

Abstract
The central players in most cellular events are assemblies of macromolecules. Structural and functional characterization of these assemblies requires knowledge of their subunit stoichiometry and intersubunit connectivity. One of the most direct means for acquiring such information is so-called native mass spectrometry (MS), wherein the masses of the intact assemblies and parts thereof are accurately determined. It is of particular interest to apply native MS to the study of endogenous protein assemblies—i.e., those wherein the component proteins are expressed at endogenous levels in their natural functional states rather than the overexpressed (sometimes partial) constructs commonly employed in classical structural studies, whose assembly can introduce stoichiometry artifacts and other unwanted effects. To date, the application of native MS to the elucidation of endogenous protein complexes has been limited by the difficulty in obtaining pristine cell-derived assemblies at sufficiently high concentrations for effective analysis. To address this challenge, we present here a robust workflow that couples rapid and efficient affinity isolation of endogenous protein complexes with a sensitive native MS readout. The resulting workflow has the potential to provide a wealth of data on the stoichiometry and intersubunit connectivity of endogenous protein assemblies—information that is key to successful integrative structural elucidation of biological systems.

Most biological processes and cellular events are accomplished by assemblies of macromolecules that form dynamic hierarchies of functional modules.1 Mapping the protein interaction networks that form these modules is yielding important insights into cellular function. These data are being gleaned through focused studies of individual functional modules as well as from large-scale genetic and protein interactome projects.2,3 One particularly informative approach is affinity isolation of endogenously interacting proteins with subsequent “bottom-up” mass spectrometric (MS) identification of the participant proteins.4 Because these native assemblies are disrupted prior to the protein identification step, it is usual to lose information about the heterogeneity of the populations of assembled interactors, the assembly masses, as well as their subunit stoichiometries. This lost information is crucial for determining the molecular architecture of macromolecular assemblies by integrative structural methods5,6 and for modeling the dynamics and behavior of functional modules within the cell. Although subunit stoichiometry can be determined by peptide-based MS methods such as label-free quantification7 or by spiking in a labeled protein comprised of concatenated reference peptides,8 it is desirable to have available methods that can directly measure the mass of intact, affinity-isolated “endogenous” protein complexes. Here, “endogenous” refers to assemblies isolated from their natural cellular environment wherein the component proteins are expressed at normal levels in their natural functional states It is particularly desirable to have available direct methods that can examine and elucidate such endogenous protein assemblies rather than the overexpressed (often partial) constructs that are commonly employed in classical structural studies. Such constructs may be prone to stoichiometry artifacts and other unwanted effects.9
One such method is native MS, which facilitates mass measurement of non-covalent macromolecular assemblies, thereby providing direct evidence on their stoichiometry and intersubunit connectivity.10,11 Although the method has been applied with spectacular success to increasingly large assemblies,12 application of native MS to the measurement of endogenous protein complexes has been limited. For example, only a handful of the estimated several hundred endogenous protein complexes from budding yeast (Saccharomyces cerevisiae)2,3,13 have been successfully analyzed by native MS.7,14–25 Clearly, there is a huge gap between the small number of protein complexes that have been successfully interrogated by native MS versus the vast space of complexes for which direct stoichiometry and interaction data are so critically needed for integrative structural modeling.5,6
One of the main challenges for successful native MS analysis of endogenous protein complexes is the need to capture sufficiently pristine cellular protein complexes and to prepare them at high enough concentrations in electrospray (ESI)-compatible solutions to obtain a useful MS spectrum. Typical native MS experiments have required the availability of relatively pure protein complexes with concentrations exceeding a few hundred nanomolar in volatile buffer solutions such as ammonium acetate.26 These requirements have often led to the use of slow, sometimes inefficient procedures requiring large amounts of starting cellular material for sample preparation. In response to the need for increasingly facile and effective procedures, we present a workflow that couples rapid, efficient affinity capture with sensitive native MS analysis, and demonstrate its efficacy via analysis of three exemplary protein complexes.
EXPERIMENTAL SECTION
Cell culture, cryolysis and affinity isolation
Tagged budding yeast strains were cultured using standard procedures, harvested, flash-frozen in liquid nitrogen and cryomilled using a planetary ball mill (Retsch) as previously described.27 The resulting cryomilled cell powder was stored indefinitely at −80 °C until sample processing. Typically, we obtain 2–2.5 g of cell powder per 1L of yeast culture grown to midlog phase. Affinity isolations were performed using antibody-conjugated magnetic beads as previously detailed27,28 (see Supporting Information). The protein complexes bound to the magnetic beads were then eluted either by addition of peptide (PEGylOx) or protease cleavage.
