

Abstract
Aims:
In 1955, Professor Morio Kasai first performed a hepatic portoenterostomy. Since then, the procedure has changed the lives of children with biliary atresia (BA). We report our initial experience in performing “extended” Kasai portoenterostomy (KPE), a modification of the original procedure.
Materials and Methods:
Since 2013, we have used the technique of “extended KPE” and prospectively recorded data on all children undergoing this operation. Data on demographics, clinical features, liver function tests, and perioperative cholangiogram findings were collected. Outcome of KPE was measured by Jaundice Disappearance Rate (JDR) and Native Liver Survival Rate (NLSR). We present our preliminary results from a 30-month period (February 2013 to May 2015).
Results:
Thirty-one children underwent KPE during this period (19 males) and only 1 child had biliary atresia splenic malformation (BASM). The mean age at KPE was 73 ± 24 days. Five (16.1%) children were more than 90 days old at the time of KPE. Fourteen children cleared jaundice (JDR 45.2%). Eleven (35.5%) children developed episodes of cholangitis, of whom 8 had early cholangitis (within 3 months of the operation). The proportion of children who survived with their own liver 6 months after KPE (NLSR) was 84.2%. Of those children older than 90 days, 2 cleared jaundice and have survived with their native livers for more than 16 months.
Conclusion:
In our preliminary report of 31 children, we conclude that the extended KPE leads to increased jaundice clearance and improved NLSR in children with BA.
KEY WORDS: Biliary atresia (BA), jaundice clearance, Kasai portoenterostomy (KPE), native liver survival
INTRODUCTION
In 1955, Professor Morio Kasai operated on a 72-day old infant with biliary atresia (BA).[1] He found that there were no extrahepatic bile ducts and on further dissection encountered significant bleeding from the hepatic hilum. To achieve hemostasis, he is said to have placed the duodenum over the hilum. Postoperatively, the baby passed yellow stools and went on to clear jaundice. He modestly named this procedure hepatic portoenterostomy and published on it in a journal called Shijitsu in 1959.[2] The procedure has since changed the lives of children with BA and the surgical community referred to it as Kasai portoenterostomy (KPE), acknowledging the surgeon-scientist who pioneered it. With a KPE alone, 45% of children have been known to reach their teenage years with their own livers and up to 15% have a truly symptom-free long-term survival.[3] Sequential treatment strategy involving KPE as first line followed by liver transplantation (LT) has been known to lead to 90% survival.[4]
Since it was first described, KPE has undergone several modifications by Professor Kasai and others. We started to perform one of the modifications, the “extended” KPE, in 2013 because of its anatomical accuracy in identifying biliary ductules in the hilum. We report our initial experience with it.
MATERIALS AND METHODS
Since 2013, we used the technique of “extended KPE” and prospectively recorded data on all children undergoing this operation. The exclusion criterion was children with BA who presented with synthetic liver cell failure (ascites, low albumin, uncorrectable coagulopathy). The data collected included demographics, clinical features, liver function tests, and perioperative cholangiogram findings. All KPE operations were performed by a single surgeon. The technique used was as follows.
All children undergo a limited laparotomy through a 4-cm incision in the right upper quadrant one finger-breadth above the umbilicus. The liver is inspected for gross evidence of fibrosis and nodularity. At the time of surgery, the appearance of the liver did not influence our decision to proceed with KPE because this was based exclusively on the absence of synthetic liver failure as previously defined. Inspection of the liver is followed by identification of the gall bladder (GB) or its remnant, which is held under traction with a suture and cannulated. The contrast, diatrizoic acid (Urograffin), is injected after being diluted 1:1 with normal saline. The cholangiogram images are recorded and inspected to confirm the diagnosis and to identify the type of BA (Ohi classification). In case there is no GB remnant, cholangiogram is not done.
