

Abstract
Objective
To investigate whether cholesterol efflux to high-density lipoprotein (HDL) via ATP-binding cassette transporter G1 (ABCG1) modulates the interaction of caveolin (Cav) 1 and endothelial NO synthase (eNOS).
Methods and Results
ABCG1 promotes cholesterol and 7-oxysterol efflux from endothelial cells (ECs) to HDL. It was previously reported that ABCG1 protects against dietary cholesterol-induced endothelial dysfunction by promoting the efflux of 7-oxysterols to HDL. Increased cholesterol loading in ECs is known to cause an inhibitory interaction between Cav-1 and eNOS and impaired NO release. In human aortic ECs, free cholesterol loading promoted the interaction of Cav-1 with eNOS, reducing eNOS activity. These effects of cholesterol loading were reversed by HDL in an ABCG1-dependent manner. HDL also reversed the inhibition of eNOS by cholesterol loading in murine lung ECs, but this effect of HDL was abolished in Cav-1–deficient murine lung ECs. Increased interaction of Cav-1 with eNOS was also detected in aortic homogenates of high-cholesterol diet–fed Abcg1−/− mice, paralleling a decrease in eNOS activity and impaired endothelial function.
Conclusion
The promotion of cholesterol efflux via ABCG1 results in a reduced inhibitory interaction of eNOS with Cav-1.
Keywords: ABCG1, caveolin-1, cholesterol, high density lipoprotein, nitric oxide synthase
Endothelial dysfunction is a key feature of early atherosclerotic lesions in both human and animal models.1–3 Plasma high-density lipoprotein (HDL) cholesterol levels are inversely related to the incidence of atherothrombotic disease.4,5 A part of the atheroprotective effect of HDL may be related to its role in preserving endothelial function.6,7 In humans, HDL levels are correlated with flow-mediated vasodilation responses of the brachial artery6,7 and with decreased coronary vasoconstrictor responses.8,9 HDL may have a specific role in reversing decreased endothelial NO synthase (eNOS) activity in human endothelial cells (ECs) treated with oxidized low-density lipoprotein (LDL)10 or in reversing the decrease in eNOS-dependent vascular relaxation induced by high-cholesterol diets (HCDs).11,12
Two ATP-binding cassette transporters (ABCA1 and ABCG1) have a major role in inducing cellular cholesterol efflux13–15 and are known to be expressed in ECs.12,16 ABCA1 mediates cholesterol efflux to lipid-poor apolipoprotein (apo) A-1 but only modestly increases cholesterol efflux to HDL.15,17,18 In contrast, ABCG1 promotes macrophage cholesterol efflux to HDL but not to lipid-poor apoA-1.15,19–21 ABCG1 has a specific role in promoting the efflux of 7-oxysterols,22 a dietary oxysterol that is detected at high levels in human atherosclerotic plaques and is abundant in oxidized LDL.23 HDL treatment of human ECs prevents 7-oxysterol–induced reactive oxygen species production.12 Furthermore, ABCG1 and HDL reversed a decreased level of the active dimeric form of eNOS by promoting the efflux of 7-oxysterols, resulting in protection against endothelial dysfunction in animals fed HCDs.12
Specific interactions between the cholesterol-binding protein caveolin (Cav) 1 and signal-transducing proteins, including eNOS and various kinases, are known to repress the catalytic activity of these enzymes.24,25 Several studies showed that the increase in Cav-1 abundance induced by LDL cholesterol promotes its inhibitory interaction with eNOS, resulting in decreased NO production.26 Cholesterol/oxysterol-mediated impairment of NO production may be involved in the pathogenesis of endothelial dysfunction induced by hypercholesterolemia and dietary sterols through a variety of mechanisms, including decreased eNOS expression, reduced eNOS dimer levels, or reduced eNOS substrate availability.12,27,28 Indeed, by reducing circulating LDL cholesterol or directly inhibiting cholesterol synthesis in ECs, statins can reverse endothelial dysfunction. This may involve decreased Cav-1 expression and promotion of NO release through destabilization of the inhibitory Cav-1/eNOS interaction.29 Endothelial Cav-1 also plays an important role in modulating eNOS activity in vivo and accelerates atherogenesis in apoe−/−cav-1−/− mice.30 Although ABCG1 has a major role in inducing cholesterol efflux in ECs,12 its role in reversing the inhibitory Cav-1/eNOS interaction has not been explored. The present study was undertaken to test the hypothesis that ABCG1, by promoting efflux of cholesterol from ECs, modulates the interaction of eNOS with Cav-1.
