |
| 1 pathogenic variant and 1 VUS | 17 | 4.0 |
| 2 VUSs | 13 | 3.1 |
| 1 pathogenic variant | 168 | 39.4 |
| 1 VUS | 76 | 17.8 |
| No findings | 127 | 29.8 |
To search for pathogenic alleles potentially missed by Sanger analysis, we performed exon targeted array comparative genomic hybridization (aCGH) analysis of ACADVL for 131 individuals in the NBS or pNBS cohort. For approximately 73% of the patients in this group, concurrent Sanger sequence analysis identified a single pathogenic variant or VUS within ACADVL. In all cases, aCGH analysis failed to detect evidence of a copy number variation (CNV) within ACADVL.
In a further attempt to detect evidence of missed pathogenic alleles, we correlated follow-up quantitative plasma acylcarnitine data with the genotype for 53 patients within the NBS and pNBS cohort (Fig. 4A). The average age at the time of quantitative acylcarnitine analysis was 30 days of life (min = 8 and max =120). Patients with two pathogenic variants had C14:1 levels >1 μmol/L. Nearly all patients with zero or one variant had C14:1 levels below this threshold, with ∼62% having values below the upper limit of normal (C14:1 <0.196 μmol/L).
One notable exception, PAT0165, was a carrier for a single VUS (c.1268C>T (p.S423L)), but had a plasma [C14:1] = 1.88 μmol/L at the initial testing. The levels of C14:1 in this patient were the highest detected in a carrier in our cohort and were in the range seen in molecularly confirmed affected individuals (Fig. 4A, indicated by *). aCGH analysis failed to detect evidence of an ACADVL CNV. This patient was followed clinically for a number of years. Plasma acylcarnitine analysis was completed on seven separate occasions, and C14:1 fluctuated between normal and mild elevations (Fig. 4B).
3.3 Clinical outcomes for patients harboring homozygous p.V283A pathogenic variants
The prevalence of the p.V283A allele makes it an important candidate for further characterization. We detected at least one copy of this pathogenic variant in ∼10% of patients with an abnormal NBS, thus making it the most common variant detected, by a wide margin (Supplementary Table S2). Structural modeling indicates that Val283 is located outside of the catalytic site and is embedded near the periphery of the protein (Fig. 3). This change does not have an obvious consequence on structure/function and may act by reducing protein stability.
The p.V283A allele is likely of European origin. A subset of patients in our dataset provided self-identified ethnicity. The majority of patients with the p.V283A allele identified as European Caucasian (26 of 35), with the remainder identifying as European Caucasian plus another ethnicity (5 of 35), Hispanic only (3 of 35), or African only (1 of 35). European predominance of the p.V283A allele is also supported by findings in the ExAC database wherein 165 of 173 p.V283A alleles were identified in people reporting European ethnicity.
Clinical information was obtained for seven unrelated individuals homozygous for the p.V283A allele and is summarized in Table 3. In all cases, an abnormal newborn screening result alone prompted further investigation for VLCADD. In addition to molecular analysis, follow-up testing included quantitative plasma acylcarnitine analysis which detected C14:1 ranging from normal levels to significant elevations depending on the patient and time of testing (Fig. 5). Some patients also had enzymatic studies completed on peripheral leukocytes or fibroblasts, and in all cases, the results were interpreted as diagnostic for VLCADD. This is consistent with previously reported enzymatic studies using lymphocytes, which found 11% and 12% residual activity in two individuals homozygous for p.V283A and 35-64% residual activity in five individuals heterozygous for p.V283A [13, 23].
Few neonatal symptoms were reported in this cohort. Hypoglycemia was noted in two patients. For PAT0472, hypoglycemia was detected prior to receiving NBS results, and this symptom was successfully treated by supplementing breast milk with formula. In PAT0613, mild hypoglycemia was detected as a result of follow-up testing instigated by an abnormal NBS result. Other symptoms noted in the neonatal period are listed in Table 3 and may be unrelated to VLCADD. Hyperbilirubinemia was seen in two patients, possibly indicating hepatic dysfunction. Echocardiogram studies were completed on all patients within the first year of life, and in all cases failed to find evidence of cardiomyopathy. PAT0135 had a small atrial septal defect detected by echocardiogram at 31 days of life. This finding was not detected during follow-up echocardiogram analysis one year later, and subsequent annual exams up to the age of four have also failed to detect cardiac abnormalities. Other features associated with VLCADD such as hepatomegaly and myopathy were not detected in any patient in this cohort.