Once the diagnosis of BA is confirmed, the incision is extended to about 15 cm dividing both rectii. The liver is then exteriorized by dividing the left triangular ligament. The abdominal cavity is inspected for features of biliary atresia splenic malformation syndrome (BASM) such as polysplenia, absent retrohepatic vena cava, malrotation, and preduodenal portal vein. The remnant GB is mobilized from its bed and excised along with the remnants of the extrahepatic biliary tree distal to the cystic duct in the hepatoduodenal ligament. The dissection is continued in the hepatoduodenal ligament by ligating and dividing the fibrotic tissue surrounding the vessels. Electrocautery is not used for dissection because of the possibility of thermal injury to microductules at the portal plate. The common hepatic artery with its right and left branches is dissected and isolated. The right branch is traced to its bifurcation into anterior and posterior branches in the right corner of the portal plate. Next, the main portal vein is identified. Its right and left branches are dissected and isolated with vessel loops to help retract it inferiorly in order to expose the lower limit of the portal plate. Small branches encountered from the right and left portal vein supplying the caudate lobe are meticulously ligated and divided. The left branch is traced upward to its confluence with the umbilical vein (round ligament) in the Rex recess. To reach this point, it is sometimes necessary to divide the liver bridge surrounding the round ligament. Branches of the left portal vein to segment 4 can be seen and they mark the limit of dissection at the left corner of the portal plate. The fibrotic portal plate is excised down to the Glisson's capsule using scissors from the bifurcation of the right hepatic artery to the Rex recess [Figure 1]. After excision of the portal plate, hemostasis is secured using saline adrenaline (1:100,000 dilution) soaked gauze.
Figure 1.
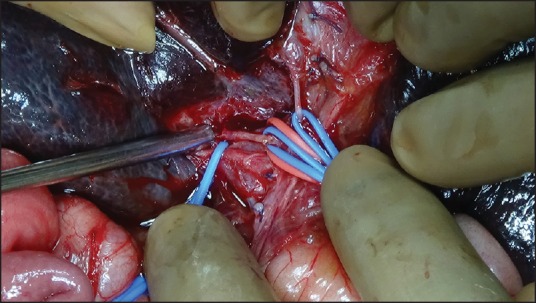
Liver hilum in a child with BA after excision of the fibrotic portal plate
A 60-cm Roux-en-Y loop of jejunum is created for anastomosis and brought to the supracolic compartment through a retrocolic window. The lateral-most sutures of the anastomosis are placed superficial and lateral to the entry points of right and left ducts to avoid damage to the microductules. The posterior layer of sutures is placed first. Since the dissection exposes the vessels, some of these sutures in the lower limit of the portal plate are placed on the adventitia of the vessels. The bowel enterotomy is performed after this layer of sutures are in place as this allows for precise placement of sutures low on the caudate lobe and prevents the spill of enteric contents. After the enterotomy is made, the posterior wall sutures remain buried. The anterior wall is anchored to the liver parenchyma of the quadrate lobe. Thus the hepaticojejunostomy forms a funnel around the hilum.
Postoperative care and follow up is standardized for all patients. Postoperatively, feeds are started on day 5 and intravenous antibiotics are continued for 7 days. After discharge, prophylactic oral antibiotics (cephalexin and ofloxacin in 2-week rotating cycles) are given for 6 months along with ursodeoxycholic acid and supplemental vitamins. Follow-up is on a monthly basis for 6 months and quarterly thereafter. All children undergo upper gastrointestinal (GI) endoscopy 1 year after KPE.
Outcome of KPE is measured by Jaundice Disappearance Rate (JDR) and Native Liver Survival Rate (NLSR). JDR is defined as a serum total bilirubin level below 2 mg/dL within 3 months postoperatively. NLSR is defined as the number of children who have retained their own livers over a defined period of time.
We present our preliminary results from a 30-month period (February 2013 to May 2015).
This study was done after obtaining prior approval from the Institutional Review Board and clearance from the Ethics Committee.
RESULTS
Thirty-one children underwent KPE during this period. During the study period, there were no other children who met the exclusion criteria. Of those who underwent KPE, 19 were males with a mean age of 73.3 ± 24.0 days. Their mean weight was 4.8 ± 0.9 kg. Only 1 child had BASM and this did not affect the operative procedure. The mean preoperative total bilirubin was 11.5 ± 3.8 mg/dL, preoperative gamma glutamyltransferase (GGT) was 538 IU/L (55-1755 IU/L), preoperative aspartate transaminase (AST) was 212 IU/L (68-790 IU/L), and platelet count was 438,196.8 ± 178,231.7 cells/mm3. Aspartate-platelet ratio index (APRI) was 1.0 (0.3-7.6). All children had type III BA based on the peroperative cholangiogram findings. Intraoperatively, none of the children had excessive bleeding from the extensive hilar dissection.
There was one (3.2%) postoperative complication in a child who had adhesive obstruction needing laparotomy and adhesiolysis. There were no postoperative mortality in our series.