Methods
Materials
eNOS antibody was purchased from BD Transduction (Lexington, Ky); Cav-1 antibody from Cell Signaling (Danvers, Mass); and ABCG1 and scavenger receptor BI antibodies from Santa Cruz (Santa Cruz, Calif). β-Actin antibody, methyl-β-cyclodextrin, Ca2+ ionophore (A23187), cholesterol, 7-ketocholesterol, 7β-hydroxycholesterol, and 25-hydroxycholesterol were purchased from Sigma-Aldrich (St. Louis, Minn); and 27-Hydroxycholesterol from Steraloids (Wilton, NH). HDL (density, 1.06–1.21 g/mL) was isolated by preparative ultracentrifugation, as previously described.12 Methyl-β-cyclodextrin cholesterol (molar ratio, 2.5:1) was prepared as previously described.31
Cell Culture and Transfection
Human aortic ECs (HAECs)12 and murine lung ECs (MLECs)32 were cultured as previously described. All small and interfering RNAs (siRNAs) were purchased from Invitrogen (Carlsbad, Calif) and Santa Cruz (Santa Cruz, Calif). The HAECs or MLECs were transfected with siRNA using Lipofectamine RNAiMAX reagent from Invitrogen, as previously described.12
Immunoprecipitation and Immunoblotting
Mouse aortas were obtained from wild-type, Abcg1−/−, Abca1−/−, and Abca1−/−Abcg1−/− mice as previously described.12 Mouse aortas and ECs were homogenized in an octyl glucopyranoside–containing buffer (60-mmol/L octyl glucopyranoside, 50-mmol/L Tris-chloride [pH, 7.4], 150-mmol/L NaCl, 1-mmol/L EDTA, 1-mmol/L sodium orthovanadate [Na3VO4], and protease inhibitor cocktail from Roche [Indianapolis, Ind]). Homogenates were incubated with Cav-1 polyclonal antibody. After 16 hours at 4°C, magnetic beads coupled with protein G (Dynabeads Protein G from Invitrogen Dynal AS, Oslo, Norway) were added to the supernatant for a further 1-hour incubation at 4°C. Bound immune complexes were captured by the magnets (DynaMag-Spin; Invitrogen) and washed with octyl glucopyranoside buffer. The immunoprecipitates were eluted in Laemmli sample buffer from Biorad (Hercules, Calif).
Sodium Carbonate Extraction Followed by Sucrose Gradient Fractionation
A detergent-free method was used, as previously described.33,34 Briefly, the cell homogenates prepared in hypotonic buffer (50-mmol/L sodium carbonate [Na2CO3]; pH, 11) were adjusted to 42.5% sucrose with 85% sucrose in 2-[morpholino] ethanesulfonic acid (Mes)-buffered saline (25-mmol/L Mes [pH, 6.5] or 150-mmol/L NaCl) and placed at the bottom of an ultracentrifuge tube. A 5% to 30% discontinuous sucrose gradient was formed and ultracentrifuged at 35 000 rpm for 16 hours in an SW40 rotor (Beckman, Palo Alto, Calif). Gradient fractions, 1 mL, were collected from the top of the tube to yield 12 fractions; and each fraction (fractions 1–12) was used for Western blot and cholesterol mass analyses.
Western Blotting and NOS Activity Assay
Aortic and cell lysates and the fractions obtained by sucrose gradient fractionation were processed for Western blotting, as previously described.12
The NO synthesizing activity was determined by quantifying the rate of the conversion of [3H]l-arginine to [3H]l-citrulline with commercially available kits (Calbiochem-Novabiochem, EMD Biosciences, La Jolla, Calif), as previously described.12,35
Statistical analysis was performed using the t test. Bonferroni posttests were also used. Results are represented as mean±SEM.
Results
Effects of Cholesterol and Oxysterols on Cav-1/eNOS Interaction in HAECs
A specific role of ABCG1 in the efflux of 7-oxysterols was previously reported22; also, 7-oxysterols, but not cholesterol, can induce eNOS dimer disruption.12 To further evaluate the roles of HDL and ABCG1 on eNOS activity, we tested the effects of different oxysterols (ie, 7-ketocholesterol, 7α-hydroxycholesterol, 7β-hydroxycholesterol, 25-hydroxycholesterol, 27-hydroxycholesterol, and cholesterol [each 40 μg/mL, except for 7-ketocholesterol and 7β-hydroxycholesterol, which were 10 μg/mL]) on eNOS activity in HAECs. Cholesterol and all tested oxysterols significantly decreased eNOS activity (Figure 1A, open bars). HDL treatment prevented the reduction of eNOS activity induced by cholesterol/oxysterols (Figure 1A, filled bars). The data suggest that the efflux of cholesterol and oxysterols have an important role in maintaining eNOS activity, although cholesterol loading did not affect eNOS dimer or phosphorylated eNOS levels.12
Figure 1.
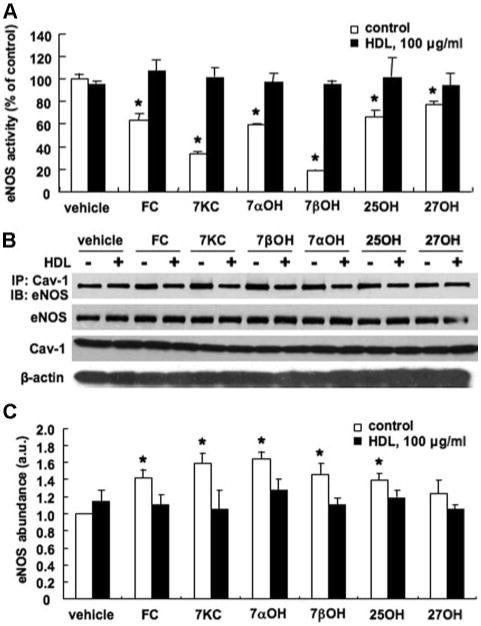
Effects of cholesterol and different oxysterols on NOS activity and Cav-1/eNOS interaction in HAECs. The HAECs were incubated with cholesterol or different oxysterols (each 40 μg/mL, except for 7-ketocholesterol and 7β-hydroxycholesterol [10 μg/mL]) in the absence (control: open bars) or presence (HDL: filled bars) of HDL, 100 μg/mL, for 16 hours. Vehicle indicates no preincubation with cholesterol/oxysterols; FC, cholesterol; 7KC, 7-ketocholesterol; 7αOH, 7α-hydroxycholesterol; 7βOH, 7β-hydroxycholesterol; 25OH, 25-hydroxycholesterol; and 27OH, 27-hydroxycholesterol. A, eNOS activity. B, Western blot analysis of amount of eNOS coimmunoprecipitated (IP) with Cav-1, total eNOS, Cav-1, and β-actin. C, Quantification of eNOS IP with Cav-1. AU indicates arbitrary unit. The results are represented as mean±SEM of 3 individual experiments. *P<0.05 vs. vehicle control.