The seven patients in the homozygous p.V283 A cohort were all identified by NBS within the last decade and therefore long-term clinical outcomes were not available. The patient's age at their most recent clinical consultation ranged from 8 weeks to 5.5 years. During this time, all VLCADD patients received clinical management including medium chain triglyceride (MCT) containing oil or formula. Dietary fat restriction was recommended for patients after weaning and some, but not all, received oral supplemental carnitine. After the neonatal period, only one adverse event potentially related to VLCADD was reported in our cohort. PAT0044 was noted to be noncompliant with MCT consumption, though the patient maintained a fat restricted diet. At 2 years of age, PAT0044 had an episode of poor feeding lasting 10 hours and awoke with pale blue lips possibly indicating hypoglycemia. This resolved when given juice.
4. Discussion
Challenges associated with the newborn screening of VLCADD have been well documented [10, 12, 14]. Potentially confounding the situation is a lack of data on the outcomes of follow-up analyses of screen positive individuals. This is due in part to the logistical and patient-privacy issues NBS laboratories face when attempting to collect clinical information from a large cohort of patients seen by potentially hundreds of different providers in their state. As a large molecular reference laboratory that has experience providing molecular and biochemical testing for thousands of NBS screen positive patients, we are in a unique position to provide an outside perspective on NBS outcomes.
Limitations of this analysis should be considered when interpreting the results. First, our dataset has a regional bias and therefore may not represent the molecular outcomes for all state NBS testing laboratories in the US. The majority of specimens in our NBS and pNBS cohort came from Texas (56.3%) and the remainder came from 29 other states with a small number (8 patients) coming from a foreign country. Importantly, the carrier rates seen in the Texas NBS population did not differ appreciably from that reported in NBS patients located elsewhere-52.7% vs. 49.7%, respectively. Therefore, data reported herein is likely to be generally reflective of NBS outcomes across the US. Second, it is possible that patients with severe phenotypes receive molecular analysis less frequently than patients with more moderate biochemical or clinical phenotypes. The American College of Medical Genetics and Genomics (ACMGG), advocates for sequence analysis for positive newborn screen results when follow-up plasma acylcarnitine analysis (ACP) is normal, but considers additional testing optional when follow-up ACP is also positive (https://www.acmg.net/StaticContent/ACT/Algorithms/Visio-C14-1_DM.pdf). Failure to molecularly confirm all affected individuals may explain why we found that only 13.0% of screen positive individuals had two variants in ACADVL. However, this does not appear likely, based on our clinical experience. Among the metabolic clinicians authoring this manuscript, the consensus practice is to order molecular testing even when the follow-up ACP supports the initial positive NBS result. Therefore, the high carrier rate and low molecular confirmation rate we report are unlikely to be an artifact of biased sampling wherein biochemically severe cases are excluded.
We report that carrier status is the most common finding following a positive newborn screen, totaling 57% of all cases seen by our laboratory when including both pathogenic variants and VUSs. This result is consistent with a recent publication describing the experiences of a large multistate NBS consortium where carrier status was reported in 45% of patients, although specific molecular results were not available in all cases for comparison [9]. One possibility for this high apparent carrier rate is that the second pathogenic allele was missed by Sanger sequence analysis. We could find at least one example in the literature of a patient with a large deletion in ACADVL who was missed by Sanger sequence analysis [24]. Alternatively, variants affecting deep intronic or promoter sequence may also be missed by the Sanger analysis we provide. Results from our array CGH analysis as well as follow-up acylcarnitine studies failed to identify clear examples of pathogenic alleles missed by Sanger testing, although acylcarnitine analysis on a small subset of individuals were suggestive of a missed pathogenic allele. The key point is that cryptic pathogenic alleles missed by standard Sanger analysis do not appear to be frequent in the general US population and therefore, the most common result for patients that fail an NBS for VLCADD is in fact carrier status.
Given the relatively high frequency of the p.V283A allele in screen positive individuals, a clear understanding of the clinical outcomes associated with this pathogenic variant are critical to improving patient outcomes. Each year, 8 new patients homozygous for the p.V283A allele are predicted to be born in the US when assuming Hardy-Weinberg equilibrium and using the ExAC population allele frequency data (MAF = 0.143%) and the 2013 US birth rate - 3,932,181 (www.cdc.gov). Many more are expected to be compound heterozygous for pathogenic variants in trans with p.V283A. In our dataset, ∼10% of all individuals screen positive for VLCADD had one or more p.V283 A alleles.