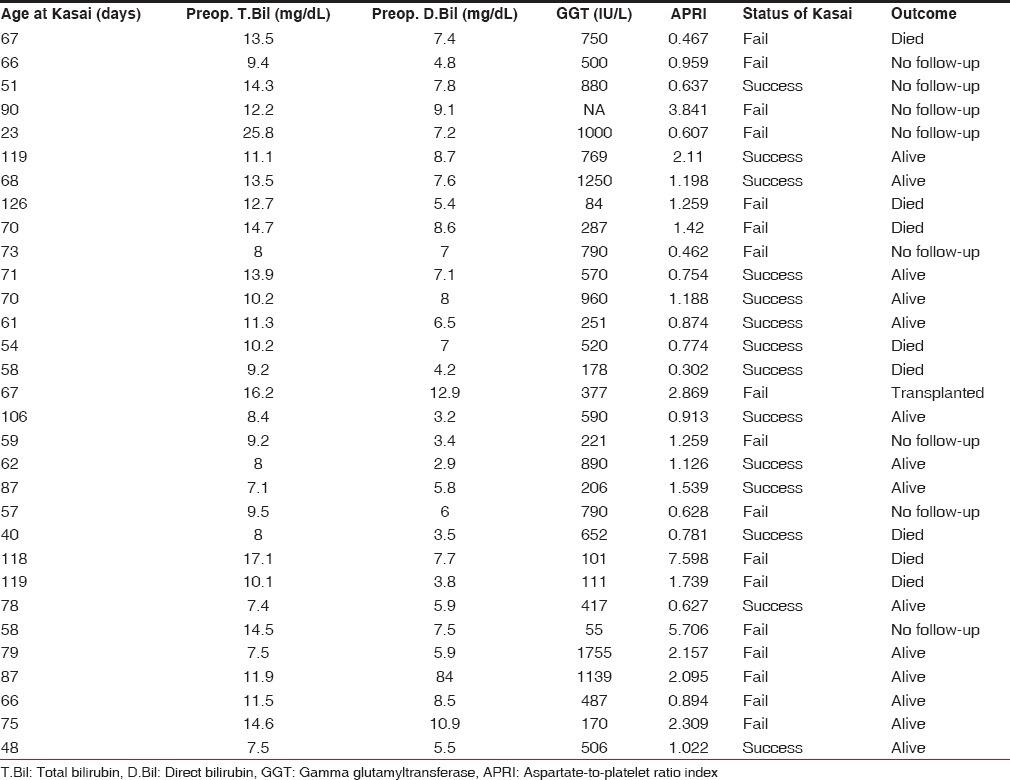
Fourteen children cleared jaundice (JDR 45.2%), of whom 3 died 6-12 months after the KPE from portal hypertension and variceal bleeds not responding to sclerotherapy. The families of these children could not afford transplantation [Table 1].
Table 1.
Patient data
Out of 17 children who did not clear jaundice, 7 were lost to follow-up, 5 died of liver cell failure as they could not afford transplantation, 4 are alive of whom 1 is awaiting transplant, and 1 underwent liver transplantation and is currently well.
Sixteen out of 19 (8 were lost to follow-up and 4 have less than 6 month follow-up) children are alive with their own liver 6 months after KPE (84.2% NLSR). Of these, 3 children fulfil criteria for transplantation.
Five children were over 90 days old at the time of KPE. Two of them cleared jaundice and are alive with their native livers for more than 16 months.
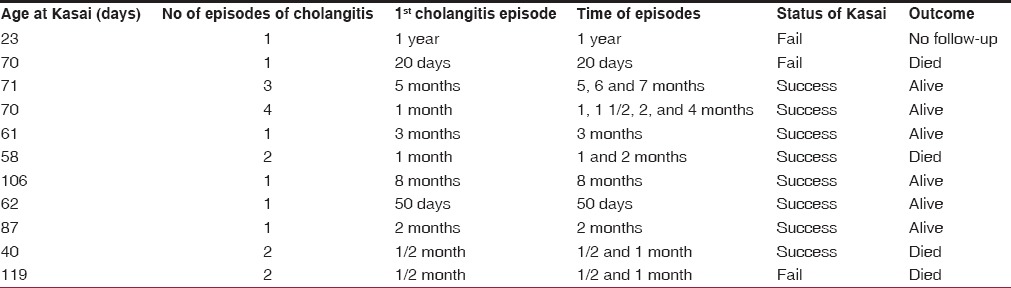
Eleven (35.5%) children developed 19 episodes of cholangitis. Of these, 8 children had cholangitis within 3 months of the operation. The other 3 children had cholangitis 6-12 months after the operation [Table 2]. Portal hypertension was seen in 7 children and all of them had cleared jaundice.