Previous studies have shown that high LDL cholesterol levels promote an inhibitory interaction of eNOS with Cav-1, resulting in a decrease in NO production.26 Therefore, by reducing cholesterol and oxysterols in ECs, HDL might also reverse endothelial dysfunction by promoting cholesterol and oxysterol efflux and consequently increasing NO release through the destabilization of the inhibitory Cav-1/eNOS complex. To test this hypothesis, we incubated HAECs with cholesterol/oxysterols and HDL. The data demonstrated that cholesterol and oxysterols increased Cav-1/eNOS interaction (Figure 1B and C, open bars). HDL treatment reversed cholesterol/oxysterol-induced Cav-1/eNOS interaction (Figure 1B, filled bars). Total eNOS and Cav-1 levels were not affected by either cholesterol or oxysterols (Figure 1B).
Effects of HDL and ABCG1 on Cav-1/eNOS Interaction in HAECs
Next, we examined the effects of different concentrations of HDL on increased Cav-1/eNOS interaction and reduced eNOS activity by cholesterol loading in HAECs. HDL treatment reduced both the increase in Cav-1/eNOS interaction (Figure 2A) and the reduction in eNOS activity (Figure 2B) in a similar concentration-dependent manner (between 25 and 100 μg/mL). HDL treatment did not affect total eNOS or Cav-1 levels (Figure 2A). The incubation of HAECs with cholesterol and HDL simultaneously similarly reversed the Cav-1/eNOS interaction as sequential incubation with cholesterol and HDL (supplemental Figure I; available online at http://atvb.ahajournals.org).
Figure 2.
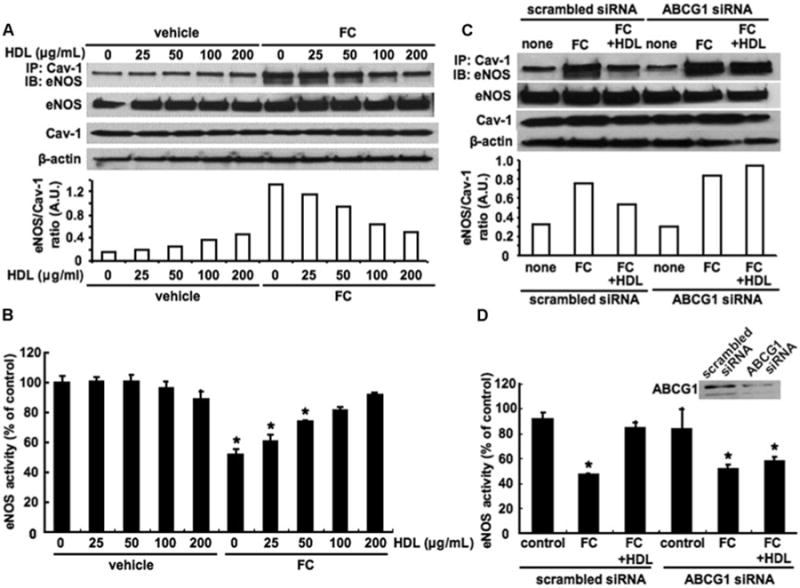
Effects of HDL and ABCG1 expression on eNOS activity and Cav-1/eNOS interaction in HAECs. A and B, The HAECs were incubated with cholesterol, 40 μg/mL, and HDL, 0 to 200 μg/mL, for 16 hours. C and D, The HAECs were transfected with scrambled or ABCG1 siRNA. Forty-eight hours after transfection, HAECs were incubated with cholesterol, 40 μg/mL, in the presence or absence of HDL, 100 μg/mL, for 16 hours. A and C, Western blot analysis of amount of eNOS coimmunoprecipitated (IP) with Cav-1, total eNOS, Cav-1, and β-actin. The ratio of eNOS in the caveolin-1 immunoprecipitate to total caveolin-1 is quantified. A.U. indicates arbitrary unit. B and D, eNOS activity. The results are represented as mean±SEM of 3 individual experiments. *P<0.05 vs vehicle control.
To assess the role of ABCG1 in the ability of HDL to reduce Cav-1/eNOS interaction, we knocked down the expression of ABCG1 by siRNA. ABCG1 knockdown abolished the ability of HDL to reduce cholesterol-mediated Cav-1/eNOS interaction (Figure 2C) and the corresponding increase in eNOS activity (Figure 2D).
Effects of Cav-1 on eNOS Activity in MLECs
Next, we examined whether Cav-1 deficiency would abolish the sterol-mediated decrease in eNOS activity and the ability of HDL to activate eNOS. We used wild-type and cav-1−/− MLECs for these experiments because we could not achieve a greater than 50% Cav-1 knockdown in HAECs using several different Cav-1 siRNAs. Under basal conditions, we found that Cav-1 deficiency induced eNOS activity by approximately 100% compared with the controls (data not shown). Fernández-Hernando et al30 found a similar increase in eNOS activity in apoe−/− cav-1−/− MLECs compared with apoe−/− MLECs. Because the eNOS activity in wild-type MLECs was relatively low under basal conditions, we added the Ca2+ ionophore (A23187) that mediates the dissociation of eNOS from Cav-1 to induce eNOS activity.26 Therefore, the Ca2+ ionophore only increased eNOS activity in wild-type MLECs. In wild-type MLECs, cholesterol loading reduced eNOS activity, and this was completely reversed by HDL (Figure 3), similar to our observations in HAECs. In cav-1−/− MLECs, eNOS activity was not reduced by cholesterol loading and HDL had no ability to increase eNOS activity in cholesterol-loaded cav-1−/− MLECs (Figure 3). Thus, Cav-1 deficiency abolished the ability of HDL to increase eNOS activity.