Nondenaturing elution
PEGylOx preparation and elution was performed as described previously.29 For peptide elution, 15 μL of 2 mM PEGylOx in 20 mM Tris pH 8.0, 100 mM NaCl, 2 mM EDTA, 5% EtOH, and 0.01% Tween-20 was added to the beads containing the bound protein complexes and elution was achieved by gentle rotation for 15 min at room temperature. For elution by protease release, the beads containing bound protein complexes were incubated with 0.5–2 μg of the protease (i.e., 1 μg protease/g of frozen cell powder) in 10–30 μL of protease digestion buffer (HRV 3C protease: 20 mM HEPES pH 7.4, 150 mM NaCl, 0.05% Tween-20, 1 mM DTT or TEV protease: 50 mM Tris pH 8, 0.5 mM EDTA, 1 mM DTT, 100 mM NaCl, 0.05% Tween-20). Incubation was performed for one hour at 4 °C with gentle rotation. Depending on the engineered cleavage site available, either the His-tagged HRV 3C protease (1 μg/μL stock; EMD Biosciences) or the His-tagged AcTEV protease (1 μg/μL stock; Life Technologies) was used.
Removal of elution reagent and buffer exchange
Depletion of the PEGylOx elution reagent and buffer exchange were performed using a Zeba micro desalting spin column with 40-kDa molecular weight cutoff (MWCO) (Thermo Fisher Scientific). First, the column was equilibrated four times with 50 μL each of the desired native MS buffer by centrifugation at 1,500×g for 1 min each at room temperature. Then, the PEGylOxeluted sample (volume 10–13 μL) was loaded onto the column, spun for 2 min, and collected. For protease depletion, the protease-eluted sample was collected and the beads were washed with 10–30 μL filtration buffer (FB: 20 mM HEPES pH 7.4, 0.01% Tween-20). The wash was pooled with the sample and the volume adjusted to 150 μL with FB. The mixture was then loaded onto a 0.5 mL centrifugal filter (Microcon) with 100-kDa MWCO (Ultracel YM-100 from Millipore), pre-washed twice with FB. The Microcon was centrifuged for 5 min at 12,000 rpm at 4 °C. Afterwards, 150–200 μL FB was added and another round of centrifugation was performed until the final volume was less than 20 μL. Buffer exchange into the respective native MS buffer was performed similar to what was outlined for PEGylOx removal, except that it was performed at 4 °C instead of at room temperature.
Native MS analysis
An aliquot (2–3 μL) of the sample was loaded into an in-house fabricated gold-coated quartz capillary and sprayed using a static nanospray source into the Exactive Plus EMR instrument (Thermo Fisher Scientific).30 Typical MS parameters include capillary temperature, 100 °C–150 °C; instrument resolution setting, 8,750 or 17,500; total number of scans, 100 (see Supporting Information for more details).
RESULTS AND DISCUSSION
Overall Experimental Workflow
Previously published methodologies for sample preparation of endogenous protein complexes from yeast have employed mechanical cell lysis using glass beads followed by multiple affinity isolation and chromatographic steps (Table S-1). In contrast, our workflow (Figure 1) employs a highly optimized affinity capture methodology27,28,31,32 that enables efficient recovery of tagged endogenous protein assemblies. The cells expressing the tagged target protein complex are cultured, harvested, flash-frozen in liquid nitrogen (LN), and then mechanically fractured and milled at LN temperatures until the cells are reduced into a micron-sized powder. This cryomilling step maximizes the efficiency and speed of solvent extraction of the protein complexes, while preserving their native environment prior to the solvent extraction step; cryolysis has been demonstrated to consistently preserve the oligomeric and functional states of the proteins at the moment of flash-freezing in LN.27,28,31 In addition, the milled frozen cell powder can be stored at −80 °C almost indefinitely and aliquots can be weighed out depending on the scale or needs of the experiment. This convenient stopping point decouples the largely non-perturbative (at the level of the protein assemblies) preparation of the cellular material from subsequent affinity isolation, buffer exchange and native MS analysis steps. Since all these latter steps can potentially perturb native protein assemblies, convenient tests can be made on small aliquots of the frozen powder to assess the relative levels of perturbation under different conditions in order to optimize these steps.
Figure 1.

Workflow for affinity isolation of endogenous protein complexes coupled to native MS.
The frozen cell powder is rapidly thawed into an appropriate extraction buffer containing protease inhibitors to minimize protein degradation, yielding a crude lysate that is rapidly pre-cleared by centrifugation. Magnetic beads conjugated with the affinity capture reagent are then added to the supernatant for a single-step affinity isolation with incubation times as short as 30 min—sufficient for capturing >90% of the tagged protein on the beads together with its associated interactors while minimizing nonspecific protein-protein interactions, which we have shown tends to build up over time.27,28 The use of non-permeable (2.7 μm diameter) magnetic beads facilitates small-scale isolations using quick and efficient washing steps as well as subsequent rapid elution into minimal volumes (≈10 μL), which maintain the concentration of the target protein complexes at suitably high levels for native MS analysis.
Here, we have concentrated our efforts on the widely used affinity tag protein A from Staphylococcus aureus (SpA) because of its high affinity for the Fc-domain of IgG and the ready availability of extensive collections of genomically SpA-tagged yeast strains.2,3 The genomically tagged genes are under the control of their endogenous promoters ensuring that the tagged gene products are expressed at their native levels. These tags, which are mostly C-terminal, are generally not observed to interfere with the function of the tagged protein. The affinity capture reagent conjugated to the magnetic beads is simply bulk IgG from rabbit serum with its advantages of high affinity, ready availability, and low cost. Native elution methods for the SpA/IgG-based affinity isolation system have been developed for structural studies such as cross-linking and electron microscopy and include incubation with a competitive peptide29,33 or protease release through a cleavage site that is incorporated together with the affinity tag.34,35 Here, we tested both types of nondenaturing elution strategies and optimized subsequent steps after elution prior to native MS analysis.