Table 2.
Cholangitis and outcome
DISCUSSION
The Kasai operation is the initial treatment for children with BA. It continues to retain its relevance because up to 38% of children can survive with their own livers for 30 years after KPE.[3] It delays the need for transplant in a significant proportion of children, making it a safer proposition, and postpones the need for immunosuppression. In his original operation, Professor Kasai resected the fibrous biliary remnant from the hepatic hilum with dissection and retraction of the branches of the right and left hepatic artery and portal vein.[2] The anastomosis of jejunum was to the connective tissue of the hepatoduodenal ligament posteriorly and the liver parenchyma anteriorly. With the passage of time, there have been several modifications to this technique. The extended Kasai operation calls for an extensive dissection of the fibrotic portal plate. The rationale for the extensive dissection comes from the normal anatomy of the portal plate where the right hepatic duct enters the liver between the bifurcation of the right hepatic artery and the left duct enters medial to the confluence of left branch of portal vein and umbilical vein. All along this line, ductules might be present and hence it is important to include this entire area in the hepaticojejunostomy [Figure 2]. Hashimoto et al. proposed a further modification to this technique that involved partially trimming the parenchymal remnant around the hilum with a cavitron ultrasonic aspirator (CUSA) without directly applying it to the remnant. Traction sutures were placed on the remnant portal plate and it was divided with sharp dissection using knife or scissors. They reported a 77% jaundice clearance without the need for LT or reoperation.[5] Similar modifications have also been reported, with very high jaundice clearance rates (81%).[6]
Figure 2.
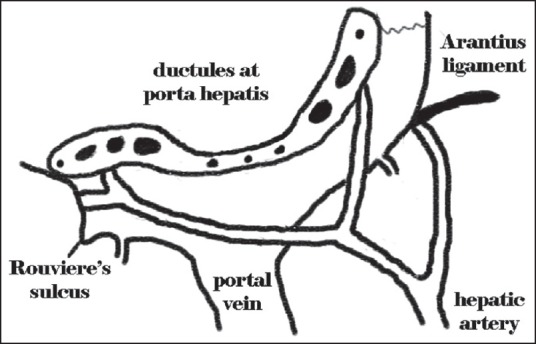
Illustration showing the distribution of the ductules at the porta hepatis
In our technique, we divide the ligamentum venosum and dissect it down to its junction with the left portal vein. We preserve the branches of the left portal vein to segment 4 but divide the fine branches to the caudate lobe. Thus the left portal vein can be retracted laterally and inferiorly, exposing the ductal plate clearly. A further extensive dissection in this region has been proposed toward the dorsal aspect of the right anterior segmental portal vein branch.[5] Extensive dissection of the portahepatis requires excellent exposure and was achieved in our patients by mobilizing the liver and dislocating it externally. We did not have any complications from this step. Other series have also used this technique.[7]
Several modifications in the Roux-en-Y loop have been proposed to prevent postoperative ascending cholangitis. These include creating antireflux valves, external conduits, and double-barrelled conduits. None of these modifications have gained acceptance, nor have they made an impact on the incidence of postoperative cholangitis.[8,9] The incidence of cholangitis in our series was 35% comparable to most series that report 30-60%.[10] The majority of the children who had cholangitis in our series had successful KPE and 6 out of 11 children continue to be free of jaundice. Large series have reported that the outcome of the KPE was worse and the probability of cirrhosis was higher following episodes of cholangitis.[11,12]
Portal hypertension is secondary to fibrosis in the liver and may be present even at the time of KPE.[13] Despite clearance of jaundice and normal liver function tests, portal hypertension can progress after KPE. Local treatment of esophageal varices and portosystemic shunts are an option in these children. Portal hypertension (as evidenced by the presence of esophageal varices and/or ascites) with normal serum bilirubin levels was seen in 7 children in our series. Three of these children did not respond to sclerotherapy and died 6 months to 1 year after KPE.