Figure 3.
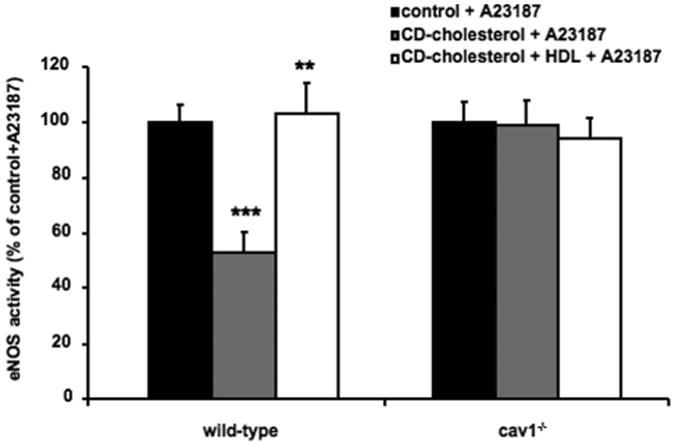
Effect of Cav-1 on cholesterol- and HDL-mediated modulation of eNOS activity in MLECs. Wild-type and cav1−/− MLECs were incubated without (control) and with methyl-β-cyclodextrin (CD) cholesterol or with CD cholesterol (both 2-mmol/L cholesterol) and HDL, 100 μg/mL, for 3 hours. Then, cells were incubated with the Ca2+ ionophore (A23187) for 3 minutes and lysed; and eNOS activity was assessed on the lysate. *P<0.05 vs the wild-type control, and **P<0.05 vs the wild-type CD cholesterol.
eNOS, Cav-1, and ABCG1 Plasmalemmal Distribution in HAECs
Next, we examined the roles of HDL and ABCG1 on eNOS, Cav-1, and cholesterol plasmalemmal distribution in HAECs. eNOS was predominantly found in intermediate buoyant membranes (fractions 6–7), where it colocalized with Cav-1 in HAECs (Figure 4A). Free cholesterol (FC) loading caused a shift in eNOS and Cav-1 toward lighter-density fractions (fractions 4–6 and fractions 4–8, respectively), coinciding with FC distribution (Figure 4B). HDL treatment caused a redistribution of eNOS toward more dense fractions and away from Cav-1, especially in cholesterol-loaded cells, and a decrease in cholesterol content in cholesterol-loaded cells (Figure 4C and D). ABCG1 could be seen in heavy-density fractions, especially in cholesterol-loaded cells (fractions 9–12) and did not colocalize with eNOS or Cav-1 (Figure 4B). The distribution of ABCG1 and β-actin were not affected by either FC loading or HDL. To assess the role of ABCG1 in Cav-1/eNOS redistribution by HDL, we knocked down the expression of ABCG1 by siRNA (Figure 4E and F). In ABCG1 siRNA-transfected FC-loaded HAECs, the ability of HDL to cause redistribution of eNOS into more dense fractions and away from Cav-1 was markedly attenuated (Figure 4F). Methyl-β-cyclodextrin treatment, which removes plasma membrane cholesterol from raft/caveolae,36 dramatically reduced cholesterol levels and caused a marked shift in eNOS and Cav-1 distribution into heavy-density fractions (fractions 8–12) (Figure 4G). These experiments suggest that HDL tends to dissociate Cav-1 and eNOS in the plasmalemma under FC-loading conditions only in the presence of ABCG1. Most likely, ABCG1-mediated cholesterol efflux to HDL explains these findings. However, ABCG1 does not colocalize with Cav-1 in gradient fractions.
Figure 4.
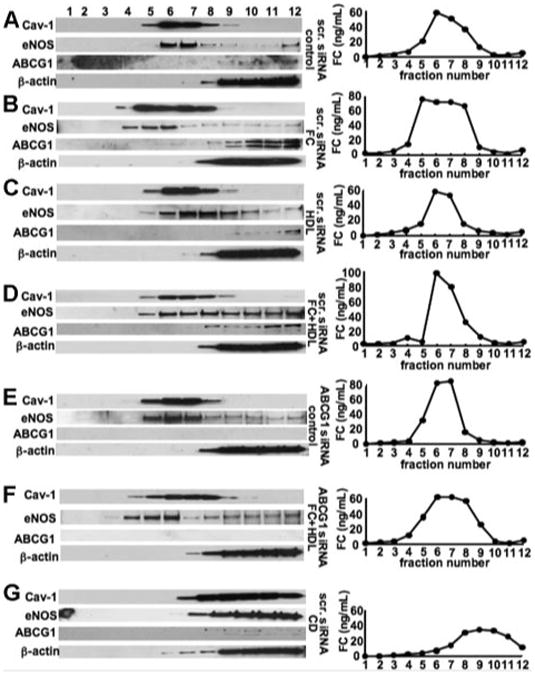
Cav-1, eNOS, ABCG1, -actin, and cholesterol plasmalemmal distribution in HAECs. A–D, The HAECs were transfected with scrambled siRNA (scr. siRNA). Forty-eight hours after transfection, HAECs were incubated with medium alone (A); cholesterol, 40 g/mL (B); HDL, 100 g/mL (C); or cholesterol + HDL (D) for 16 hours. E and F. The HAECs were transfected with ABCG1 siRNA. Forty-eight hours after transfection, HAECs were incubated with medium alone (E), or cholesterol, 40 g/mL, and HDL, 100 g/mL (F). G, The HAECs were transfected with scrambled siRNA. Forty-eight hours after transfection, HAECs were incubated with 10-mmol/L methyl-β-cyclodextrin (scr. siRNA, methyl-β-cyclodextrin [CD]) for 1 hour. Western blotting for Cav-1, eNOS, ABCG1, and β-actin (left) and cholesterol concentrations (right) from sucrose-gradient fractions isolated from HAECs was performed as described. The results are represented from 1 experiment performed independently 3 times. **P<0.01 vs the wild-type CD cholesterol; ***P<0.001 vs the wild type control.