After elution, the sample must be desalted and exchanged into a native MS-compatible buffer such as ammonium acetate.36 It also proves necessary to remove the eluting reagent, which has to be added in high molar excess to be effective.29,33 Furthermore, the elution reagent removal and buffer exchange steps all involve sample interaction with the surfaces of membranes, tube walls, and resins, with the undesirable potential for substantial adsorptive losses. Therefore, it is crucial to passivate these surfaces with surface-active agents such as detergents even though these nonvolatile, sticky substances can interfere with subsequent ESI-MS analysis at higher concentrations.37
Sample loss during post-elution handling can be minimized through judicious use of detergent without compromising the MS response
Prior to native MS analysis, it is usually necessary to exchange the sample elution buffer to a volatile buffer that is compatible with ESI-MS (Figure 1). To do this, we compared a number of buffer exchange columns and determined that the Zeba desalting microspin columns (Thermo Fisher Scientific) performed best in our hands, and that addition of a detergent such as Tween-20 was crucial for efficient sample recovery (Figure 2). Because detergents generally interfere with the ESI-MS response, we determined a working range of Tween-20 concentrations that yielded both minimal sample loss and minimal interference during native MS analysis. These studies were carried out with two test samples: (i) IgG (Mr ≈150 kDa), which represents a class of classically “non-sticky” glycoproteins and (ii) an affinity isolated seven-member Nup84 complex (Mr ≈600 kDa), which represents a more “sticky” protein assembly. These samples were buffer-exchanged into 150 mM ammonium acetate in the presence of varying concentrations of Tween-20 (Figure 2). With no Tween-20 in the buffer, more than 80% of the input IgG and virtually 100% of the Nup84 complex were lost to the desalting column. However, with increasing concentrations of Tween-20 the recovery was observed to increase until maximum recovery was achieved at Tween concentrations at and above 0.001%. We then investigated the effect of Tween-20 on the mass spectra obtained from these protein complexes. Our prior experience with Tween-20 on native MS using a Waters Synapt Q-TOF demonstrated sufficient interference as to make the mass spectra virtually unusable (data not shown), so we were pleasantly surprised when we found that we could obtain well-resolved native MS with the Exactive Plus EMR Orbitrap mass spectrometer. Indeed, with appropriate tuning, we observed negligible native MS interference from Tween-20 with concentrations up to 0.01% (Figures S-1). Thus, we chose to carry out all our elution reagent removal and buffer exchange steps in the presence of Tween-20 with concentrations ranging between 0.001% and 0.01%.
Figure 2.
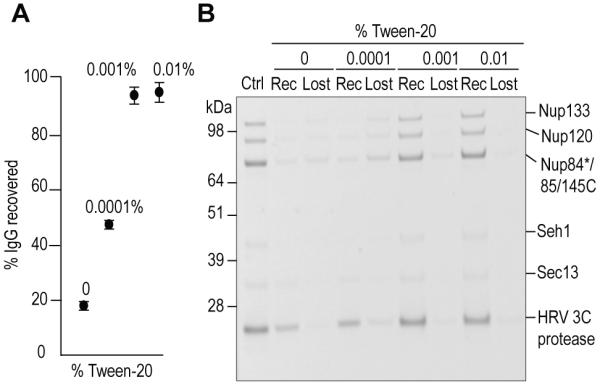
Effect of increasing Tween-20 concentration on sample retention during buffer exchange. (A) IgG recovery from buffer exchange (n=6). Protein quantification was based on gel band intensities. For more details, see Supporting Information and Figure S-1. (B) SDS-PAGE separation of buffer-exchanged Nup84 complex obtained from affinity isolation and elution by HRV 3C protease.
Elution with a competitive peptide
A disulfide bond-constrained 13-amino acid peptide (termed FcIII) was evolved to bind competitively to the SpA-IgG binding interface.38 PEGylOx, a modified FcIII peptide with four PEG units at its N-terminus has been shown to competitively elute SpA-tagged protein complexes under nondenaturing conditions in 15 min at room temperature.29 Because fairly high concentrations (2 mM) of PEGylOx are needed for efficient elution of native protein complexes, it proves important to have an effective means for its later removal prior to the MS step. We found that high concentrations of this 1.7-kDa peptide can be rapidly and effectively depleted by buffer exchange using a desalting spin column with 40-kDa MWCO,29 making subsequent native MS analysis feasible.
We analyzed the yeast GINS complex (Figure 3) to assess the efficacy of our workflow that incorporates peptide elution (Figure 1, left). GINS complexes, which are an essential component of the eukaryotic DNA replication machinery,39 are expected to be present in S. cerevisiae in modest abundance (≈1,000 copies per cell40,41). Affinity capture was performed using the GINS component Psf2 with a 26-kDa C-terminal SpA tag bearing three complete IgG-binding domains and one almost complete IgG-binding domain.42,43 For this procedure, 1 g of frozen grindate was used. Psf2 assembles into the GINS complex with Psf1, Psf3 and Sld5,39,44 as confirmed by SDS-PAGE (Figure 3A) and bottom-up LC-MS analysis (Tables S-2 and S-4). Ctf4 was also observed and has been shown to directly associate with the GINS complex throughout the cell cycle.45
Figure 3.