The outcome of KPE has been well documented in large series of patients from the French National Registry, Japanese Biliary Atresia Registry, and the Registry from England and Wales [Table 3]. The JDR was 57% in most series. NLSR (10 years) was the highest in the Japanese series and the series from England and Wales at 53% and 40%, respectively.[4,14] The French national study reported that the outcome of KPE improved with centralization of services and the need for transplantation was reduced by as much as 24%.[15] Although there is no registry in India, some centers have also reported good outcomes for KPE.[16,17,18] In the National consensus report, it was recommended that KPE should be performed only in centers that do at least 6 of these operations a year.[19] This is of particular importance in the developing world where the cost of liver transplantation is prohibitively high. Centralization of BA management will significantly reduce the need for transplantation.[20]
Table 3.
Reports from national registries
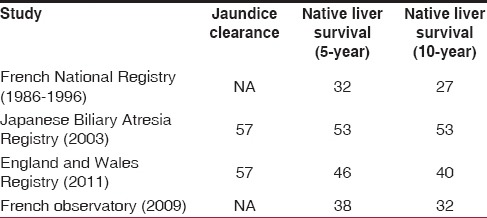
We have used the extended KPE technique from 2013 and since then, our centers have performed approximately 15 KPEs every year. We have a JDR of 45.2%, comparable to most series. All KPEs in our centers are performed by a single surgeon.
Even in older children, there is a significant benefit in undergoing KPE, with some series showing a JDR of 48% and a 10-year NLSR of up to 45%.[14,21,22] Nio et al. have demonstrated 30-year NLSR of 25% in children who underwent KPE in 91-120 days and 7% in those who underwent KPE in 121-150 days.[21] Thus there is a significant benefit to performing KPE after 90 days of age in “seniors” with BA. Two out of 5 children in our series operated on after 90 days are alive and well.
The only contraindications for KPE are liver cell failure, portal hypertension, and uncorrectable coagulopathy. These children have rapid decompensation in the postoperative period, resulting in a very high morbidity and mortality.
The NLSR in our series is 83% at 6 months, but this is a very short period of follow-up and hence we are unable to draw significant conclusions from this. Additionally, 7 children were lost to follow-up because they failed to clear jaundice and we are not aware of the status of these children. Despite India currently being the fastest-growing economy in the world, a lot of our patients cannot afford expensive health care solutions such as liver transplantation. Hence a significant number of children are lost to follow-up or die, as is the case in our series. Under the circumstances, it is extremely important to standardize care and surgical technique and form centers of excellence so that the benefit of KPE can be maximized.
CONCLUSION
In our preliminary report of 31 children, we conclude that the extended KPE leads to increased jaundice clearance and improved NLSR in children with BA. As this study continues, we are prospectively collecting data and analyzing clinical and histological factors that affect the outcome of KPE, and we look forward to reporting results with larger numbers of patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Garcia AV, Cowles RA, Kato T, Hardy MA. Morio Kasai: A remarkable impact beyond the Kasai procedure. J Pediatr Surg. 2012;47:1023–7. doi: 10.1016/j.jpedsurg.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kasai M, Suzuki S. A new operation for non-correctable biliary atresia-hepatic portoenterostomy. Shijitsu. 1959;13:733–9. [Google Scholar]
- 3.Hadzic N, Davenport M, Tizzard S, Singer J, Howard ER, Mieli-Vergani G. Long-term survival following Kasai portoenterostomy: Is chronic liver disease inevitable? J Pediatr Gastroenterol Nutr. 2003;37:430–3. doi: 10.1097/00005176-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Davenport M, Ong E, Sharif K, Alizai N, McClean P, Hadzic N, et al. Biliary atresia in England and Wales: Results of centralization and new benchmark. J Pediatr Surg. 2011;46:1689–94. doi: 10.1016/j.jpedsurg.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto T, Otobe Y, Shimizu Y, Suzuki T, Nakamura T, Hayashi S, et al. A modification of hepatic portoenterostomy (Kasai operation) for biliary atresia. J Am Coll Surg. 1997;185:548–53. doi: 10.1016/s1072-7515(97)00104-x. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Hashimoto T, Kondo S, Sato Y, Hussein MH. Evaluating patients’ outcome post-Kasai operation: A 19-year experience with modification of the hepatic portoenterostomy and applying a novel steroid therapy regimen. Pediatr Surg Int. 2010;26:825–30. doi: 10.1007/s00383-010-2637-y. [DOI] [PubMed] [Google Scholar]
