

Abstract
Interleukin 17 (IL-17)-producing helper (TH17) and inducible regulatory CD4+ T (iTreg) cells emerge from an overlapping developmental program. In the intestines, the vitamin A metabolite retinoic acid (RA) is produced at steady state and acts as an important cofactor to induce iTreg cell development while potently inhibiting TH17 development. Here, we found that IL-1 was required to fully override RA-mediated Foxp3 expression and induce protective TH17 responses. Through induction of an NF-κB-dependent repression of SOCS3 expression, IL-1 increased the amplitude and duration of STAT3 phosphorylation induced by TH17-polarizing cytokines, leading to an altered balance of STAT3–STAT5 binding to shared consensus sequences in developing T cells. Thus, IL-1 signaling differentially modulated STAT activation downstream of cytokine receptors to control TH17–iTreg developmental fate.
INTRODUCTION
TH17 and induced regulatory CD4+ T (iTreg) cells emerge from a shared developmental axis1. While transforming growth factor-β (TGF-β) contributes to developmental programming of both subsets, the pro-inflammatory cytokine interleukin 6 (IL-6) favors TH17 development at the expense of iTreg cell development2–6. Conversely, retinoic acid (RA), a vitamin A metabolite produced by intestinal stromal cells and dendritic cells (DCs) that express retinaldehyde dehydrogenases (RALDHs)7, acts in concert with TGF-β to promote Foxp3+ expression and Treg cell development while potently inhibiting TH17 development8–12. A substantial percentage of TH17 cells resident in intestinal lamina propria have expressed Foxp3 at some point during their development, indicating a dynamic relationship between Rorγt+ TH17 and Foxp3+ Treg cells developing in the intestines5. Whereas IL-6 signaling induces STAT3 phosphorylation that is required for Rorγt expression and TH17 development, the actions of RA are at least partially dependent on IL-2, which induces STAT5 phosphorylation that is required for Foxp3 expression and iTreg cell development, and which suppresses TH17 development9,13,14. A number of DNA binding sites targeted by STAT3 in TH17 lineage gene loci can also bind STAT5, providing a mechanism for competitive antagonism of these trans-acting factors downstream of IL-6 and IL-2 receptor signaling15.
While IL-6 is important for reciprocally promoting TH17 cell development and repressing iTreg development, it effects in countering RA-mediated inhibition of TH17 development are incomplete9,14. Thus, even at saturating doses of IL-6, TH17 development is suppressed by RA, suggesting that additional signals might be required to override dominant RA signaling in the gut. IL-1 is another pro-inflammatory cytokine that promotes TH17 development while subverting Treg cell development2,16, and has been shown to be important in the development of TH17 cells in the gut, at least at steady-state17. However, other reports have suggested that IL-1β–IL-1R signaling is dispensable in differentiation of intestinal TH17 cells at steady-state, based on studies of MyD88-deficient mice, which have impaired IL-1R and TLR signaling18,19. The role of IL-1 signaling during pathogen-induced intestinal inflammation has not been well studied. Therefore, both the mechanism of IL-1 in promoting intestinal TH17 development as well as the role of IL-1 in TH17 development during acute intestinal inflammation, particularly in disease models where TH17 cells are protective, remain largely undefined20,21.
In this study, we sought to define the role of IL-1 in regulating the developmental balance of TH17 and Treg cells, with emphasis on its effects in the intestines during challenge with the enteropathogenic bacterium, Citrobacter rodentium, which elicits a TH17 pathway response that is required for host protection3,21,22. Our results indicate that IL-1 signaling is required to enhance IL-6 signaling to fully override RA-mediated Foxp3 expression and induce protective TH17 responses. Through an NF-κB–dependent mechanism, IL-1 repressed the expression of suppressor of cytokine synthesis (SOCS3), a feedback inhibitor of JAK-induced tyrosine phosphorylation of STAT3. Inhibition of SOCS3 expression by IL-1 resulted in an increased amplitude and duration of STAT3 tyrosine phosphorylation downstream of signaling by TH17-polarizing cytokines, without altering IL-2–induced STAT5 signaling tyrosine phosphorylation. IL-1 signaling therefore altered the balance of STAT3/STAT5 binding to shared consensus sequences in developing T cells, including the CNS2 element in the Foxp3 locus that regulates stability of Foxp3 expression, as well as target sequences in the Il17a–Il17f locus. Thus, IL-1 signaling differentially modulates STAT activation downstream of cytokine receptors to control TH17–iTreg cell developmental fate.
RESULTS
IL-1β reverses RA-induced inhibition of TH17 differentiation
IL-6 counteracts the effects of RA-mediated suppression of TH17 cell development, albeit incompletely9. In the course of examining the role for IL-1β in promoting TH17 cell development, we found that, in contrast to IL-6, IL-1β completely reversed the impairment of TH17 cell differentiation observed when DCs from mesenteric lymph nodes (MLNs) were used to activate naïve CD4+ T cells (Fig. 1a,b). Moreover, IL-1β was comparable to the retinoic acid receptor (RAR) inhibitor, LE450, in blocking the effects of RA. Accordingly, addition of IL-1β overrode the inhibition of TH17 differentiation by RA, irrespective of RA concentration (Fig. 1c,d). This result was not due to down-regulation of RAR or RXR receptor subunits, as all family members were either unchanged or modestly increased by IL-1 signaling, and occurred despite partial RA-mediated down-modulation of IL-1R1, which was highly expressed by developing TH17 cells relative to TH0 cells (Supplementary Fig. 1).
FIGURE 1. IL-1β counteracts RA-dependent inhibition of TH17 cell development.
