

Abstract
Context: Hemophagocytic syndrome (HPS) is mainly characterized by massive infiltration of bone marrow by activated macrophages and often presents with pancytopenia. Thrombotic microangiopathy (TMA) is also present with thrombocytopenia and renal involvement. Both conditions could coexist with each other and complicate the condition.
Evidence Acquisition: Directory of Open Access Journals (DOAJ), EMBASE, Google Scholar, PubMed, EBSCO, and Web of Science with keywords relevant to; Hemophagocytic syndrome, macrophage activation syndrome, interferon-gamma and thrombotic microangiopathy, have been searched.
Results: Viral infection, rheumatologic disease and malignancies are the main underlying causes for secondary HPS. calcineurin inhibitors and viral infections are also the main underlying causes of TMA in transplant recipients. In this review, we discussed a 39-year-old male who presented with pancytopenia and renal allograft dysfunction. With the diagnosis of HPS induced TMA his renal condition and pancytopenia improved after receiving intravenous immunoglobulin (IVIG) and plasmapheresis therapy.
Conclusions: HPS is an increasingly recognized disorder in the realm of different medical specialties. Renal involvement complicates the clinical picture of the disease, and this condition even is more complex in renal transplant recipients. We should consider the possibility of HPS in any renal transplant recipient with pancytopenia and allograft dysfunction. The combination of HPS with TMA future increases the complexity of the situation.
Keywords: Hemophagocytic syndrome, Macrophage activation syndrome, Interferon-gamma, Thrombotic microangiopathy
Implication for health policy/practice/research/medical education:
Hemophagocytic syndrome (HSP) (macrophage activation syndrome) is an increasingly recognized disorder in the realm of different medical specialties. Renal involvement complicates the clinical picture of the disease, and this condition even is more complex in renal transplant recipients. We should consider the possibility of HPS in any renal transplant recipient with pancytopenia and allograft dysfunction.
1. Context
Hemophagocytic syndrome (HPS) also known as macrophage activation syndrome or hemophagocytic lymphohistiocytosis (HLH), is a massive infiltration of bone marrow and lymphoid organ by activated macrophages and often presents with fever, pancytopenia, adenopathies, hepatosplenomegaly, hyperferritinemia and hypertriglyceridemia (1). Clinical signs are variable, nonspecific and often evolves in a subacute course within 1-4 weeks (2). Primary HPS occurs in children with inherited dysfunction of the immune response, particularly natural killer (NK) cells and cytotoxic T-cell the condition subsequently leads to cytokine elevation and mononuclear-phagocytes activation (2,3). Secondary HPS is often triggered by infectious, autoimmune, and neoplastic diseases, and sometimes more than one underlying cause dose exist (2).
2. Evidence Acquisition
For this review, we used a variety of sources by searching through PubMed/Medline, Scopus, EMBASE, EBSCO and directory of open access journals (DOAJ). The search was conducted, for articles published from January 1, 1970 up to September 29, 2011, using combinations of the following key words and or their equivalents; Hemophagocytic syndrome, macrophage activation syndrome, interferon-gamma and thrombotic microangiopathy.
3. Mechanisms and diagnostic criteria
HPS’s staring point is the inability of immune system to control the primary causative infectious lead to hyper-secretion of pro-inflammatory cytokines including; Tumor necrosis factor-alpha (TNF-a), interferon-gamma (IFN-γ). Interleukin 1, interleukin 4, interleukin 6, interleukin 8, interleukin 10, interleukin 18, and creating a condition that is named cytokine storm (4-6). Fever, cachexia, elevated serum triglyceride, high serum ferritin level, acute kidney injury (AKI) and tubular necrosis that are present in this situation all are attributed to high cytokines levels (1,4).