Affinity isolation, peptide elution and native MS analysis of the endogenous GINS assembly from budding yeast. (A) SDS-PAGE separation and Coomassie staining to assess the post-elution sample handling steps. Elution was performed with 2 mM PEGylOx, which was later removed by buffer exchange into 150 mM ammonium acetate, 0.01 % Tween-20. (B) The native MS spectrum of the endogenous yeast GINS complex and (C) the peak series for the Ctf4 trimer. For the full spectra, see Figure S-2. (D) Spectrum showing HCD activation of the GINS complex.
Native MS characterization of this affinity-isolated GINS complex yielded a well-resolved charge-state distribution centered at m/z 6,000 (Figure 3B) corresponding to a mass of 131,094 ± 5 Da—i.e., the mass of the complex consisting of Psf1, Psf2, Psf3, and Sld5 at unit stoichiometry. A separate peak series corresponding to the Ctf4 trimer was also observed, albeit at relatively low signal intensity (Figure 3C). Indeed, Ctf4 has been previously shown to constitutively form a homotrimer, which serves as a platform for multivalent interactions with other replisome assemblies, including the GINS complex.46 Here, its observation as a separate trimer indicates that it most likely dissociated from the GINS complex subsequent to the affinity isolation step, during the treatment prior to electrospray or during the electrospray process, but not in the gas phase.
Inducing collision-activated gas-phase dissociation generates charge-stripped subcomplexes with lower charge-states and highly charged ejected subunits.47,48 This can be achieved in the EMR by increasing the HCD voltage, which is applied to all ions exiting the transport multipole (all-ion activation) as there is no prior precursor mass selection possible in this particular instrument. Increasing the HCD voltage offset from 75 V to 200 V yielded two charge-stripped subcomplexes (Psf1/Psf2/Sld5 and Psf2/Psf3/Sld5) together with the corresponding ejected subunits Psf3 and Psf1, respectively (Figure 3D). Additional subcomplexes (Psf1/Psf3/Sld5 and Psf2/Sld5) were also observed at lower signal intensity (Table S-3). From these results, we are able to derive an subunit connectivity map (Figures 3B and 3D), which is consistent with the structures of the homologous human GINS complex.49 This interaction map is also consistent with that previously found from native MS analysis of the human GINS complex,50 with the exception of the Psf2/Psf3 interaction in the Psf2/Psf3/Sld5 heterotrimer observed in this study. The measured mass errors for the complex and subcomplexes ranged from 0.002% to 0.1%, and the masses of the two dissociated subunits (Psf1 and Psf3) agreed with the predicted masses to within 30 ppm (Table S-3).
Elution with HRV 3C protease
The HRV 3C protease is a 22-kDa cysteine protease, which acts with high specificity and is active at 4 °C.51 We tested two commercially available HRV 3C proteases and found that the His-tagged version required less enzyme-per-substrate ratio than the GST-tagged version (Figure S-3). This could be due to the dimerization of the GST-tagged version, which reduces its effective protease concentration. In addition, the higher molecular weight (≈45 kDa) of the GST-tagged version makes post-elution protease removal more challenging. For these reasons, we opted to use the His-tagged HRV protease in the protease elution leg of the workflow (Figure 1, right).
We optimized the conditions for native elution using this protease in terms of starting amount of frozen grindate, elution volume, and temperature, determining that 1 μg protease was sufficient to completely release captured protein complexes on affinity isolation beads exposed to 1 g of resuspended frozen grindate within one hour at 4 °C (Figure S-4). Because this protease release step was most effective in small volumes (typically 10 μL), the concentration of protease used was high (5 μM); thus, to avoid interference in native MS analysis, it is important to remove the protease from the sample prior to the ESI-MS step. We were able to remove most of the 22-kDa protease without incurring significant losses of the protein complexes through the use of a 0.5-mL filter concentrator with a 100-kDa MWCO and just two wash steps of 150–200 μL each (Figure 4A). Rapid and efficient buffer exchange into a native MS-compatible buffer containing ammonium acetate and Tween-20 was then achieved by the small desalting spin column with 40-kDa MWCO, identical to that used for PEGylOx removal (Figure 4A). Note that in previously published protocols (Table S-1), the eluting protease was removed either by a second round of affinity isolation using a different tag in the target protein complex or by size-exclusion chromatography. These extra steps dilute the samples, extend sample handling times, and can lead to significant sample losses.
Figure 4.