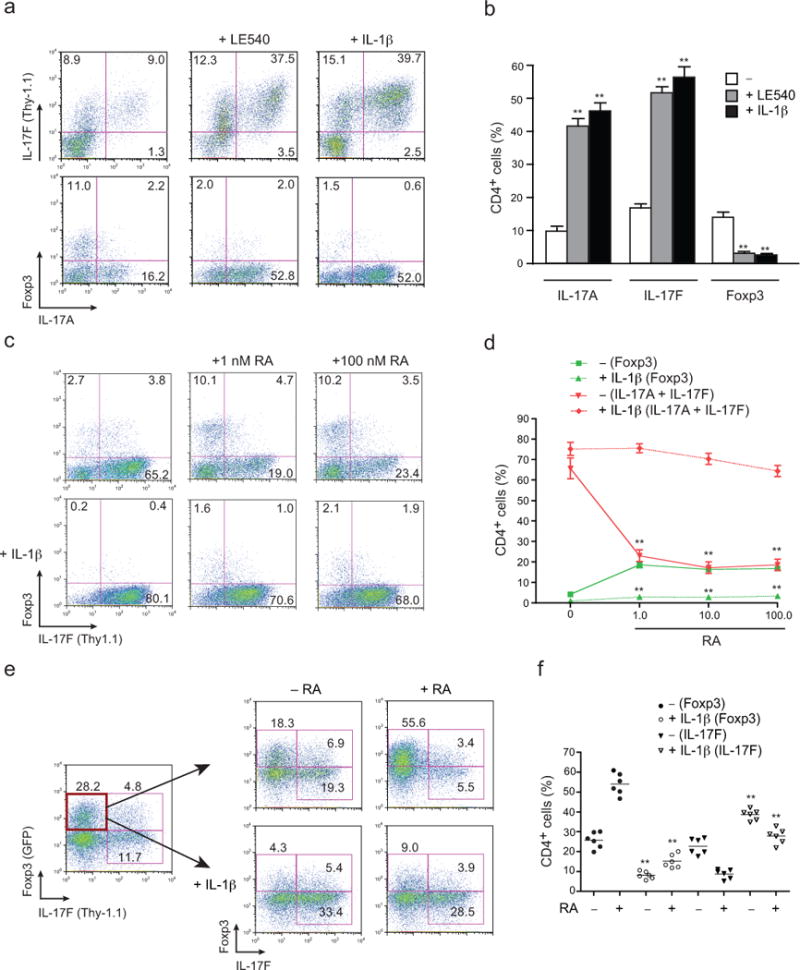
(a) Naïve CD4+ T cells (CD4+CD25−CD62Lhi CD44lo) from Il17fthy1.1 mice were activated with soluble anti-CD3 on DCs isolated from mesenteric lymph nodes (MLNs) of IL-1β–deficient (Il1b−/−) mice under TH17 polarizing conditions, with the indicated additions of an at-RA inhibitor (LE540; 1 μM) or IL-1β (20 ng/ml). Cells recovered on day 4 were stained for surface CD4 and Thy1.1 (IL-17F), and intracellular IL-17A and Foxp3, and analyzed by flow cytometry. Numbers are percentages of cells in each quadrant. (b) Pooled data from a showing frequencies of IL-17A, IL-17F (Thy1.1) and Foxp3 single producers among CD4+ T cells. (c) Expression of Thy1.1 (IL-17F) and Foxp3 from naïve CD4+ T cells from Il17fthy1.1 mice activated with soluble anti-CD3 and Il1b−/− DCs under TH17 polarizing conditions with or without IL-1β, in presence or absence of indicated concentration of at-RA at day 4. (d) Pooled data from c showing total frequencies of IL-17A–, IL-17F (Thy1.1)– and Foxp3–expressing CD4+ T cells. (e) Naïve CD4+ T cells from Il17fthy1.1.Foxp3gfp mice were activated with plate-bound anti-CD3 and soluble anti-CD28 under TH17 polarizing conditions. On day 4 of primary culture the Thy1.1−GFP+ fraction of cells were sorted by flow cytometry and further cultured under TH17 conditions with or without IL-1β (20 ng/ml), in the presence or absence of at-RA (1 nM). On day 4 of secondary culture, frequencies of IL-17F (Thy1.1) versus Foxp3-GFP+ cells were determined after surface staining of Thy1.1 (IL-17F) and flow cytometry. Numbers are percentages of cells in the each quadrant. (f) Pooled data from secondary cultures from e showing frequencies of Foxp3+ (GFP) and IL-17F+ (Thy1.1) CD4+ T cells. Data are representative of one of three similar independent experiments (a); pooled from three independent experiments with nine samples (n = 9) per group (b); representative of one of three similar independent experiments (c); pooled from three experiments with twelve samples (n = 12) per group (d); representative of one of two independent experiments (e); or pooled from two independent experiments with six samples (n = 6) per group (f). Data are means and s.e.m. in b,d,f. **P < 0.01 (two-tailed unpaired t-test).
In extension of these studies, we examined the effects of IL-1 in reversing the expression of Foxp3 supported by RA signaling in developing TH17 cells (Fig. 1e,f). Using TH17 cells derived from naïve precursors of dual reporter mice (Il17fThy1.11Foxp3gfp), Foxp3+IL-17F− cells were sorted and re-stimulated under TH17 polarizing conditions in the absence or presence of RA addition. In the absence of added RA, a significant fraction of sorted Foxp3+ cells lost Foxp3 expression and expressed IL-17F. This transition was significantly inhibited by RA, which supported retention of Foxp3 and suppressed expression of IL-17. Similar to its effects on developing TH17 cells, IL-1 signaling overrode the effects of RA to support Foxp3 expression and IL-17 repression. Thus, IL-1 receptor signaling overrides RA-mediated induction of iTreg cell programming to promote and stabilize TH17 programming, thereby shifting the iTreg–TH17 balance in favor of TH17 cell development.