Diagnosis of HPS or HLH is based on the presence of at least 5 or more of the following conditions; fever >38.3°C, splenomegaly, pancytopenia that defines two of the followings (hemoglobin <90 g/dl, platelets <100 000/μl, and neutrophils <1000/μl), hypertriglyceridemia and/or hypofibrinogenemia and hemophagocytosis diagnosed in bone marrow, spleen, or lymph nodes biopsy. Immunologic markers include low or absent NK-cell activity (assessed by chromium 51 or granzyme B proteolytic activity). Key diagnostic immunologic markers are increased plasma level of CD163, as a marker of macrophage activation, and CD25 as an interleukin 2 receptor (sIL-2R), is increased is in 79% of adult with HPS (7,8). The bone marrow aspiration is the best investigation site and identifies mature histiocytes ingesting other blood cells in 84% of conditions (9). Liver is another frequently involved organ and 60% of patients have abnormal liver tests. Splenic involvement and rupture, ascites, veno-occlusive disease, pulmonary involvement and encephalopathy are other rare reported conditions in HPS. Renal involvement in HPS will be discussed later and it is a predictor of poor prognosis (10,11).
3.1. Hemophagocytic syndrome in adults
Secondary HPS was first described at 1979 in 19 patients interestingly, thirteen of of these patients were renal transplant recipients. In transplant recipients, HPS often triggers by an infection, mainly viral, including adenovirus, human herpes virus 8 (HHV-8), Epstein-Barr virus (EBV), cytomegalovirus (CMV) (12,13), HHV-6 (14), parvovirus B19 (15,16) and BK polyomavirus (17), herpes viruses account for 62% of viral cases of HPS, and EBV is known as the most prominent of them (2). Tuberculosis, toxoplasmosis, leishmaniosis, babesiosis, Rickettsia spp, and Staphylococcus spp and Escherichia coli have been reported as the causative infections triger for initiation and development of HPS (18-20). Among the different features of tuberculosis the role of extra pulmonary tuberculosis is more prominent (21). Pneumocystis jiroveci, Toxoplasma gondii, and fungi infection are other possible triggers (2). HPS has been reported in chronic hepatitis B or hepatitis C virus infections (22). In addition, lymphoma and sarcoma should also be considered as another possible trigeres in transplant recipients. T-cell lymphoma is related to EBV infection and metacentric Castleman disease is also associated with herpesvirus-8 infection interestingly both of these conditions could be trigeres for HPS development. In rheumatologic disorders associated-HPS, such as systemic lupus erythematous (SLE), adult-onset Still’s, systemic vasculitis, and inflammatory bowel disease, also, various infections are the main underlying trigger (2,23). Excessive cytokine secretion by tumor cells could be the main trigger in malignancy associated HPS (2,24,25).
Here we describe a renal transplant recipient with HPS and TMA with renal involvement. A 39-year-old male was admitted for a 2-week onset of fever, weakness and anemia. He received a second living unrelated renal transplantation 9 months ago and was on triple immunosuppressive therapy with tacrolimus (3 mg/day), mycophenolate mofetil (1.5 mg/day) and prednisolone (5 mg/day). He received multiple sessions of plasma exchange, and intravenous immunoglobulin (IVIG) before transplantation and induction therapy with rituximab, basiliximab, and anti-thymocyte globulin (ATG). One month before admission, his serum creatinine level was 1.2 mg/dl, however it was raised to 3.3 mg/ dl in the next admission. Physical examination revealed a blood pressure of 90/70 mm Hg, body temperature 38.5°C and patient was alert. Hepatosplenomegaly or lymphadenopathy was not detected. Laboratory examination revealed hemoglobin; 6 mg/dl, white blood cell; 19 000/μl (60% polymorphonuclear cells), platelets count; 35 000/μl, normal prothrombin (PT) and partial thromboplastin (PTT) times. Peripheral blood smear showed high proportion of schistocytes (Figure 1). Reticulocyte count was 2.4%, fibrinogen level; 3.8 mg/dl (2–4 mg/dl) and level of D-dimer was in normal range, serum lactate dehydrogenase (LDH); 850 (105–333 IU/l), serum ferritin; 2634 (12–150 ng/ml), triglyceride; 8100 mg/dl. Urine dipstick proteinuria was trace. A 24-hour urine protein excretion was 300 mg/day. Renal allograft ultrasonography disclosed a normal size and texture. Workup for infective etiology, including urinary tract infection and pulmonary infection was negative. Serologic study for CMV, EBV and parvovirus B19 was negative. The serum PCR examinations were also negative for above infections. Furthermore, serologic study for toxoplasmosis was also negative. Bone marrow aspiration revealed an increase in histiocytes with widespread hemophagocytosis. With the diagnosis of TMA and HPS, patient received IVIG, and 2 liter/day therapeutic plasma exchange (TPE) with fresh frozen plasma replacement. After eight cycles, patient’s condition improved clinically. LDH decreased and platelet increased to 120 000/μl. At this time a renal biopsy was conducted. In light microscopy, we found endothelial cell swelling resulted in glomerular capillary narrowing and obliteration. In some parts the capillary lumen was occluded by fibrin thrombi which was indicative of TMA (Figure 2). The disease course stabilized gradually. Patient discharged with clinical and laboratory improvement and good general condition. The last serum creatinine was 1.9 mg/dl. This case history is an example of difficulties on diagnosis and therapeutic options of these patients.