Affinity isolation, protease elution and subsequent native MS analysis of the endogenous Nup84 complex from budding yeast. (A) SDS-PAGE separation and Coomassie staining to assess the post-elution sample handling steps. Elution was achieved by cleavage with the HRV 3C protease, later removed by filtration. Subsequent buffer exchange was performed with 500 mM ammonium acetate, 0.01% Tween-20. The native MS spectrum of the Nup84 complex with (B) low and (C) high in-source activation. The structural model for the Nup84 holocomplex is also shown based on integrative structural studies.34,35
We then tested the overall optimized protocol for the right leg of our workflow (Figure 1) using the Nup84 subcomplex (≈2,000 copies/cell), which forms the outer rings of the yeast nuclear pore complex (NPC)—the sole mediator of molecular transport between the cytoplasm and the nucleus.35,52,53 Nup84 was tagged at the C-terminus with the same SpA construct as that used for Psf2-SpA except that it was preceded by 10 amino acids bearing the cleavage site for HRV 3C protease.35 Using this Nup84-HRV-SpA strain, we affinity-isolated the Nup84 complex from 2 g of frozen grindate obtained from 1 L of yeast culture grown to midlog phase. SDS-PAGE analysisof aliquots (10% of the sample) from each major step in the workflow (Figure 4A) showed negligible losses. Importantly, we used 0.01% Tween-20 in the protease depletion and buffer exchange steps. Bottom-up LC-MS analysis of the buffer-exchanged sample confirmed capture of the seven known components of the Nup84 complex (Tables S-2 and S-5).
Native MS analysis of the sample with minimal in-source activation yielded five main ion series (Figure 4B). The highest charge-state series centered at m/z 9,227 (48+), which deconvolutes to a mass of 442,890 ± 50 Da and corresponds to a heterohexameric complex comprised of Nup84, Nup85, Nup145C, Nup120, Seh1, Sec13 at unit stoichiometry. A separate ion series for Nup133 (measured mass of 133,192 ± 4 Da) was also observed. Its charge-state distribution indicates a native-like state, suggesting that it dissociated in solution and not in the gas phase. Nup133 has been shown to be the most labile of the seven Nup84 subcomplex components, readily dissociating from the subcomplex during affinity isolation.35 Of the two other main ion series observed, one has a mass of 385,340 ± 20 Da corresponding to a pentameric subassembly consisting of one copy each of Nup85, Nup145C, Nup120, Seh1, and Sec 13 and another a mass of 123,850 ± 10 Da matching the Nup85/Seh1 dimer. The Nup84 subunit by itself was also observed (84,463 ± 1 Da) (Table S-3).
Ramping the in-source dissociation parameter to the maximum (from 50 V to 200 V) and slightly increasing the trapping gas generated more peak series corresponding mainly to dissociated subcomplexes and subunits (Figure 4C). In addition to the five peak series observed from Figure 4B, two additional heterotrimers were detected with masses 244,191 ± 13 Da, corresponding to Nup120/Nup85/Seh1, and 204,810 ± 12 Da, corresponding to Nup145C/Nup85/Seh1 (Table S-3). The latter was observed with a higher charge-state distribution than predicted for the native-like state (about 31+),54 indicating that dissociation and partial unfolding had occurred in solution and/or during the electrospray process. An ion series for Nup120 was also observed with charge-state distribution centered at 33+, which is higher than what is predicted for native-like state (about 23+),54 indicating that it likely arose from gas-phase dissociation. However, a search for the peak series corresponding to the charge-reduced assemblies resulting from Nup120 ejection did not yield any matches, likely due to their low signal intensities.
In terms of intersubunit connectivity, Nup85 and Seh1 interact strongly and the Nup85/Seh1 dimer associates both with Nup145C and with Nup120. These observations are all consistent with protein domain mapping, cross-linking and integrative structural investigation of the endogenous Nup84 complex from budding yeast.34,35 Overall, comparison with the expected calculated masses shows that the measured masses fall between 0.01% and 0.05% for the complexes, subcomplexes and Nup133, and below 40 ppm for the dissociated component proteins, namely Nup84 and Nup120 (Table S-3).
Elution of TAP-tagged protein complexes
The TAP tagging strategy involves an affinity tag construct with an engineered TEV protease cleavage site between two affinity handles—i.e., the SpA tag and the calmodulin-binding protein (CBP).4,55 It is noteworthy that the TAP tag has only two repeats of the synthetic Z-domain derived from SpA,4 and that in our hands, the SpA tag described above (with almost four full repeats42) outperforms the TAP tag as an affinity reagent.29 Typically, the TAP method involves initial purification by SpA/IgG binding, release by incubation with TEV protease,56 and a second-stage purification using the CBP, although it is noteworthy that in the present work we use only the TEV cleavage step. To test the workflow that we optimized for the HRV 3C protease elution, we affinity-isolated the yeast exosome assembly (5,000 copies/cell)40 using a TAP-tagged Csl4 strain. The exosome is involved in ribonucleolytic processing and exhibits 3′-5′ exonuclease activity.57,58 We affinity-isolated the exosome complex from 0.5 g of frozen grindate, and determined that an equivalent of 1 μg of TEV protease per 1 g of cryogrindate was sufficient to yield full cleavage of the tagged protein with a one hour incubation at 4 °C (Figure S-5).