IL-1R1 deficiency alters iTreg– TH17 balance during infection
To explore the role of IL-1 signaling in modulating the iTreg–TH17 balance in vivo, we examined infection by the intestinal pathogen, C. rodentium, protection against which is mediated by the TH17 pathway3,21,22. It has been suggested from studies of steady-state intestinal TH17 development that conflicting reports on the role of IL-1 have arisen due to use of phorbol ester (PMA) and ionomycin stimulation of recovered T cells ex vivo17,23. We therefore used wild-type and IL-1R1-deficient Il17fThy1.11Foxp3gfp dual reporter mice to directly examine gene expression ex vivo without requirement for PMA plus ionomycin or anti-CD3 stimulation-induced recall24. Because Il17f is expressed early in TH17 development, at which time it is dominant over Il17a expression24, the Il17fThy1.1 reporter model provides a sensitive read-out of early TH17 gene expression. Moreover, the Il17fThy1.1 reporter model enables specific depletion of IL-17F–producing cells in vivo by administration of anti-Thy1.1 mAb25.
Anti-Thy1.1 mAb-mediated depletion of IL-17F–producing cells in C. rodentium-inoculated Il17fThy1.1 reporter mice during the peak of infection (3–7 days post-infection; ref.21, and data not shown) resulted in impaired bacterial clearance and heightened injury of the intestinal mucosa (Fig. 2a,b and Supplementary Fig. 2a,b). Infection of mice deficient for IL-1 receptor 1 (Il1r1−/−) showed similarly impaired host protection with a concomitant decrease in colonic IL-17A production but not production of interferon-γ (IFN-γ)(Fig. 2c,d). Those results are similar to our previous findings using mice deficient for IL-17 receptor A (IL-17RA) and consistent with a role for IL-1 in promoting TH17 pathway–dependent host protection21. Depletion of IL-17-producing cells in Il1r1−/−Il17fThy1.1 mice did not significantly alter this result, suggesting that a major effect of IL-1R1 deficiency during this period of the infection was mediated by its actions on these cells. Production of IL-22 in the infected colon was reduced in IL-1R1-deficient mice, but only at the peak time point (d7 post-infection; Supplementary Fig. 2c).
FIGURE 2. IL-1R signaling is required for host-protective TH17 and Treg–TH17 cell balance in vivo.
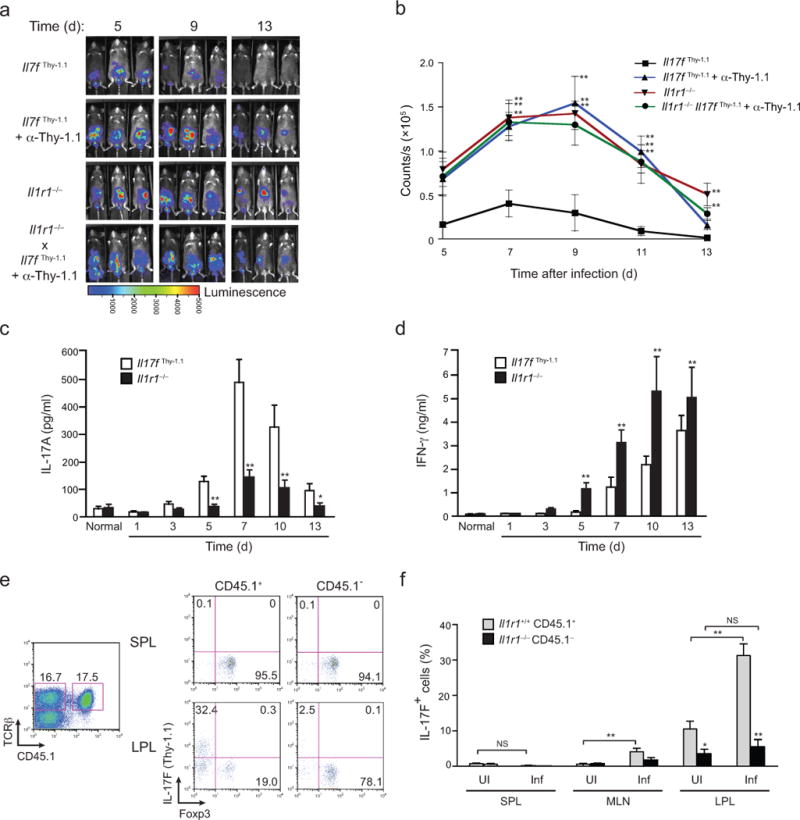
(a) Serial whole-body imaging of untreated Il17fthy1.1 mice, Il17fthy1.1 mice treated with depleting anti-Thy1.1 mAb or Il1r1−/− mice or Il1r1−/− mice treated with depleting anti-Thy1.1 mAb after inoculation with luminescent strain of C. rodentium and imaged at the indicated days post infection. (b) Colonization kinetic data from a represented as counts/sec at different time points post-infection with C. rodentium. (c,d) Quantitative ELISA of IL-17A (c) and IFNγ (d) in supernatants from cultured homogenates of colonic tissue collected from Il17fthy1.1 and Il1r1−/− mice at the indicated times after inoculation with C. rodentium. (e) 3 × 106 of donor Il1r1+/+.Il17fthy1.1 (CD45.1/CD45.2) and Il1r1−/−.Il17fthy1.1 (CD45.2) CD4+ T cells were mixed and co-transferred to recipient Tcrb−/− mice that were either uninfected or infected with C. rodentium 2 weeks post-reconstitution (see Supplementary Fig. 2d for schematic). Seven days later, expression of Thy1.1 (IL-17F) and intracellular Foxp3 by CD45.1+ and CD45.1− splenic lymphocyte (SPL) and colonic lamina propria lymphocytes (LPL) from reconstituted recipient Tcrb−/− mice was analyzed (gated on activated CD4+ T cells). Numbers in each quadrant indicate the frequency of cells. Single plot represents equal frequencies of Il1r1+/+ and Il1r1−/− TCRβ+ cells within reconstituted mice. (f) Frequencies of IL-17F (Thy1.1) cells in Il1r1+/+ CD45.1+ and Il1r1−/− CD45.1− populations of SPL, MLN and LPL of uninfected and infected recipient Tcrb−/− mice. Data are: representative of one of two similar independent (Il1r1−/− mice treated with depleting anti-Thy1.1 mAb) or one of three similar independent experiments (Il17fthy1.1 mice, Il17fthy1.1 mice treated with depleting anti-Thy1.1 mAb or Il1r1−/− mice) a; pooled from two or three independent experiments with nine to eleven mice per group b; from two independent experiments with six mice per group c,d; representative of one of two similar independent experiments e; or pooled from two independent experiments with six mice per group f. Data are means and s.e.m. in b,c,d,f. N.S.= Not significant, *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