eNOS, Cav-1, and ABCG1 Plasmalemmal Distribution in MLECs
We also examined the plasmalemmal distribution of ABCG1 in MLECs. In wild-type MLECs, Cav-1 was distributed into light buoyant membranes (fractions 1–4) and heavy membranes (fractions 8–12) (Figure 5A). eNOS was dominantly distributed into heavy membranes (fractions 8–12), and a small amount of eNOS could be seen distributed into light buoyant membranes (fraction 3) (Figure 5A). ABCG1 was found only in heavy-density fractions (fractions 9–12) (Figure 5A). In cav-1−/− MLECs, the distributions of ABCG1 and eNOS were unchanged compared with wild-type cells (Figure 5B).
Figure 5.
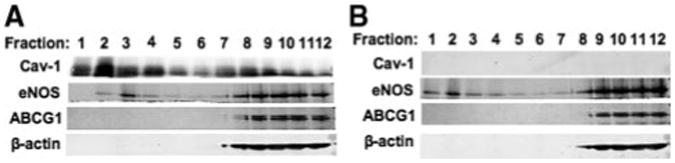
Cav-1, eNOS, ABCG1, and β-actin plasmalemmal distribution in MLECs. A and B, Western blot for Cav-1, eNOS, ABCG1, and β-actin from sucrose-gradient fractions isolated from wild-type MLECs (A) or Cav-1−/− MLECs (B) as described. The results are representative of 1 experiment performed 3 times.
Effect of ABCG1 on eNOS and Cav-1 Interaction in Mouse Aorta
It was recently documented that Abcg1−/− mice fed either an HCD or a Western-type diet exhibited a marked decrease in endothelium-dependent vasorelaxation and that this correlated with decreased levels of the active dimeric form of eNOS.12 To further evaluate the role of ABCG1 in endothelial function, we investigated the impact of ABC transporter deficiency on eNOS and Cav-1 interaction, using samples from a previously published study.12
In response to the HCD, eNOS/Cav-1 interaction was dramatically increased (Figure 6, left). Total Cav-1 levels were increased by the HCD (Figure 6, left). Using the same samples, we observed in our previous study that total eNOS levels were slightly (approximately 28%) decreased in aortas from Abcg1−/− mice on the HCD.12 Together with the data presented herein, this indicates that the ratio of Cav-1–bound eNOS, after correction for 28% decreased total eNOS, to total Cav-1 was increased in Abcg1−/− mice (Figure 6), paralleling the decreased eNOS activity in a previous study.12 For the Western-type diet, Abca1−/−, Abcg1−/−, and Abca1−/− Abcg1−/− mice exhibited increased Cav-1–bound eNOS levels, whereas total Cav-1 levels were similar (Figure 6, right). Also, eNOS levels were similar as assessed in the same samples in a previous study.12 The increase in Cav-1–bound eNOS levels was most prominent in Abcg1−/− and Abca1−/− Abcg1−/− mice (Figure 6, right), closely paralleling the decrease in eNOS activity previously found in arterial rings from these mice.12
Figure 6.
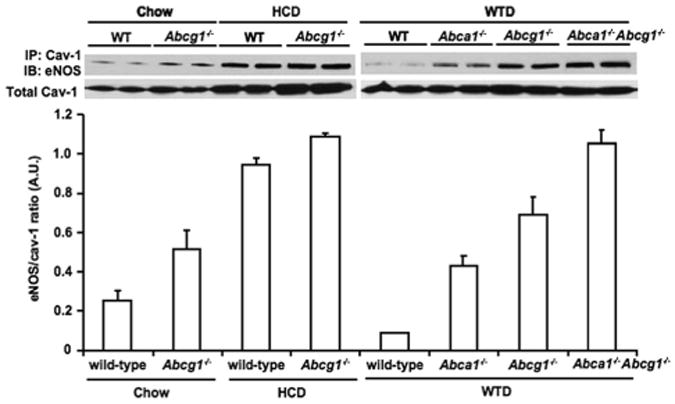
Effects of ABC transporter deficiency and different diets on Cav-1/eNOS interaction in mouse aorta. Western blot analysis on amount of eNOS coimmunoprecipitated (IP) with Cav-1 (top) and Cav-1 in the total lysate (bottom). The HCD includes 1.25% cholesterol, 7.5% cocoa butter, and 0.5% sodium cholate; and the Western-type diet (WTD), 0.25% cholesterol and 21% milk fat. Chow indicates chow diet. Mice (n=4–5 in each group) were fed either diet for 11 to 12 weeks.12 The immunoprecipations and Western blots were performed on 2 pools per mouse per genotype and represent 1 experiment that was performed 3 times. The ratio of eNOS in the caveolin-1 immunoprecipitate, after correction for total eNOS, to total caveolin-1 is quantified. A.U. indicates arbitrary unit; WT, wild type.