Figure 5A shows the SDS-PAGE analysis of aliquots (10% of each sample) from each major step in the workflow. Again, key to the success of these steps was the inclusion of 0.01% Tween-20. Trypsin digestion and subsequent LC-MS analysis of the resulting sample demonstrated the presence of the known components of the exosome, namely the nine main subunits (Csl4-CBP, Mtr3, and Rrp4/40/41/42/43/45/46) together with the catalytic Dis3 subunit forming the so-called Exo-10 assembly, the nuclear-specific associated factors Rrp6, Lrp1, and Mtr4, as well as the cytoplasm-specific associated proteins Ski7, Ski2, Ski3, and Ski8 (Tables S-2 and S-6).
Figure 5.

Affinity isolation, protease elution and subsequent native MS analysis of the endogenous exosome assembly from budding yeast. (A) SDS-PAGE separation and Coomassie staining to assess the post-elution sample handling steps. Elution was achieved by cleavage with the TEV protease, later removed by filtration. Buffer exchange into 400 mM ammonium acetate, 0.01 % Tween-20 was then performed. (B) Representative native MS spectrum of the affinity-isolated exosome complex and the corresponding peak assignments, except for the 205-kDa subcomplex marked with *, which matched three possible subassemblies (see Table S-3).
From a representative native MS spectrum for the affinity isolated exosome (Figure 5B), the most intense cluster of peaks centered at m/z 9,500 corresponds to a measured mass of 403,235 ± 15 Da, which matches the expected mass of the Exo-10 complex within 0.05%. Another peak series corresponds to Exo-10 with the loss of Csl4-CBP (366,115 ± 10 Da) in solution or during the electrospray process at the front of the instrument. Upon gas-phase activation, both these complexes dissociated with ejection of Rrp40 (Figure 5B). Additional peaks with charge-state distributions that indicate native-like states point to subcomplexes also originating from dissociation in solution or during the electrospray process. The difference between the two measured masses (271,032 ± 13 Da and 243,412 ± 3 Da) corresponds to the mass of Mtr3 (27,620 Da, Table S-3), leading us to assign these masses to Mtr3/Dis3/Rrp4/41/42/45 and Dis3/Rrp4/41/42/45 sub-complexes, respectively (Figure 5B). Most of the exosome subcomplexes observed from in-solution and gas-phase dissociation have been consistently observed in previous native MS studies.15,16 Overall, the mass errors of the assemblies and subassemblies were measured at or below 0.06% (Table S-3). Additional peak series were assigned to associated compartment-specific factors that were bound to the exosome during affinity isolation but that likely dissociated during the post-elution handling steps or native MS analysis. The cytoplasmic heterotetrameric Ski complex (2 copies of Ski8 and one copy of Ski2 and Ski3) was observed (398,229 ± 9 Da) with a charge-state distribution similar to that characterized in an earlier native MS study.19 Finally, the 122-kDa nuclear-specific Mtr4 and the cytoplasmic-specific 85-kDa Ski7 were also detected (Figure 5B, Table S-3).
In addition, we observed satellite peaks corresponding to mass shifts of 320-350 Da on the intact exosome and its subassemblies (Figure S-6, Table S-3). Despite experiments that employed more stringent washes and buffer exchange steps, the presence of these satellite peaks remained unchanged. Thus, we infer that these satellite peaks result from adduction of presently unknown moieties or addition of unknown post-translational modification(s).
Comparison of the two nondenaturing elution modes
We have tested two modes of nondenaturing elution using (i) incubation with a competitive peptide and (ii) protease cleavage. The advantage of the peptide-based PEGylOx release is its high elution efficiency and speed (30 min together with peptide removal and buffer exchange). However, we have observed PEGylOx adduction in some of the protein complexes that we have characterized (e.g., the exosome assembly shown in Figure S-7), indicating that even low residual amounts of the nonvolatile peptide can cause heterogeneity and signal attenuation during native MS analysis.
Considering protease elution, an extensive library of TAP-tagged yeast strains with TEV cleavage sites40 are commercially available. When strains with appropriate cleavage sites are not available, homologous recombination or other DNA-insertion techniques are straightforward to implement. Generally, we do not observe peak series corresponding to the 22-kDa HRV 3C protease or 28-kDaTEV protease in our native MS analyses (Figures 4B, 4C and 5B), indicating that these have been efficiently removed by the post-elution filtration step. However, even if residual protease is left prior to native MS, only minimal interference is expected and the corresponding peak series can still be readily identified and characterized.