We observed a transient increase in Foxp3+CD4+ T cells in the large intestine and draining lymph nodes of wild-type mice during infection (days 3–5; Supplementary Fig. 3a,b), as well as a shift in the balance of IL-17+ versus Foxp3+ cells recovered from the large intestines of infected IL-1R1–deficient mice (Supplementary Fig. 3c,d). Therefore, we posited that infection-induced IL-1 signaling in CD4+ T cells might be required to overcome Treg programming that is favored in intestinal tissues by production of RA, as was observed ex vivo. To test this hypothesis, congenically marked CD4+ T cells derived from wild-type (CD45.1+) or IL-1R1–deficient (CD45.1−) Il17fThy1.1 reporter mice were co-transferred into T cell receptor β-deficient (Tcrb−/−) recipients, which were either untreated (uninfected) or inoculated with C. rodentium (infected), and the frequencies of Foxp3+ and IL-17F+ cells assessed (Fig. 2e,f and Supplementary Fig. 2d). Although the large majority of transferred T cells were unreactive to C. rodentium antigens, assessment of the frequencies of Foxp3+ and IL-17F+ CD4+ T cells among the pool of recently activated cells in the lamina propria of the large intestine (LPL) showed a marked shift towards IL-17F expression by wild-type T cells relative to that of IL-1R1–deficient T cells (>6-fold), with a reciprocal decrease in the frequency of Foxp3+ T cells (>2.5-fold). In contrast, there were no significant differences in frequencies of Foxp3+ or IL-17F+ cells recovered from recipient spleens. These findings are consistent with a defect in iTreg to TH17 transition of activated T cells in the absence of IL-1 signaling.
In vivo RA blockade compensates for IL-1 signaling deficiency
To further examine the possible transition of Foxp3+ precursors into IL-17–producing effector cells, we tracked the fates Foxp3+IL-17F− CD4+ T cells isolated from infected wild-type and IL-1R1–deficient dual reporter mice (Il17fThy1.11Foxp3gfp) following their transfer into infected TCRβ–deficient recipients (Fig. 3a,b and Supplementary Fig. 3e). In accord with a role for IL-1 signaling to promote the silencing of Foxp3 and induction of IL-17, there was a significant increase in the frequencies of Foxp3-expressing T cells recovered from recipients of IL-1R1–deficient Foxp3+ T cells compared to recipients of wild-type Foxp3+ T cells, with a reciprocal deficit in IL-17–expressing T cells. We then determined whether impaired TH17 development in IL-1R1-deficient mice was due to a failure to overcome RA-mediated repression. Infected IL-1R1-deficient mice were treated with an RAR antagonist or vehicle following inoculation with C. rodentium (Fig. 3c,d). Blockade of RAR signaling in infected IL-1R1-deficient mice, but not in wild-type mice, significantly reversed the deficit of IL-17–producing T cells in both the LPL and MLN, but not in spleen, and resulted in decreased bacterial loads that were associated with protection from mucosal injury and reduced weight loss (Fig. 3e,f and Supplementary Fig. 4). Collectively, these findings identify an important role for pathogen-induced IL-1 signaling in overriding iTreg cell programming favored by RA-producing intestinal DCs to induce protective TH17 responses T cells to microbial antigens.
FIGURE 3. In absence of IL-1β, in vivo blockade of RA facilitates iTreg to TH17 cell conversion during enteropathogenic bacterial infection.