Discussion
A principal antiatherogenic property of HDL is thought to be its ability to promote cholesterol efflux from macrophages; recent studies have highlighted the key roles of ABCA1 and ABCG1 in reversing macrophage foam cell formation37 and atherosclerosis.38,39 HDL has also exerted a variety of beneficial actions that are independent of macrophage cholesterol efflux. For example, HDL inhibits LDL oxidation, smooth muscle cell migration, and platelet aggregation; it also reverses endothelial dysfunction.40–42 It was recently shown that ABCG1 and, to a lesser extent, ABCA1 help to preserve endothelial eNOS activity in mice fed HCDs.12 The ability of ABCG1 to preserve endothelial function appeared to be at least partly related to its role in promoting the efflux of 7-oxysterols,12 which are detected at high levels in human atherosclerotic plaques and are abundant in oxidized LDL.23 This effect was associated with decreased reactive oxygen species formation and preservation of the active dimeric form of eNOS.12 To further understand the beneficial effects of HDL and ABCG1 on endothelial function, the present study was undertaken to test the hypothesis that ABCG1, by promoting efflux of cholesterol and oxysterols from ECs, also modulates the inhibitory interaction of eNOS with Cav-1. We have shown that ABCG1 and HDL can reverse the Cav-1/eNOS interaction induced by cholesterol loading, resulting in increased eNOS activity.
Although 7-oxysterols have a prominent role in producing endothelial dysfunction, cholesterol accumulation also inhibits endothelium-dependent vasorelaxation.11,12,43 Indeed, both FC and 7-oxysterols could increase the inhibitory interaction of eNOS with Cav-1 and could cause impaired NOS activity (Figure 1), whereas only treatment with 7-oxysterols led to eNOS dimer disruption.12 It was recently reported that ABCG1 is highly expressed in the endothelium of nonatherosclerotic aorta and showed accumulation of cholesterol and 7-oxysterols in ECs of high cholesterol diet–fed mice.12 Figure 6 shows that the inhibitory interaction of Cav-1 with eNOS was increased in aortic homogenates of HCD or Western-type diet–fed Abcg1 −/− mice, paralleling endothelial dysfunction.12 Similar to some previous studies, we found an increase in Cav-1 levels in response to the HCD (Figure 6); however, we did not find such an effect in cholesterol-loaded HAECs. Thus, multiple different mechanisms can lead to the formation of inhibitory Cav-1/eNOS complex formation.
ABCG1 is highly expressed in HAECs,16 and ABCG1 and HDL had a major role in promoting the efflux of cholesterol and 7-oxysterols from HAECs.12 The expression of other proteins mediating cholesterol efflux, such as ABCA1 and the scavenger receptor BI, was low or downregulated by cholesterol treatment, respectively.8 We found low levels of ABCA1-mediated efflux to lipid-free apoAI- or scavenger receptor BI–mediated cholesterol efflux to HDL in HAECs.16 In this study, FC loading caused a shift in eNOS distribution into lighter-density fractions coinciding with FC distribution; this effect was reversed by HDL treatment in an ABCG1-dependent manner (Figure 4). ABCG1 distribution was distinct from Cav-1 (Figures 4 and 5), and cholesterol efflux to HDL via ABCG1 was not affected by Cav-1 expression (supplemental Figure II and supplemental Figure III). In addition, the distribution of ABCG1 was unaffected by FC loading. Taken together, our data suggest that the promotion of cholesterol efflux by ABCG1 leads to a redistribution of cholesterol from caveolae to noncaveolae domains, where ABCG1 localizes, consequently reducing caveolae cholesterol and reversing the inhibitory eNOS/Cav-1 interaction.
LDL cholesterol loading of ECs is known to increase the Cav-1/eNOS interaction, a process that was unaffected by treatment with antioxidants26; whereas eNOS dimer disruption, induced by 7-oxysterols, was reversed by antioxidants.12 Reducing circulating LDL cholesterol and/or directly inhibiting cholesterol synthesis in ECs by statin could reverse endothelial dysfunction by decreasing inhibitory eNOS/Cav-1 interaction. Interestingly, our study indicates that a similar preservation of endothelial function can be exerted by HDL by promoting cholesterol or 7-oxysterol efflux via ABCG1. A number of different mechanisms have accounted for the ability of HDL to preserve eNOS activity,12,27,28 and further studies will be required to determine their relative importance.
In humans, HDL levels are correlated with flow-mediated vasodilation responses of the brachial artery6,7 and with decreased coronary vasoconstrictor responses.8 The infusion of recombinant phospholipid/apoA-1 particles into Tangier disease heterozygotes with isolated low HDL levels reversed defective forearm blood flow measurements.9 Therapies that increase HDL levels, such as niacin and cholesteryl ester transfer protein inhibitors, are probably activating the ABCG1-mediated cholesterol/oxysterol efflux pathway in ECs and macrophages. More important, niacin therapy has improved NO-mediated vascular relaxation in humans.44 Our studies highlight the therapeutic potential of increasing HDL to correct endothelial dysfunction associated with dietary cholesterol and hypercholesterolemia.