CONCLUSION
We have described a robust and efficient workflow for coupling affinity-isolation of endogenous protein complexes with sensitive native MS readout to determine their stoichiometry and elements of intersubunit connectivity. General requirements for our protocol to work are the availability of appropriately tagged strains as well as the ability to affinity isolate the complex of interest and to stabilize these in native MS compatible buffers. Compared to previously published protocols (Table S-1), there are several noteworthy features in our workflow that address one of the key limiting factors of native MS—i.e., obtaining protein complexes in sufficiently high concentration for effective native MS detection. First, we use flash-freezing and cryolysis of cells to preserve endogenous protein-protein interactions within the native cellular milieu and minimize proteolytic damage. This freezing-cryolysis step separates the preparation of cellular material from subsequent downstream steps, allows flexibility and control of the scale and timing of the affinity-isolation step, and maximizes extraction efficiency of the desired protein assemblies. Second, we employ single-step affinity capture using antibody-conjugated magnetic beads, which facilitates rapid and efficient isolation of protein complexes as well as subsequent nondenaturing elution into small volumes so as to maintain relatively high sample concentrations. The resulting high efficiency of capture and elution enables us to use modest amounts of starting material to gain access to endogenous protein complexes that are expressed at medium to low abundance. Third, we found that adding Tween-20 at concentrations of 0.001%–0.01% prevents adsorptive losses during the elution reagent removal, buffer exchange steps, and presumably during sample loading in the nanospray capillaries, without significant signal interference during native MS analysis. Fourth, the use of the Exactive Plus EMR enabled sensitive native MS analysis. The high precision and mass accuracy of the mass measurements (Table S-3) were consistent with the good desolvation efficiency, high resolving power and minimal peak interferences observed. The tuning of the voltage offsets on the transport multipoles and ion lenses enabled mass filtering of the incoming ions, particularly for low-mass contaminants such as Tween-20 and residual PEGylOx. Finally, the rapid protocol and streamlined sample handling steps from resuspension of frozen cell powder to native MS analysis minimize both the time that the complexes spend out of their native environment and sample losses. Overall, the time required to go from frozen grindate to native MS-ready samples is 2 h for the PEGylOx-based elution and 3 h for the protease-based elution.
We envision that our overall workflow should also be applicable to other systems that use a competitive peptide or protease for nondenaturing elution (e.g., 59). We anticipate that this facile workflow will enable routine and widespread adoption of native MS for characterization of affinity-captured endogenous protein assemblies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants P41 GM109824, P41 GM103314, and R01 GM112108. We gratefully acknowledge Zachary Quinkert for sharing his expertise in native MS and for insightful discussions. We thank Dr. Roman Subbotin for providing the Psf2-SpA grindate and preliminary bottom-up MS analysis, and Zhanna Hakhverdyan for providing the Csl4-TAP cell powder. We also thank Dr. John LaCava for assistance and for providing the PEGylOx reagent.
Footnotes
Additional experimental details, supplemental figures and tables noted in the text. This material is available free of charge at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hartwell LH, Hopfield JJ, Leibler S, Murray AW. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- (2).Gavin A-C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier M-A, Hoffman V, Hoefert C, Klein K, Hudak M, Michon A-M, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- (3).Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrín-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MHY, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- (4).Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- (5).Robinson CV, Sali A, Baumeister W. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- (6).Alber F, Förster F, Korkin D, Topf M, Sali A. Annu. Rev. Biochem. 2008;77:443–477. doi: 10.1146/annurev.biochem.77.060407.135530. [DOI] [PubMed] [Google Scholar]
- (7).Politis A, Stengel F, Hall Z, Hernández H, Leitner A, Walzthoeni T, Robinson CV, Aebersold R. Nat. Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Nat. Methods. 2005;2:587–589. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- (9).Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J. FEBS J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- (10).Loo JA. Mass Spectrom. Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- (11).Heck AJR. Nat. Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- (12).Snijder J, Heck AJR. Annu. Rev. Anal. Chem. 2014;7:43–64. doi: 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- (13).Pu S, Wong J, Turner B, Cho E, Wodak SJ. Nucleic Acids Res. 2009;37:825–831. doi: 10.1093/nar/gkn1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hanson CL, Videler H, Santos C, Ballesta JPG, Robinson CV. J. Biol. Chem. 2004;279:42750–42757. doi: 10.1074/jbc.M405718200. [DOI] [PubMed] [Google Scholar]
- (15).Hernández H, Dziembowski A, Taverner T, Séraphin B, Robinson CV. EMBO Rep. 2006;7:605–610. doi: 10.1038/sj.embor.7400702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Synowsky SA, van den Heuvel RHH, Mohammed S, Pijnappel PWWM, Heck AJR. Mol. Cell. Proteomics. 2006;5:1581–1592. doi: 10.1074/mcp.M600043-MCP200. [DOI] [PubMed] [Google Scholar]