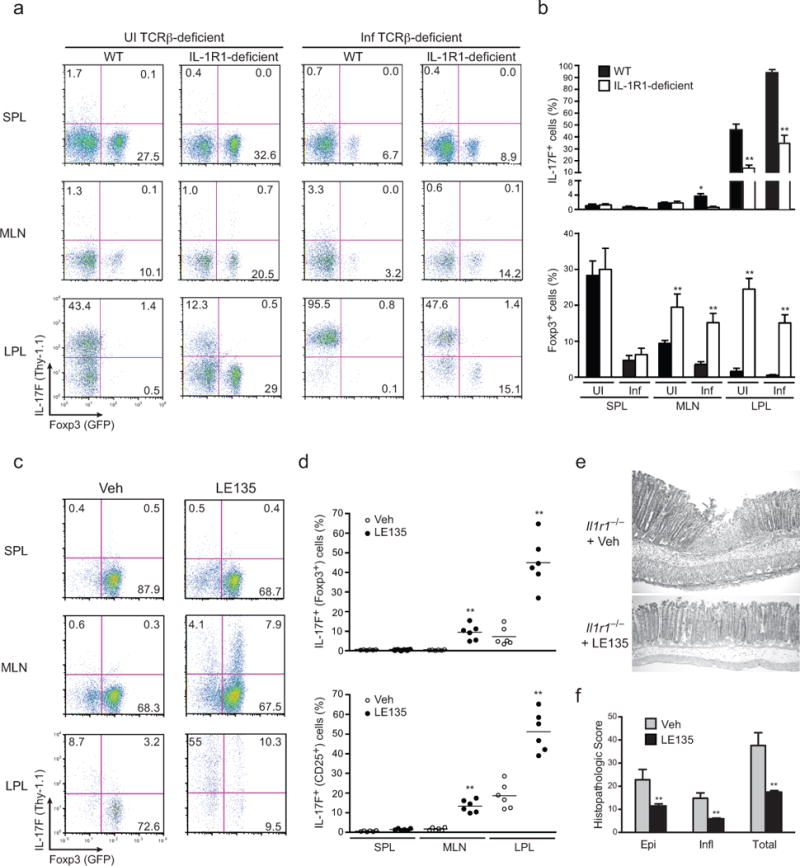
(a) 2.5 × 106 Foxp3+IL-17F− CD4+ T cells of infected donor Il1r1+/+.Il17fthy1.1.Foxp3gfp and Il1r1−/−.Il17fthy1.1.Foxp3gfp mice were collected and sorted on day 2 post-infection (PI) and transferred to recipient TCRβ–deficient (Tcrb−/−) mice that were either uninfected or infected with C. rodentium 2 days before (see Supplemental Fig. 3e for schematic). Expression of Thy1.1 (IL-17F) and Foxp3-GFP on splenic, MLN and LP isolates of uninfected recipient Tcrb−/− or d2 infected recipient Tcrb−/− was analyzed 8 days post-infection. Splenic, MLN and LP lymphocytes were isolated and analyzed without restimulation ex vivo for Thy1.1 (IL-17F) and Foxp3-GFP expression by flow cytometry after gating on TCRβ+ cells. Numbers indicate the frequencies in each quadrant. (b) Frequencies of splenic, MLN and colonic IL-17F (Thy1.1) cells and Foxp3-GFP cells as prepared in b. (c) Expression of Thy1.1 (IL-17F) and Foxp3 (GFP+) CD4+CD25+ T cells in Il1r1−/−.Il17fthy1.1.Foxp3gfp mice that were inoculated with 2 × 109 cfu C. rodentium and gavaged with vehicle alone or with retinoic acid inhibitor (LE135) on days 3–7 post infection. On d8 PI, mice were sacrificed and frequencies of Thy1.1 (IL-17F) and Foxp3 (GFP+) CD4+CD25+ T cells in spleen, MLN and lamina propria were assessed by flow cytometry without restimulation ex vivo. (d) Frequencies of Thy1.1 (IL-17F) and Foxp3 (GFP+) expression within Foxp3+ (GFP+) and activated (CD4+CD25+) fractions from splenic, MLN and lamina propria lymphocytes of vehicle treated or LE135 treated C. rodentium infected Il1r1−/−.Il17fthy1.1.Foxp3gfp mice. (e) Representative histopathology of H&E-stained colonic tissues sections derived from vehicle–treated or LE135–treated, C. rodentium–infected Il1r1−/−.Il17fthy1.1.Foxp3gfp mice at d10 PI. (f) Histopathology scoring of colons from untreated or LE135 treated, infected Il1r1−/−.Il17fthy1.1.Foxp3gfp mice processed on d10 PI. Data are: representative of one of three similar independent experiments a; pooled from three independent experiments with 10 mice per group b; representative of one of two similar independent experiments c; pooled from two independent experiment with 6 mice per group where each point indicates one mouse d; representative of one of two independent experiments e; or pooled from two independent experiments with 4 mice per group f. Data are means and s.e.m. in b,d,f. *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test
IL-1β counters RA-driven, IL-2–STAT5–mediated TH17 repression
RA suppresses TH17 cell development in favor of iTreg cell development at least in part via an IL-2–dependent mechanism9,14. To determine whether IL-1 signaling could also modulate IL-2–dependent suppression of TH17 differentiation, we examined the effect of addition of increasing concentrations of IL-2 to cultures of naïve CD4+ T cells activated by antigen under TH17 cell polarizing conditions, with or without the addition of IL-1β (Fig. 4a,b). In the absence of IL-1β addition, increasing concentrations of IL-2 suppressed frequencies of IL-17+ cells with reciprocal increases in frequencies of Foxp3+ cells as reported14. IL-1β abrogated the suppressive effects of IL-2, irrespective of the concentration of IL-2. Comparable effects of IL-1β on IL-17 expression and Foxp3 repression were obtained under DC-free conditions of TH17 polarization, wherein the induction of higher concentrations of endogenously produced IL-2 favor less robust TH17 development and higher expression of Foxp3 (Supplementary Fig. 5, and data not shown). Notably, IL-23 signaling had no comparable effect on the TH17–iTreg cell balance during the early stages of differentiation (3–4 days) when IL-1 signaling induced an enhanced TH17 response, and addition of both IL-23 and IL-1β did not augment the effect of IL-1β alone (Supplementary Fig. 5b,c, and data not shown). So, in line with its effects to override RA-mediated repression of TH17 differentiation, IL-1β antagonized the effects of IL-2 and this effect was independent of a requirement for IL-23 co-signaling.