Outcomes for patients harboring a p.V283A allele in trans with a second pathogenic mutation have shown surprising phenotypic heterogeneity. Lethal neonatal onset hypoglycemia was reported in a patient with compound heterozygous pathogenic variants, p.V283A and c.342+ 1G>C [25]. Whereas an individual harboring compound heterozygous pathogenic variants, p.V283A and c.1376g>a (p.R459Q), remained asymptomatic into her 30's despite receiving no treatment for VLCADD and while maintaining an active lifestyle that might be expected to exacerbate her condition; she was eventually brought to clinical attention as a result of an abnormal newborn screen result for her child [11]. As has been suggested by previous molecular studies, perhaps the key difference between these two patients is that the second pathogenic variant was a null allele in the severe case and a missense allele in the mild case, presumably with some residual activity [6]. With this in mind, we might expect homozygous p.V283A individuals to have a relatively mild presentation. Indeed, our clinical follow-up of seven homozygous p.V283 A patients found that hypoglycemia was the primary concern, but with appropriate clinical management, C14:1 levels can normalize and most patients can remain asymptomatic, at least during the first few years of life.
5. Conclusions
This report is the first of its kind to share the complete dataset from a large molecular reference laboratory that handles ACADVL sequence analysis of hundreds of VLCADD screen positive individuals. A number of specific clinical recommendations can be drawn from the analysis of this dataset. (i) The following variants should be considered likely pathogenic based on analyses reported here and using the ACMG criteria for variant interpretation: c.1273G>A (p.A425T), c.1001T>G (p.M334R), c.538G>A (p.A180T), c.640T>G (p.F214V), c.1076C>T (p.A359V), c.1019G>T (p.G340V), c.889_891delGAG (p.E297del), and c.1103A>C (p.Q368P) [22] (ii) Del/dup analysis should be considered a low-yield test in cases involving a positive NBS for VLCADD without additional supporting clinical or biochemical findings. This test should be reserved for patients with strong biochemical/clinical evidence for VLCADD and who have received equivocal ACADVL sequence analyses. (iii) Patients homozygous for the most common pathogenic variant discovered by NBS, c.848T>C (p.V283A), can be expected to have a more benign clinical course than the classic neonatal-onset cardiomypathic form of VLCADD. With standard VLCADD treatment, patients are likely to remain asymptomatic during the first years of life. Hypoglycemia may be a concern, especially during the neonatal period.
Supplementary Material
Highlights.
>200 unique variants were identified in ∼700 patients NBS screen positive for VLCADD
Carrier status was the most common NBS outcome
8 VUSs were reclassified to likely pathogenic using in silico approaches
∼10% of NBS screen positive patients had at least one copy of the p.V283A allele
7 unrelated homo p.V283A patients had mild or no clinical findings early in life
Acknowledgments
The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. The authors would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). This work was funded, in part, by the T32 GM07526-37 Medical Genetics Research Fellowship Program (M.J.M.). L.C.B. is supported by a the National Urea Cycle Disorders Foundation Fellowship and a fellowship from the Urea Cycle Disorders Consortium (UCDC; U54HD061221), which is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
Abbreviations
- VLCAD
very long chain acyl-coA dehydrogenase
- VLCADD
very long chain acyl-coA dehydrogenase deficiency
- VUS
variant of unknown clinical significance
- NBS
newborn screening
Footnotes
Conflict of Interest: All authors declare that there is no conflict of interest. Dr.'s Elsea, Sutton, Sun, Craigen, Zhang, and Wong are faculty in the department of Molecular and Human Genetics at the Baylor College of Medicine (BCM) and also directors within the Medical Genetics Laboratories (MGL). The MGL is jointly owned by BCM and Miraca Life Sciences and provides a number of clinical tests on a fee-for-service basis including those described in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aoyama T, Souri M, Ueno I, Kamijo T, Yamaguchi S, Rhead WJ, Tanaka K, Hashimoto T. Cloning of human very-long-chain acyl-coenzyme A dehydrogenase and molecular characterization of its deficiency in two patients. Am J Hum Genet. 1995;57:273–283. [PMC free article] [PubMed] [Google Scholar]