Figure 1.
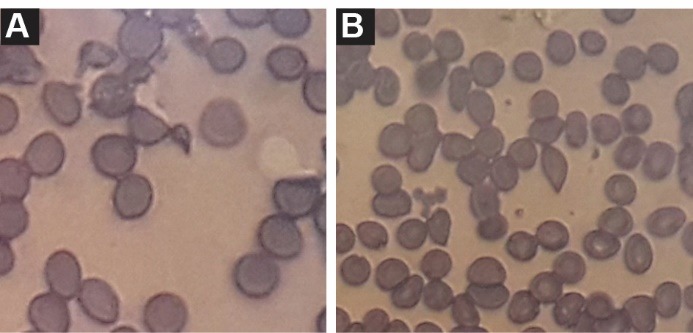
Peripheral blood smear (10*40) shows helmet cells (schistocytes) in the central lesions of both slide field that is compatible with thrombotic microangiopathy.
Figure 2.
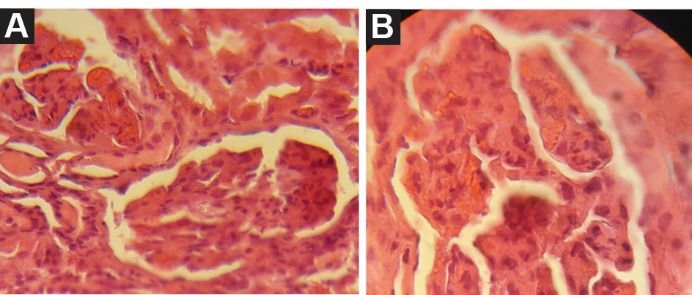
Renal biopsy , A) (40*10 ) and B) (100*10), glomerular capillary loops are constricted with fibrin thrombi and endothelial swelling, without any cellular infiltration.
Renal transplant recipients are very prone to HPS. Their immunosuppression and deregulated immune system are risk factors for both opportunistic infections and malignancies. Despite this situation, HPS is a rare condition. However, it is possible that this syndrome is under diagnosed among this population. Sign and symptoms of HPS easily overlooked by the signs and symptoms of underlying infection. Post-transplant HPS usually happens during the early period when patients are in highest immunosuppressed condition. Polyclonal antithymocyte antibodies and history of acute rejection treatment are predisposing conditions (26,27). The underlying viral, bacterial, and protozoa infections are the main contributor contributors (28). Posttransplantation lymphoproliferative disorders and solid tumors should be considered as important underlying causes (28).
There is a high mortality rate among renal transplant recipients with HPS (26-46). In a cohort study conducted by Karras et al (28) 8 of 17 renal transplant recipients died and 4 of the 9 survivors lost the allograft. However, underlying infectious disease, malignancies, immunosuppressive regimen side effects and liver involvement could be the risk factors of HPS associated mortality and morbidity among these patients (25). Differential diagnosis of the HPS from a severe presentation of the underlying infection, malignancy or autoimmune disease is an important diagnostic challenge (29). Neurological and psychiatric presentations of HPS happen in one fourth of patients. They are heterogeneous and include coma, seizures, meningitis, cerebral hemorrhage, mood disorders, delirium or psychosis (2,23). In renal recipient patients with unexplained fever and neurological symptoms, the possibility of toxoplasmosis should also be considered. Interestingly, the toxoplasma infection by itself could be a triger for HPS development and this combination creates diagnostic difficulties (2).