FIGURE 4. IL-1β counteracts RA-driven IL-2/STAT5–dependent repression of TH17 development.
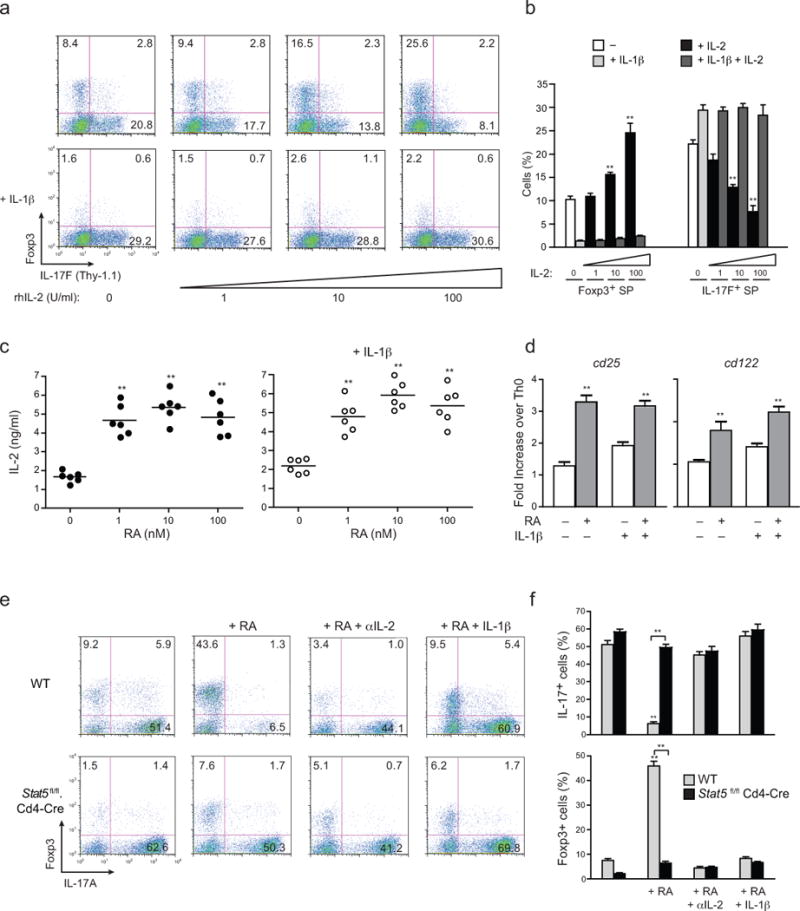
(a) Expression of Thy1.1 (IL-17F) and Foxp3 from naïve OTII-Tg CD4 T cells (CD4+CD25−CD62LhiCD44lo) from OTII.Il17fthy1.1 mice activated with OVA peptide and IL-1β–deficient DCs for 4 days under TH17 polarizing conditions, with or without IL-1β addition, and with or without addition of the indicated doses of rhIL-2 at day 4. (b) Frequencies of Foxp3 and IL-17F single-producers from OTII-Tg CD4+ T cells polarized as in a at indicated concentrations of rhIL-2 (c) Quantitative IL-2 ELISA from supernatants of naïve CD4+ T cells from Il17fthy1.1 mice that were activated with plate-bound anti-CD3 and soluble anti-CD28 under TH17 polarizing conditions in absence (left) or presence (right) of IL-1β addition, with or without addition of addition of RA at the indicated concentrations (1–100 nM). (d) Expression of Cd25 and Cd122 transcripts by quantitative RT-PCR from naïve CD4+ T cells from Il17fthy1.1 mice that were activated with plate-bound anti-CD3 and soluble anti-CD28 under TH17 polarizing conditions in absence or presence of IL-1β, with or without addition of RA (1 nM) at 60 h. Expression values are normalized to TH0 controls. (e) Expression of IL-17A and Foxp3 from MACS-purified CD8−CD4+ thymocytes isolated from WT B6 or Stat5fl/fl.Cd4-Cre mice that were activated with soluble anti-CD3 and Il1b−/− splenic DCs under TH17 polarizing condition (IL-6+TGF-β with or without the indicated additions of RA (1 nM), anti-IL-2 (10 μg/ml) and IL-1β (20 ng/ml) for 4 days. (f) Frequencies of IL-17A+ and Foxp3+ from CD4+ T cells from WT B6 or Stat5fl/fl.Cd4-Cre mice as treated in (e). Data are: representative of one of three similar independent experiments a; pooled from three independent experiments with nine samples per group (n = 9) b; pooled from two independent experiments with six samples (n = 6) c; representative of two independent experiments with 6 samples per group (n = 6) where individual data points represent each sample d; representative of one of three similar independent experiments e; or pooled from three independent experiments with ten samples (n = 10) per group f. Data are means and s.e.m. in b,c,d,f). **P < 0.01 (two-tailed unpaired T-test).