- 2.Strauss AW, Powell CK, Hale DE, Anderson MM, Ahuja A, Brackett JC, Sims HF. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc Natl Acad Sci U S A. 1995;92:10496–10500. doi: 10.1073/pnas.92.23.10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hale DE, Batshaw ML, Coates PM, Frerman FE, Goodman SI, Singh I, Stanley CA. Long-chain acyl coenzyme A dehydrogenase deficiency: an inherited cause of nonketotic hypoglycemia. Pediatr Res. 1985;19:666–671. doi: 10.1203/00006450-198507000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33:501–506. doi: 10.1007/s10545-009-9001-1. [DOI] [PubMed] [Google Scholar]
- 5.Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Roschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32:488–497. doi: 10.1007/s10545-009-1125-9. [DOI] [PubMed] [Google Scholar]
- 6.Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, Strauss AW, Comeau AM, Eaton RB, Grady GF. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47:1945–1955. [PubMed] [Google Scholar]
- 8.Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33:521–526. doi: 10.1007/s10545-010-9076-8. [DOI] [PubMed] [Google Scholar]
- 9.Merritt JL, 2nd, Vedal S, Abdenur JE, Au SM, Barshop BA, Feuchtbaum L, Harding CO, Hermerath C, Lorey F, Sesser DE, Thompson JD, Yu A. Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab. 2014;111:484–492. doi: 10.1016/j.ymgme.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Schymik I, Liebig M, Mueller M, Wendel U, Mayatepek E, Strauss AW, Wanders RJ, Spiekerkoetter U. Pitfalls of neonatal screening for very-long-chain acyl-CoA dehydrogenase deficiency using tandem mass spectrometry. J Pediatr. 2006;149:128–130. doi: 10.1016/j.jpeds.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 11.McGoey RR, Marble M. Positive newborn screen in a normal infant of a mother with asymptomatic very long-chain Acyl-CoA dehydrogenase deficiency. J Pediatr. 2011;158:1031–1032. doi: 10.1016/j.jpeds.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 12.Boneh A, Andresen BS, Gregersen N, Ibrahim M, Tzanakos N, Peters H, Yaplito-Lee J, Pitt JJ. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88:166–170. doi: 10.1016/j.ymgme.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Spiekerkoetter U, Sun B, Zytkovicz T, Wanders R, Strauss AW, Wendel U. MS/MS-based newborn and family screening detects asymptomatic patients with very-long-chain acyl-CoA dehydrogenase deficiency. J Pediatr. 2003;143:335–342. doi: 10.1067/S0022-3476(03)00292-0. [DOI] [PubMed] [Google Scholar]
- 14.Browning MF, Larson C, Strauss A, Marsden DL. Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis. 2005;28:545–550. doi: 10.1007/s10545-005-0545-4. [DOI] [PubMed] [Google Scholar]
- 15.Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, Garganta C, Ficicioglu C, Cederbaum S, Harding C, Boles RG, Matern D, Chakraborty P, Feigenbaum A. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:85–90. doi: 10.1016/j.ymgme.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, Strauss AW, Wanders RJ, Spiekerkoetter U. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics. 2006;118:1065–1069. doi: 10.1542/peds.2006-0666. [DOI] [PubMed] [Google Scholar]
- 17.Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH. Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis. 1999;22:302–306. doi: 10.1023/a:1005587617745. [DOI] [PubMed] [Google Scholar]
- 18.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE, ALQAC Molecular Subcommittee of the ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.McAndrew RP, Wang Y, Mohsen AW, He M, Vockley J, Kim JJ. Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. J Biol Chem. 2008;283:9435–9443. doi: 10.1074/jbc.M709135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ALQA Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmann L, Haussmann U, Mueller M, Spiekerkoetter U. VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis. 2012;35:269–277. doi: 10.1007/s10545-011-9391-8. [DOI] [PubMed] [Google Scholar]
- 24.Pervaiz MA, Kendal F, Hegde M, Singh RH. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J Hum Gen. 2011;17:29–32. doi: 10.4103/0971-6866.82190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coughlin CR, 2nd, Ficicioglu C. Genotype-phenotype correlations: sudden death in an infant with very-long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2010;33(Suppl 3):S129–131. doi: 10.1007/s10545-009-9041-6. [DOI] [PubMed] [Google Scholar]
- 26.Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, Hug G, Strauss AW. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99:1337–1343. doi: 10.1161/01.cir.99.10.1337. [DOI] [PubMed] [Google Scholar]
- 27.Chien YH, Lee NC, Chao MC, Chen LC, Chen LH, Chien CC, Ho HC, Suen JH, Hwu WL. Fatty Acid oxidation disorders in a chinese population in taiwan. JIMD Rep. 2013;11:165–172. doi: 10.1007/8904_2013_236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.