Management of HPS in renal transplant recipients requires immunosuppressive dose reduction and introduction of specific antiviral treatment. High-dose polyvalent immunoglobulin is a beneficial treatment (30). These patients often receive steroids and cyclosporine as their immunosuppression regimen, as this regimens also has been proposed for treatment of HPS. However, the mortality of HPS in renal transplant recipients is high (30).
Interestingly, the highest frequency of HPS occurs in umbilical cord blood derived hematopoietic stem cell transplantation (HSC) and it happens in 17% of the cases. In kidney transplanted patients, HPS happens only in 0.4%–2% of recipients (2). Allograft rejection by itself could be a trigger for HPS evolution (2). Despite profound workup, some patients with HPS remains underdiagnosed (2). Diagnosis of post-transplant HPS is not easy, pancytopenia may be caused by immunosuppressive drugs, and hypertriglyceridemia is frequent in these patients, particularly, in those receiving high doses of steroids, sirolimus or everolimus. High levels of serum ferritin level is helpful, however, its elevation could be detected in other inflammatory conditions (26,30).
3.2. Renal involvement in HPS
AKI is the most common renal disorder in HPS followed by nephrotic syndrome. In general, the prognosis of HPS related renal involvement is not good. AKI in HPS could be as a part of the multiorgan failure due to increased capillary permeability and pre-renal condition. It can also be related to the nephrotoxic effects of antimicrobial therapy. Acute tubular necrosis is often associated with interstitial inflammation. Glomerular involvement is less common in HPS. Nephrotic syndrome could be due to minimal-change disease or collapsing glomerulopathy. TMA renal involvement in HPS is a very rare condition (31-33).
Tubular destruction is the result of inflammatory cytokines damage (34-36). The serum level of tumor necrosis factor α (TNFα) in HPS patients is very high, and might have some roles in tubular necrosis and glomerular involvement (37). TNFα band to its receptor (TNFR1) mediates renal damage through increasing granulocyte infiltration and activation of apoptotic signaling kinase-1 (ASK1) in tubular cells (38,39). The idea of pro-inflammatory cytokines renal epithelial damage is supported by the clinical observation of AKI in the absence of circulatory disorders, referred to the direct tubular toxic effect of high serum levels of IL-6 and TNFα (35,40,41). TNFα can disorganize the podocytes actin cytoskeleton leads to increase glomerular permeability to albumin. Genetic background affects the degree and type of podocyte lesion in this condition (16). HPS-associated glomerulopathy is a rare condition and often is associated with sever nephrotic syndrome combined with renal dysfunction that may need dialysis during the course of HPS. Collapsing glomerulopathy was the most common pathologic feature that only was observed among black patients and minimal-change disease was the second common finding (40,41). TNFα and TNFRs are also important determinants of renal transplant rejection. Expression of TNFRs is up-regulated in normal kidney, and based on the type of receptor predeominancy (i.e. TNFR1 or TNFR2) inflammation progresses or halts (39,42,43). Collapsing glomerulopathy and nephrotic syndrome are less common than tubular damage. However, it is very important diagnosis in HPS related renal involvement. The development of proteinuria is correlated with the systemic and organ-specific signs and cytokine burst. Glomerular involvement is not related to immune deposits, rather, it is a podocyte injury because of actin cytoskeleton degeneration and genetic background also influence the condition. Interestingly serum TNFα in 3 patients with HPS and glomerulopathy was more than 10 times the maximal normal value (28,31,44).
TMA is another type of glomerular involvement in HPS (2,7). TMA is a more general term for both thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). It is a multisystem disorder characterized by consumptive microangiopathic hemolytic anemia, thrombocytopenia, with various degrees of neurologic symptoms, and renal impairment. Simultaneous existence of HPS and TMA has been confirmed in a few reports (15,45). Renal involvement in TMA could be the consequence of cytokine induced endothelial injury (15,45). There is also a reported case of HPS triggered by plasma exchange that was started for treatment of TTP, and the condition was explained by high amounts of cytokines released from neutrophils during their passage to the plasma exchange materials (46,47). Furthermore, there are two reported cases of HPS-associated with preeclampsia. Interestingly, the pathologic feature of the glomerular endotheliosis and capillary loop occlusion in preeclampsia are very similar to the renal lesions by TMA (33,47).