The similar effects of IL-1 signaling on reversal of RA– and IL-2–mediated inhibition of TH17 cell differentiation were consistent with our finding that production of IL-2 by T cells was enhanced ~3-fold by addition of RA at all concentrations tested (Fig. 4c). This result indicated that RA-mediated repression of TH17 cell differentiation was due, at least in part, to enhanced IL-2 production. Consistent with the actions of IL-2 to induce heightened expression of the inducible components of the IL-2 receptor, addition of RA also up-regulated IL-2Rα (CD25) and IL-2Rβ (CD122) (Fig. 4d). Notably, neither the enhanced IL-2 production nor up-regulation of IL-2 receptor components by RA was significantly affected by IL-1β, suggesting that the principal effect of IL-1β in blocking the effects of RA was likely due to interference of IL-2 signaling downstream of receptor binding of IL-2. Accordingly, blockade of IL-2 signaling reversed the reciprocal effects of RA on the TH17-iTreg cell developmental balance similarly to that induced by IL-1β (Fig. 4e,f). Further, using CD4+ T cells deficient in expression of STAT5 (and thus IL-2 signaling), the effects of RA to repress IL-17 expression in favor of Foxp3 expression were comparably impaired and were not further augmented by IL-1β signaling (Fig. 4e,f). Thus, although RA acts to enhance IL-2 production and expression of the inducible components of the IL-2 receptor by T cells, the reversal of RA-mediated effects by IL-1β signaling appears to be mediated primarily through interference of IL-2 signaling and not through altered expression of IL-2 or its receptor.
IL-1β enhances pSTAT3 via NF-κB–dependent repression of Socs3
To explore mechanisms by which IL-1 might interfere with RA-mediated modulation of IL-2 signaling, we examined the tyrosine phosphorylation of STAT5 following stimulation of T cells under TH17 polarizing conditions, with or without addition of RA or RA plus IL-1β (Fig. 5a). Consistent with its effects to enhance expression of IL-2 and its receptor, RA enhanced tyrosine phosphorylation of STAT5 in TH17 cells, which was not significantly altered by concurrent IL-1 signaling. In contrast, RA had no effect on IL-6–induced tyrosine phosphorylation of STAT3, whereas addition of IL-1 resulted in a substantial increase in tyrosine-phosphorylated (pY)-STAT3. The effect of IL-1 signaling resulted in both increased and more sustained STAT3 tyrosine phosphorylation (Fig. 5b), and was not limited to IL-6–induced pY-STAT3, as similar results were obtained when STAT3 was activated downstream of signaling by IL-21 (Supplementary Fig. 6) or IL-23 (Fig. 5c). Therefore, while IL-1 signaling failed to directly repress the RA-mediated enhancement of IL-2–induced pY-STAT5, it did alter the pY-STAT5/pY-STAT3 ratio through its enhancement of IL-6–, IL-21– and IL-23–induced pY-STAT3, establishing an indirect mechanism by which IL-1 acts in concert with each of the STAT3-activating TH17 pathway cytokines to override RA-mediated, IL-2–dependent repression of TH17 development.
FIGURE 5. NF-κB-dependent SOCS3 repression by IL-1β enhances amplitude and duration of STAT3 phosphorylation.
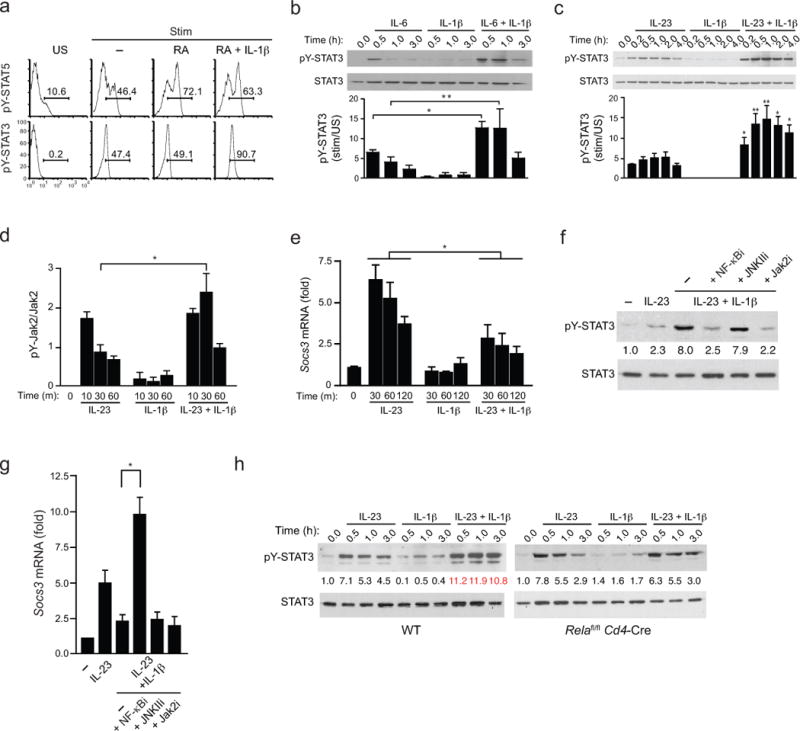
(a) Intracellular pSTAT5 and pSTAT3 analysis from naïve CD4+ T cells from WT B6 mice that were activated with plate-bound anti-CD3 and soluble anti-CD28 under TH17 polarizing conditions (IL-6 +TGF-β with or without the indicated additions of RA and IL-1β for 4 days. Prior to analysis, IL-6 and IL-2 were added to cultures for 20 min and intracellular pSTAT5 and pSTAT3 were determined by flow cytometry. (b) Expression of phospho-tyrosine(705)-STAT3 (pY-STAT3) from naïve CD4+ T cells cultured under TH17 polarizing conditions for 4–5 days, then restimulated with IL-6 alone, IL-1β alone, or both for indicated time periods. Cell lysates were harvested, immunoblotted with antibody directed against phospho-tyrosine(705)-STAT3 (pY-STAT3) or total STAT3 (top). (c) Expression of phospho-tyrosine(705)-STAT3 (pY-STAT3) by immunoblot from naïve CD4+ T cells that were cultured under TH17 polarizing conditions for 4 days, then restimulated with IL-23 alone, IL-1β alone, or both for indicated time periods and assessed as in b. (d) Quantitative analysis of active phosphorylated form of Jak2 from naïve CD4+ T cells that were cultured for under TH17-polarizing conditions and activated as in c, then lysed and subjected to ELISA analysis that quantified both the active phosphorylated form of Jak2 [Jak2(pYpY1007/1008)] and total Jak2. pYpY-Jak2 values were normalized to total Jak2 expression and data are expressed as fold change over unstimulated TH17 cells where unstimulated cells were assigned a value of 1. (e) Expression of Socs3 transcripts