Supplementary Material
Figure I. Effect of simultaneous and consecutive incubation with cholesterol and HDL on cav-1/eNOS interaction in HAECs. HAECs were incubated with cholesterol (40 μg/ml) for 16 h in the presence of increasing concentrations of HDL (0-100 μg/ml) (left) or were incubated with cholesterol (40 μg/ml) followed by incubation with increasing concentrations of HDL (0-100 μg/ml) (right). An immunoprecipitation using cav-1 antibody was performed and a Western blot for eNOS was carried out on the immunoprecipitate.
Figure II. Effect of caveolin-1 on cholesterol efflux from MLECs. Wild-type (WT; white bars), caveolin-1 knockout (Cav-1 ko; grey bars) and caveolin-1 transgenic (Cav-1 tg; black bars) MLECs were incubated with cholesterol (40 μg/mL) and without (control) or with apoAl (10 μg/mL) or with HDL (100 μg/mL) for 16 h, after which cholesterol mass in the media and cells was measured using gas-chromatography and the % cholesterol efflux was calculated. *P<0.05, control vs HDL.
Figure III. Effect of ABCG1, ABCA1, and SR-BI on cholesterol efflux from MLECs. Wild-type MLECs were transfected with scrambled siRNA, ABCG1 siRNA, ABCA1 siRNA, or SR-BI siRNA, and incubated with cholesterol (40 μg/mL) without or with HDL (100 μg/mL) for 16 h, after which cholesterol mass in the media and cells was measured using gas-chromatography and the % cholesterol efflux was calculated. P<0.05 indicates significant difference between the HDL condition in the scrambled siRNA transfected cells vs the ABCG1 siRNA transfected cells.
Acknowledgments
Sources of Funding: This study was supported by postdoctoral fellowship 09POST2110109 from the American Heart Association (Dr Westerterp) and fellowship WdL/TD/09-036 from the Saal van Zwanenbergstichting, the Netherlands (Dr Koetsveld).
Footnotes
Disclosures: None.
References
- 1.Zeiher AM, Drexler H, Wollschlager H, Just H. Modulation of coronary vasomotor tone in humans: progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation. 1991;83:391–401. doi: 10.1161/01.cir.83.2.391. [DOI] [PubMed] [Google Scholar]
- 2.Bossaller C, Habib GB, Yamamoto H, Williams C, Wells S, Henry PD. Impaired muscarinic endothelium-dependent relaxation and cyclic guanosine 5′-monophosphate formation in atherosclerotic human coronary artery and rabbit aorta. J Clin Invest. 1987;79:170–174. doi: 10.1172/JCI112779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forstermann U, Mtigge A, Alheid U, Haverich A, Frdlich JC. Selective attenuation of endothelium-mediated vasodilation in atherosclerotic human coronary arteries. Circ Res. 1988;62:185–190. doi: 10.1161/01.res.62.2.185. [DOI] [PubMed] [Google Scholar]
- 4.Gordon DJ, Rifkind BM. High-density lipoprotein: the clinical implications of recent studies. N Engl J Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 5.Assmann G, Gotto AM., Jr HDL cholesterol and protective factors in atherosclerosis. Circulation. 2004;109(suppl III):III-8–III-14. doi: 10.1161/01.CIR.0000131512.50667.46. [DOI] [PubMed] [Google Scholar]
- 6.Li XP, Zhao SP, Zhang XY, Liu L, Gao M, Zhou QC. Protective effect of high density lipoprotein on endothelium-dependent vasodilatation. Int J Cardiol. 2000;73:231–236. doi: 10.1016/s0167-5273(00)00221-7. [DOI] [PubMed] [Google Scholar]
- 7.Kuvin JT, Patel AR, Sidhu M, Rand WM, Sliney KA, Pandian NG, Karas RH. Relation between high-density lipoprotein cholesterol and peripheral vasomotor function. Am J Cardiol. 2003;92:275–279. doi: 10.1016/s0002-9149(03)00623-4. [DOI] [PubMed] [Google Scholar]
- 8.Zeiher AM, Schachlinger V, Hohnloser SH, Saurbier B, Just H. Coronary atherosclerotic wall thickening and vascular reactivity in humans: elevated high-density lipoprotein levels ameliorate abnormal vasoconstriction in early atherosclerosis. Circulation. 1994;89:2525–2532. doi: 10.1161/01.cir.89.6.2525. [DOI] [PubMed] [Google Scholar]
- 9.Bisoendial RJ, Hovingh GK, Levels JH, Lerch PG, Andresen I, Hayden MR, Kastelein JJ, Stroes ES. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation. 2003;107:2944–2948. doi: 10.1161/01.CIR.0000070934.69310.1A. [DOI] [PubMed] [Google Scholar]
- 10.Uittenbogaard A, Shaul PW, Yuhanna IS, Blair A, Smart EJ. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem. 2000;275:11278–11283. doi: 10.1074/jbc.275.15.11278. [DOI] [PubMed] [Google Scholar]
- 11.Deckert V, Lizard G, Duverger N, Athias A, Palleau V, Emmanuel F, Moisant M, Gambert P, Lallemant C, Lagrost L. Impairment of endothelium-dependent arterial relaxation by high-fat feeding in ApoE-deficient mice. Circulation. 1999;100:1230–1235. doi: 10.1161/01.cir.100.11.1230. [DOI] [PubMed] [Google Scholar]
- 12.Terasaka N, Yu S, Yvan-Charvet L, Wang N, Mzhavia N, Langlois R, Pagler T, Li R, Welch CL, Goldberg IJ, Tall AR. ABCG1 and HDL against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Invest. 2008;118:3701–3713. doi: 10.1172/JCI35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuura F, Wang N, Chen W, Jiang XC, Tall AR. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J Clin Invest. 2006;116:1435–1442. doi: 10.1172/JCI27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yvan-Charvet L, Matsuura F, Wang N, Bamberger MJ, Nguyen T, Rinninger F, Jiang XC, Shear CL, Tall AR. Inhibition of cholesteryl ester transfer protein by torcetrapib modestly increases macrophage cholesterol efflux to HDL. Arterioscler Thromb Vasc Biol. 2007;27:1132–1138. doi: 10.1161/ATVBAHA.106.138347. [DOI] [PubMed] [Google Scholar]