3.3. Therapeutic options
Elimination of triggers (mainly infections) is crucial for treatment of adult patients with HPS. High-dose polyvalent IVIG is beneficial in infection, autoimmune, and transplant related HPS (48). The main predictive factor of response is its early administration particularly when ferritin level is high (49,50). C-reactive protein, erythrocyte sedimentation rate (ESR), improvement of hypertriglyceridemia, and hyperferritinemia may be useful indicators of disease activity. Steroids and cyclosporine have been proposed as a treatment for HPS (31,50). Biological treatments such as rituximab, infliximab, and etanercept (Enbrel) have been proposed for adults patients who did not respond to cyclosporine and IVIG, (51) tacrolimus, and etoposide also have been proposed in refractory cases (52).
Use of anti-TNFα drugs, anti-IL1receptor (Anakinra) antibody, and anti-IL6 (tocilizumab) has been proposed for rheumatoid arthritis and adult Still’s induced HPS (53). B-cell depleting drugs (rituximab, belimumab) have been proposed for SLE, and EBV-related HPS with or without associated lymphoma.
Use of cyclosporine in those patients who did not respond to IVIG and alemtuzumab, has been used in three adult patients with HPS with reliable results (54,55). For treatment of HPS in allograft recipients, at first we should try to find the underlying triger and target it, and in most of the condition the triger is infection. When there is a combination of HPS and TMA addition of therapeutic plasma exchange is useful. Graft nephrectomy has been proposed for kidney transplant recipients with life-threatening HPS resistant to different therapies (53).
4. Conclusions
HPS is an increasingly recognized disorder in the realm of different medical specialties. Renal involvement complicates the clinical picture of the disease, and this condition even is more complex in renal transplant recipients. We should consider the possibility of HPS in any renal transplant recipient with pancytopenia and allograft dysfunction. The combination of HPS with TMA future increases the complexity of the situation. However these conditions and their combination may not be diagnosed, if unsuspected.
Authors’ contribution
Primary draft by HE, EM, BM and MRA. Editing the final manuscript by MMS.
Conflicts of interest
The authors declared no competing interests.
Funding/Support
None.
Please cite this paper as: Esmaili H, Mostafidi E, Mehramuz B, Ardalan MR, Mohajel-Shoja MA. An update on renal involvement in hemophagocytic syndrome (macrophage activation syndrome). J Nephropathol. 2016;5(1):8-14. DOI: 10.15171/jnp.2016.02
References
- 1.Karras A. What nephrologists need to know about hemophagocytic syndrome. Nat Rev Nephrol. 2009;5(6):329–36. doi: 10.1038/nrneph.2009.73. [DOI] [PubMed] [Google Scholar]
- 2.Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–16. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 3.Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21(5):245–53. doi: 10.1016/j.blre.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Milner JD, Orekov T, Ward JM, Cheng L, Torres-Velez F, Junttila I. et al. Sustained IL-4 exposure leads to a novel pathway for hemophagocytosis, inflammation, and tissue macrophage accumulation. Blood. 2010;116(14):2476–83. doi: 10.1182/blood-2009-11-255174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canna SW, Behrens EM. Not all hemophagocytes are createdequally: appreciating the heterogeneity of the hemophagocytic syndromes. Curr Opin Rheumatol. 2012;24(1):113–8. doi: 10.1097/BOR.0b013e32834dd37e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M. et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121(6):2264–77. doi: 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S. et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 8.Tamura K, Kanazawa T, Tsukada S, Kobayashi T, Kawamura M, Morikawa A. Increased serum monocyte chemoattractant protein-1, macrophage inflammatory protein-1beta, and interleukin-8 concentrations in hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(5):662–8. doi: 10.1002/pbc.21660. [DOI] [PubMed] [Google Scholar]