- (17).Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. PLoS Biol. 2006;4:1314–1323. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lorenzen K, Vannini A, Cramer P, Heck AJR. Structure. 2007;15:1237–1245. doi: 10.1016/j.str.2007.07.016. [DOI] [PubMed] [Google Scholar]
- (19).Synowsky SA, Heck AJR. Protein Sci. 2008;17:119–125. doi: 10.1110/ps.073155908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhou M, Sandercock AM, Fraser CS, Ridlova G, Stephens E, Schenauer MR, Yokoi-Fong T, Barsky D, Leary JA, Hershey JW, Doudna JA, Robinson CV. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18139–18144. doi: 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Synowsky SA, van Wijk M, Raijmakers R, Heck AJR. J. Mol. Biol. 2009;385:1300–1313. doi: 10.1016/j.jmb.2008.11.011. [DOI] [PubMed] [Google Scholar]
- (22).Geiger SR, Lorenzen K, Schreieck A, Hanecker P, Kostrewa D, Heck AJR, Cramer P. Mol. Cell. 2010;39:583–594. doi: 10.1016/j.molcel.2010.07.028. [DOI] [PubMed] [Google Scholar]
- (23).Lane LA, Fernández-Tornero C, Zhou M, Morgner N, Ptchelkine D, Steuerwald U, Politis A, Lindner D, Gvozdenovic J, Gavin AC, Müller CW, Robinson CV. Structure. 2011;19:90–100. doi: 10.1016/j.str.2010.11.009. [DOI] [PubMed] [Google Scholar]
- (24).Sakata E, Stengel F, Fukunaga K, Zhou M, Saeki Y, Förster F, Baumeister W, Tanaka K, Robinson CV. Mol. Cell. 2011;42:637–649. doi: 10.1016/j.molcel.2011.04.021. [DOI] [PubMed] [Google Scholar]
- (25).Politis A, Schmidt C, Tjioe E, Sandercock AM, Lasker K, Gordiyenko Y, Russel D, Sali A, Robinson CV. Chem. Biol. 2015;22:117–128. doi: 10.1016/j.chembiol.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hernández H, Robinson CV. Nat. Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- (27).Oeffinger M, Wei KE, Rogers R, DeGrasse J. a, Chait BT, Aitchison JD, Rout MP. Nat. Methods. 2007;4:951–956. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- (28).Cristea IM, Williams R, Chait BT, Rout MP. Mol. Cell. Proteomics. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- (29).LaCava J, Chandramouli N, Jiang H, Rout MP. Biotechniques. 2013;54:213–216. doi: 10.2144/000114012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJR. Nat. Methods. 2012;9:2–6. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- (31).LaCava J, Molloy KR, Taylor MS, Domanski M, Chait BT, Rout MP. Biotechniques. 2015;58:103–119. doi: 10.2144/000114262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hakhverdyan Z, Domanski M, Hough LE, Oroskar AA, Oroskar AR, Keegan S, Dilworth DJ, Molloy KR, Sherman V, Aitchison JD, Fenyö D, Chait BT, Jensen TH, Rout MP, LaCava J. Nat. Methods. 2015;12:553–560. doi: 10.1038/nmeth.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Strambio-De-Castillia C, Tetenbaum-Novatt J, Imai BS, Chait BT, Rout MP. J. Proteome Res. 2005;4:2250–2256. doi: 10.1021/pr0501517. [DOI] [PubMed] [Google Scholar]
- (34).Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT. Mol. Cell. Proteomics. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velazquez-Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, Rout MP. J. Cell Biol. 2012;196:419–434. doi: 10.1083/jcb.201109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Verkerk UH, Kebarle P. J. Am. Soc. Mass Spectrom. 2005;16:1325–1341. doi: 10.1016/j.jasms.2005.03.018. [DOI] [PubMed] [Google Scholar]
- (37).Loo RR, Dales N, Andrews PC. Protein Sci. 1994;3:1975–1983. doi: 10.1002/pro.5560031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).DeLano WL, Ultsch MH, de Vos AM, Wells JA. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- (39).Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, Araki H. Genes Dev. 2003;17:1153–1165. doi: 10.1101/gad.1065903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- (41).Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Nat. Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- (42).Moks T, Abrahmsén L, Nilsson B, Hellman U, Sjöquist J, Uhlén M. Eur. J. Biochem. 1986;156:637–643. doi: 10.1111/j.1432-1033.1986.tb09625.x. [DOI] [PubMed] [Google Scholar]
- (43).Sekedat MD, Fenyö D, Rogers RS, Tackett AJ, Aitchison JD, Chait BT. Mol. Syst. Biol. 2010;6:1–10. doi: 10.1038/msb.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kanemaki M, Sanchez-Diaz A, Gambus A, Labib K. Nature. 2003;423:720–724. doi: 10.1038/nature01692. [DOI] [PubMed] [Google Scholar]
- (45).Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, Labib K. EMBO J. 2009;28:2992–3004. doi: 10.1038/emboj.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinković D, Labib K, Costa A, Pellegrini L. Nature. 2014;510:293–297. doi: 10.1038/nature13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jurchen JC, Williams ER. J. Am. Chem. Soc. 2003;12:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Chem. Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- (49).MacNeill SA. Biochem. J. 2010;425:489–500. doi: 10.1042/BJ20091531. [DOI] [PubMed] [Google Scholar]
- (50).Boskovic J, Coloma J, Aparicio T, Zhou M, Robinson CV, Méndez J, Montoya G. EMBO Rep. 2007;8:678–684. doi: 10.1038/sj.embor.7401002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cordingley MG, Register RB, Callahan PL, Garsky VM, Colonno RJ. J. Virol. 1989;63:5037–5045. doi: 10.1128/jvi.63.12.5037-5045.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT. J. Cell Biol. 2000;148:635–652. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- (54).Snijder J, Rose RJ, Veesler D, Johnson JE, Heck AJR. Angew. Chemie - Int. Ed. 2013;52:4020–4023. doi: 10.1002/anie.201210197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- (56).Kapust RB, Tözsér J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- (57).Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- (58).Liu Q, Greimann JC, Lima CD. Cell. 2006;127:1223–1237. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- (59).Domanski M, Molloy K, Jiang H, Chait BT, Rout MP, Jensen TH, LaCava J. Biotechniques. 2012:1–6. doi: 10.2144/000113864. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.