- 15.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 16.O'Connell BJ, Denis M, Genest J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation. 2004;110:2881–2888. doi: 10.1161/01.CIR.0000146333.20727.2B. [DOI] [PubMed] [Google Scholar]
- 17.Wang N, Silver DL, Costet P, Tall AR. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- 18.Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 19.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem. 2005;280:30150–30157. doi: 10.1074/jbc.M505368200. [DOI] [PubMed] [Google Scholar]
- 22.Terasaka N, Wang N, Yvan-Charvet L, Tall AR. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc Natl Acad Sci U S A. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 24.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 25.Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 26.Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J Clin Invest. 1999;103:897–905. doi: 10.1172/JCI4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest. 1997;100:2153–2157. doi: 10.1172/JCI119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wever R, Stroes E, Rabelink TJ. Nitric oxide and hypercholesterolemia: a matter of oxidation and reduction? Atherosclerosis. 1998;137(suppl):51s–60s. doi: 10.1016/s0021-9150(97)00304-3. [DOI] [PubMed] [Google Scholar]
- 29.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxymethylglutaryl–coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- 30.Fernández-Hernando C, Yu J, Suárez Y, Rahner C, Dávalos A, Lasunción MA, Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10:48–54. doi: 10.1016/j.cmet.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Sun Y, Welch C, Gorelik A, Leventhal AR, Tabas I, Tall AR. Preferential ATP-binding cassette transporter A1-mediated cholesterol efflux from late endosomes/lysosomes. J Biol Chem. 2001;276:43654–43659. doi: 10.1074/jbc.M107938200. [DOI] [PubMed] [Google Scholar]
- 32.Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 34.Sowa G, Pypaert M, Sessa WC. Distinction between signaling mechanism in lipid raft vs. caveolae. Proc Natl Acad Sci U S A. 2001;98:14072–14077. doi: 10.1073/pnas.241409998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothblat GH, de la Lera–Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res. 1999;40:781–796. [PubMed] [Google Scholar]
- 37.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090–3100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connell BJ, Genest J., Jr High-density lipoproteins and endothelial function. Circulation. 2001;104:1978–1983. doi: 10.1161/hc3901.096667. [DOI] [PubMed] [Google Scholar]
- 41.Rohrer L, Hersberger M, von Eckardstein A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr Opin Lipidol. 2004;15:269–278. doi: 10.1097/00041433-200406000-00006. [DOI] [PubMed] [Google Scholar]
- 42.Mineo C, Deguchi H, Griffin JH, Shaul PW. Endothelial and actions of HDL. Circ Res. 2006;98:1352–1364. doi: 10.1161/01.RES.0000225982.01988.93. [DOI] [PubMed] [Google Scholar]
- 43.Mason RP, Walter MF, Jacob RF. Effect of HMG-CoA reductase inhibitors on endothelial function. Circulation. 2004;109(suppl II):II-34–II-41. doi: 10.1161/01.CIR.0000129503.62747.03. [DOI] [PubMed] [Google Scholar]
- 44.Kuvin JT, Ramet ME, Patel AR, Pandian NG, Mendelsohn ME, Karas RH. A novel mechanism for the beneficial vascular effects of high density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am Heart J. 2002;144:165–172. doi: 10.1067/mhj.2002.123145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure I. Effect of simultaneous and consecutive incubation with cholesterol and HDL on cav-1/eNOS interaction in HAECs. HAECs were incubated with cholesterol (40 μg/ml) for 16 h in the presence of increasing concentrations of HDL (0-100 μg/ml) (left) or were incubated with cholesterol (40 μg/ml) followed by incubation with increasing concentrations of HDL (0-100 μg/ml) (right). An immunoprecipitation using cav-1 antibody was performed and a Western blot for eNOS was carried out on the immunoprecipitate.
Figure II. Effect of caveolin-1 on cholesterol efflux from MLECs. Wild-type (WT; white bars), caveolin-1 knockout (Cav-1 ko; grey bars) and caveolin-1 transgenic (Cav-1 tg; black bars) MLECs were incubated with cholesterol (40 μg/mL) and without (control) or with apoAl (10 μg/mL) or with HDL (100 μg/mL) for 16 h, after which cholesterol mass in the media and cells was measured using gas-chromatography and the % cholesterol efflux was calculated. *P<0.05, control vs HDL.
Figure III. Effect of ABCG1, ABCA1, and SR-BI on cholesterol efflux from MLECs. Wild-type MLECs were transfected with scrambled siRNA, ABCG1 siRNA, ABCA1 siRNA, or SR-BI siRNA, and incubated with cholesterol (40 μg/mL) without or with HDL (100 μg/mL) for 16 h, after which cholesterol mass in the media and cells was measured using gas-chromatography and the % cholesterol efflux was calculated. P<0.05 indicates significant difference between the HDL condition in the scrambled siRNA transfected cells vs the ABCG1 siRNA transfected cells.