- 9.Goel S, Polski JM, Imran H. Sensitivity and specifi city of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci. 2012;42(1):21–5. [PubMed] [Google Scholar]
- 10.de Kerguenec C, Hillaire S, Molinié V, Gardin C, Degott C, Erlinger S. et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96(3):852–7. doi: 10.1111/j.1572-0241.2001.03632.x. [DOI] [PubMed] [Google Scholar]
- 11.Prendki V, Stirnemann J, Lemoine M, Lohez M, Aras N, Ganne-Carrié N. et al. Prevalence and clinical significance of Küpffer cell hyperplasia with hemophagocytosis in liver biopsies. Am J Surg Pathol. 2011;35(3):337–45. doi: 10.1097/PAS.0b013e318209c681. [DOI] [PubMed] [Google Scholar]
- 12.Ardalan M. Rare presentations of cytomegalovirus infection in renal allograft recipients. Nephrourol Mon. 2012;4(2):431–6. doi: 10.5812/numonthly.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luppi M, Barozzi P, Rasini V, Riva G, Re A, Rossi G. et al. Severe pancytopenia and hemophagocytosis after HHV-8 primary infection in a renal transplant patient successfully treated with foscarnet. Transplantation. 2002;74(1):131–2. doi: 10.1097/00007890-200207150-00023. [DOI] [PubMed] [Google Scholar]
- 14.Ossi C, Delforge ML, Jacobs F, Wissing M, Pradier O, Remmelink M. et al. Fatal primary infection due to HHV6 variant A in a renal transplant patient. Transplantation. 2001;71(2):288–92. doi: 10.1097/00007890-200101270-00021. [DOI] [PubMed] [Google Scholar]
- 15.Ardalan MR, Shoja MM, Tubbs RS, Esmaili H, Keyvani H. Postrenal transplant hemophagocytic lymphohistiocytosis and thrombotic microangiopathy associated with parvovirus b19 infection. Am J Transplant. 2008;8(6):1340–4. doi: 10.1111/j.1600-6143.2008.02244.x. [DOI] [PubMed] [Google Scholar]
- 16.Ardalan MR, Shoja MM, Tubbs RS, Jayne D. Parvovirus B19 microepidemic in renal transplant recipients with thrombotic microangiopathy and allograft vasculitis. Exp Clin Transplant. 2008;6(2):137–43. [PubMed] [Google Scholar]
- 17.Yaich S, Charfeddine K, Hsairi D, Zaghdane S, Kammoun K, Makni S. et al. BK virus-associated hemophagocytic syndrome in a renal transplant recipient. Saudi J Kidney Dis Transpl. 2014;25(3):610–4. doi: 10.4103/1319-2442.132205. [DOI] [PubMed] [Google Scholar]
- 18.Esposito L, Hirsch H, Basse G, Fillola G, Kamar N, Rostaing L. BK virus-related hemophagocytic syndrome in a renal transplant patient. Transplantation. 2007;83(3):365. doi: 10.1097/01.tp.0000248807.63325.cc. [DOI] [PubMed] [Google Scholar]
- 19.Ardalan MR. Mycobacterial disease in renal allograft recipients. J Renal Inj Prev. 2013;2(2):83–4. doi: 10.12861/jrip.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slovut D, Benedetti E, Matas A. Babesiosis and hemophagocytic syndrome in an asplenic renal transplant recipient. Transplantation. 1996;62(4):537–9. doi: 10.1097/00007890-199608270-00018. [DOI] [PubMed] [Google Scholar]
- 21.Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated hemophagocytic syndrome. Lancet Infect Dis. 2006;6(7):447–54. doi: 10.1016/S1473-3099(06)70524-2. [DOI] [PubMed] [Google Scholar]
- 22.Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010;20(2):93–105. doi: 10.1002/rmv.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukaya S, Yasuda S, Hashimoto T, Oku K, Kataoka H, Horita T. et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008;47(11):1686–91. doi: 10.1093/rheumatology/ken342. [DOI] [PubMed] [Google Scholar]