- 7.Davies MR. Facilitating the operative exposure of the portal plate in cases of biliary atresia by dislocating the whole liver onto the abdominal wall. J Pediatr Surg. 1992;27:1391–3. doi: 10.1016/0022-3468(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 8.Suruga K, Miyano T, Kitahara T, Kojima Y, Fukuda Y. Treatment of biliary atresia: A study of our operative results. J Pediatr Surg. 1981;16(Suppl 1):621–6. doi: 10.1016/0022-3468(81)90016-6. [DOI] [PubMed] [Google Scholar]
- 9.Kasai M. Treatment of biliary atresia with special reference to hepatic porto-enterostomy and its modifications. Prog Pediatr Surg. 1974;6:5–52. [PubMed] [Google Scholar]
- 10.Luo Y, Zheng S. Current concept about postoperative cholangitis in biliary atresia. World J Pediatr. 2008;4:14–9. doi: 10.1007/s12519-008-0003-0. [DOI] [PubMed] [Google Scholar]
- 11.Lunzmann K, Schweizer P. The influence of cholangitis on the prognosis of extrahepatic biliary atresia. Eur J Pediatr Surg. 1999;9:19–23. doi: 10.1055/s-2008-1072206. [DOI] [PubMed] [Google Scholar]
- 12.Wildhaber BE, Coran AG, Drongowski RA, Hirschl RB, Geiger JD, Lelli JL, et al. The Kasai portoenterostomy for biliary atresia: A review of a 27-year experience with 81 patients. J Pediatr Surg. 2003;38:1480–5. doi: 10.1016/s0022-3468(03)00499-8. [DOI] [PubMed] [Google Scholar]
- 13.Wildhaber BE. Biliary atresia: 50 years after the first Kasai. ISRN Surg 2012. 2012 doi: 10.5402/2012/132089. 132089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nio M, Ohi R, Miyano T, Saeki M, Shiraki K, Tanaka K. Japanese Biliary Atresia Registry. Five- and 10-year survival rates after surgery for biliary atresia: A report from the Japanese Biliary Atresia Registry. J Pediatr Surg. 2003;38:997–1000. doi: 10.1016/s0022-3468(03)00178-7. [DOI] [PubMed] [Google Scholar]
- 15.Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard JL, Auvert B. Prognosis of biliary atresia in the era of liver transplantation: French national study from 1986 to 1996. Hepatology. 1999;30:606–11. doi: 10.1002/hep.510300330. [DOI] [PubMed] [Google Scholar]
- 16.Narasimhan KL, Chowdhry SK, Vaiphei K, Samujh R, Mahajan JK, Thapa BR, et al. Outcome of biliary atresia from Chandigarh: Results of a prospective analysis. Indian Pediatr. 2001;38:1144–8. [PubMed] [Google Scholar]
- 17.Roy P, Chatterjee U, Ganguli M, Banerjee S, Chatterjee SK, Basu AK. A histopathological study of liver and biliary remnants with clinical outcome in cases of extrahepatic biliary atresia. Indian J Pathol Microbiol. 2010;53:101–5. doi: 10.4103/0377-4929.59194. [DOI] [PubMed] [Google Scholar]
- 18.Gupta L, Gupta SD, Bhatnagar V. Extrahepatic biliary atresia: Correlation of histopathology and liver function tests with surgical outcomes. J Indian Assoc Pediatr Surg. 2012;17:147–52. doi: 10.4103/0971-9261.102326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Indian Academy of Pediatrics. Pediatric Gastroenterology Subspecialty Chapter. Consensus report on neonatal cholestasis syndrome. Pediatric Gastroenterology Subspecialty Chapter of Indian Academy of Pediatrics. Indian Pediatr. 2000;37:845–51. [PubMed] [Google Scholar]
- 20.Davenport M, De Ville de Goyet J, Stringer MD, Mieli-Vergani G, Kelly DA, McClean P, et al. Seamless management of biliary atresia in England and Wales (1999-2002) Lancet. 2004;363:1354–7. doi: 10.1016/S0140-6736(04)16045-5. [DOI] [PubMed] [Google Scholar]
- 21.Nio M, Sasaki H, Wada M, Kazama T, Nishi K, Tanaka H. Impact of age at Kasai operation on short- and long-term outcomes of type III biliary atresia at a single institution. J Pediatr Surg. 2010;45:2361–3. doi: 10.1016/j.jpedsurg.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Davenport M, Puricelli V, Farrant P, Hadzic N, Mieli-Vergani G, Portmann B, et al. The outcome of the older (> or =100 days) infant with biliary atresia. J Pediatr Surg. 2004;39:575–81. doi: 10.1016/j.jpedsurg.2003.12.014. [DOI] [PubMed] [Google Scholar]