- 24.Dargent JL, Vermylen P, Abramowicz D, Lespagnard L, Cochaux P, Capel P. et al. Disseminated angiosarcoma presenting as a hemophagocytic syndrome in renal allograft recipient. Transpl Int. 1997;10(1):61–4. doi: 10.1007/BF02044344. [DOI] [PubMed] [Google Scholar]
- 25.Eeters P, Sennesael J, De Raeve H, De Waele M, Verbeelen D. Hemophagocytic syndrome and T-cell lymphoma after kidney transplantation: a case report. Transpl Int. 1997;10(6):471–4. doi: 10.1007/s001470050089. [DOI] [PubMed] [Google Scholar]
- 26.Yoshiaki K, Jiro M, Masaki Y, Shoichi U. Hemophagocytic syndrome in an ABO-incompatible living renal transplant recipient with acute rejection during the chronic phase. Nishinihon J Urol. 1999;61(4):341–3. [Google Scholar]
- 27.Marques ID, Caires RA, de Paula FJ, Nahas WC, David-Neto E. Rejection-triggered haemophagocytic syndrome in renal transplantation successfully treated with intravenous immunoglobulin. Clin Kidney J. 2013;6(5):530–2. doi: 10.1093/ckj/sft077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karras A, Thervet E, Legendre C. Groupe Cooperatif de transplantation d’Ile de France Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation. 2004;77(2):238–43. doi: 10.1097/01.TP.0000107285.86939.37. [DOI] [PubMed] [Google Scholar]
- 29.Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10(3):387–92. doi: 10.1097/PCC.0b013e3181a1ae08. [DOI] [PubMed] [Google Scholar]
- 30.Asci G, Toz H, Ozkahya M, Cagirgan S, Duman S, Sezis M. High-dose immunoglobulintherapy in renal transplant recipients with hemophagocytic histiocytic syndrome. J Nephrol. 2006;19(2):322–6. [PubMed] [Google Scholar]
- 31.Braun MC, Cohn RA, Kletzel M. Nephrotic syndrome accompanying familial hemophagocytic syndrome. J Pediatr Hematol Oncol. 1996;18(2):195–7. doi: 10.1097/00043426-199605000-00021. [DOI] [PubMed] [Google Scholar]
- 32.Chiang WC, Wu MS, Tsai CC, Lin SL, Tsai TJ, Hsieh BS. Thrombotic microangiopathy in hemophagocytic syndrome: a case report. J Formos Med Assoc. 2002;101(5):362–7. [PubMed] [Google Scholar]
- 33.Pérard L, Costedoat-Chalumeau N, Limal N, Hot A, Cohen J, Vauthier-Brouzes D. et al. Hemophagocytic syndrome in apregnant patient with systemic lupuserythematosus, complicated with preeclampsia and cerebral hemorrhage. Ann Hematol. 2007;86(7):541–4. doi: 10.1007/s00277-007-0277-7. [DOI] [PubMed] [Google Scholar]
- 34.Wan L, Bellomo R, Di Giantomasso D, Ronco C. The pathogenesis of septic acute renal failure. Curr Opin Crit Care. 2003;9(6):496–502. doi: 10.1097/00075198-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Ardalan MR, Nasri H, Ghabili K, Mohajel Shoja M. Acute tubular necrosis after renal allograft segmental infarction: the nephrotoxicity of necrotic material. Exp Clin Transplant. 2008;6(4):312–4. [PubMed] [Google Scholar]
- 36.Koukouritaki B, Vardaki EA, Papakonstanti EA, Lianos E, Stournaras C, Emmanouel D. TNF-a induces actin cytoskeleton reorganization in glomerular epithelial cells involving tyrosine phosphorylation of paxillin and focal adhesion kinase. Mol Med. 1999;5(6):382–92. [PMC free article] [PubMed] [Google Scholar]
- 37.Di Paola R, Genovese T, Impellizzeri D, Ahmad A, Cuzzocrea S, Esposito E. The renal injury and inflammation caused by ischemia-reperfusion are reduced by genetic inhibition of TNF-αR1: a comparison with infliximab treatment. Eur J Pharmacol. 2013;700(1-3):134–46. doi: 10.1016/j.ejphar.2012.11.066. [DOI] [PubMed] [Google Scholar]
- 38.Al-Lamki RS, Wang J, Vandenabeele P, Bradley JA, Thiru S, Luo D. et al. TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 2005;19(12):1637–45. doi: 10.1096/fj.05-3841com. [DOI] [PubMed] [Google Scholar]
- 39.Rafia S Al-Lamki, Tanya N. Mayadas TNF receptors: signaling pathways and contribution to renal dysfunction. Kidney Int. 2015;87:281–96. doi: 10.1038/ki.2014.285. [DOI] [PubMed] [Google Scholar]
- 40.Thaunat O, Delahousse M, Fakhouri F, Martinez F, Stephan JL, Noël LH. et al. Nephrotic syndrome associated with hemophagocytic syndrome. Kidney Int. 2006;69(10):1892–8. doi: 10.1038/sj.ki.5000352. [DOI] [PubMed] [Google Scholar]
- 41.McCarthy T, Sharma R, Sharma M, Li JZ, Ge XL, Dileepan KN. et al. TNF-a increases albumin permeability of isolated rat glomeruli through the generation of superoxide J Am Soc. Nephrol. 1998;9(3):433–8. doi: 10.1681/ASN.V93433. [DOI] [PubMed] [Google Scholar]
- 42.Sun L, Kanwar YS. Relevance of TNF-α in the context of other inflammatory cytokines in the progression of diabetic nephropathy. Kidney Int. 2015;88(4):662–5. doi: 10.1038/ki.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffmann U, Bergler T, Rihm M, Pace C, Krüger B, Rümmele P. et al. Upregulation of TNF receptor type 2 in human and experimental renal allograft rejection. Am J Transplant. 2009;9(4):675–86. doi: 10.1111/j.1600-6143.2008.02536.x. [DOI] [PubMed] [Google Scholar]
- 44.Chang CC, Hsiao PJ, Chiu CC, Chen YC, Lin SH, Wu CC. et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168–71. doi: 10.1016/j.cca.2014.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Ardalan MR. Review of thrombotic microangiopathy (TMA), and post-renal transplant TMA. Saudi J Kidney Dis Transpl. 2006;17(2):235–44. [PubMed] [Google Scholar]
- 46.Kfoury Baz EM, Mikati AR, Kanj NA. Hemophagocytic syndrome associated with thrombotic thrombocytopenic purpura during therapeutic plasma exchange. Therapeutic Apheresis. 2002;6(2):159–62. doi: 10.1046/j.1526-0968.2002.00362.x. [DOI] [PubMed] [Google Scholar]
- 47.Nakabayashi M, Adachi T, Izuchi S, Sugisaki A. Association of hypercytokinemia in the development of severe preeclampsia in a case of hemophagocytic syndrome. Semin Thromb Hemost. 1999;25(5):467–71. doi: 10.1055/s-2007-994952. [DOI] [PubMed] [Google Scholar]
- 48.Emmenegger U, Spaeth PJ, Neftel KA. Intravenous immunoglobulin for hemophagocytic lymphohistiocytosis. J Clin Oncol. 2002;20(2):599–601. doi: 10.1200/JCO.2002.20.2.599. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe H, Hirase N, Goda H, Nishikawa H, Ikuyama S. Oral low-dose tacrolimus therapy for refractory hemophagocytic syndrome associated with systemic lupus erythematosus. Mod Rheumatol. 2012;22(2):284–9. doi: 10.1007/s10165-011-0491-y. [DOI] [PubMed] [Google Scholar]
- 50.Orbach H, Tishler M, Shoenfeld Y. Intravenous immunoglobulin and the kidney--a two-edged sword. Semin Arthritis Rheum. 2004;34(3):593–601. doi: 10.1016/j.semarthrit.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 51.Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29(8):569–73. doi: 10.1097/MPH.0b013e3180f61be3. [DOI] [PubMed] [Google Scholar]
- 52.Sano T, Sakai H, Takimoto K, Ohno H. Rituximab alone was effective for the treatment of a diffuse large B-cell lymphoma associated with hemophagocytic syndrome. Int J Clin Oncol. 2007;12(1):59–62. doi: 10.1007/s10147-006-0627-9. [DOI] [PubMed] [Google Scholar]
- 53.Ponticelli C, Alberighi OD. Haemophagocytic syndrome—a life-threatening complication of renal transplantation. Nephrol Dial Transplant. 2009;24(9):2623–7. doi: 10.1093/ndt/gfp282. [DOI] [PubMed] [Google Scholar]
- 54.Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S. et al. Success with inflin treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. 2006;81(1):59–61. doi: 10.1002/ajh.20462. [DOI] [PubMed] [Google Scholar]
- 55.Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J. et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–9. doi: 10.1002/pbc.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]