by quantitative RT-PCR from naïve CD4+ T cells that were cultured under TH17 conditions as in c and restimulated with IL-23 and/or IL-1β for the indicated times. Transcript abundance of Socs3 were normalized against β2-microglobulin and relative expression compared to unstimulated cells was calculated using the ΔΔCt method. (f) Expression of phospho-tyrosine(705)-STAT3 (pY-STAT3) by immunoblot from naïve CD4+ T cells polarized under TH17 conditions were isolated and pre-treated with indicated signaling inhibitors for 1 h then either left unstimulated or treated with IL-23 +/− IL-1β before cell lysates were harvested and immunoblotted for pY-STAT3 and total STAT3 as in c. Average values of pooled data representing relative expression of IDVs of pY-STAT3 normalized to total STAT3 are shown (between upper and lower panels). (g) Expression of Socs3 transcripts by quantitative RT-PCR from naïve CD4+ T cells polarized under TH17 conditions were isolated, pre-treated with the indicated signaling inhibitors for 1 h, were then restimulated with IL-23 and/or IL-1β for 45 min and harvested for RNA isolation and Socs3 mRNA quantification as in e. (h) Expression of phospho-tyrosine(705)-STAT3 (pY-STAT3) by immunoblot from naïve CD4+ T cells from WT and Relafl/fl.Cd4-Cre mice were polarized and analyzed as in c. Average values of pooled data representing relative expression of IDV of pY-STAT3 normalized to total STAT3 are shown (between upper and lower panels). Data are: representative of one of two similar experiments a; representative of one of three similar independent experiments (b,c); pooled from two independent experiments with six samples (n = 6) per group d; three independent experiments with nine samples (n = 9) per group e; pooled from two independent experiments with nine samples (n = 9) per group g; representative of one of two similar independent experiments (f,h) where numbers in red (IL-23 + IL-1β) are significantly different (P < 0.01) from corresponding values for cells stimulated with IL-23 alone (mean and s.e.m. in b,c,d,e,g). * P < 0.05 and ** P < 0.01 (two-tailed unpaired t-test).
Because of its failure to impact RA-mediated effects on the iTreg–TH17 balance during early differentiation (Supplementary Fig. 5), we chose IL-23, also a known pSTAT3 inducer, to further examine mechanisms by which IL-1 co-signaling might alter pSTAT3 abundance. We initially examined IL-1 effects upstream of STAT3 tyrosine phosphorylation (Fig. 5d). Addition of IL-1 significantly increased and sustained the rapid tyrosine phosphorylation of Jak2 induced by IL-23 receptor activation. Tyrosine phosphorylation of Jak2 (pYpY-Jak2) induced by IL-23 receptor heterodimerization recruits STAT3 to induce its tyrosine phosphorylation, and is competitively inhibited by the rapid expression of SOCS3 downstream of STAT3 activation26. We reasoned, therefore, that IL-1 might modulate Jak2 and STAT3 phosphorylation through effects on expression of SOCS3. Indeed, while IL-1 signaling alone had no effect on Socs3 mRNA expression, it significantly inhibited the expression induced by IL-23 (Fig. 5e), indicating that IL-1–dependent inhibition of Socs3 expression contributed to enhanced pY-STAT3.
NF-κB and MAP kinase signaling cascades are activated downstream of IL-1 receptor ligation. We therefore examined the effects of specific inhibitors of NF-κB, JNKII, Erk and p38 on IL-1–induced modulation of pY-STAT3 and Socs3 mRNA, including a Jak2 inhibitor as a control (Fig. 5f,g and Supplementary Fig. 7). Notably, of the IL-1 signaling inhibitors examined, only NF-κB inhibitor blocked the enhancement of pY-STAT3 induced by IL-23, and was associated with reversal of the suppression of Socs3 expression. This result suggested that IL-1–induced NF-κB activation might repress SOCS3 expression as a mechanism to enhance pY-STAT3 content. Accordingly, TH17 cells derived from mice with deficiency of the NF-κB component, RelA, targeted to CD4+ T cells (Relafl/flCd4-Cre) showed no enhancement of pY-STAT3 induction by IL-1–IL-23 co-signaling (Fig. 5h). This finding is in agreement with a study in a hepatocyte cell line27, where, after a transient, modest early enhancement of induction, IL-1 repressed IL-6–induced SOCS3 expression via NF-κB. In addition to its effects on tyrosine phosphorylation of STAT3, we found that IL-1 signaling increased serine phosphorylation of STAT3 (pS727-STAT3) via the p38 kinase pathway (Supplementary Fig. 7a,b), an effect that has been shown to potentiate the actions of the STAT3 homodimer28. However, this effect was more transient and independent of a requirement of IL-23 (or IL-6) co-signaling, suggesting that any effects of STAT3 serine phosphorylation are likely to be secondary to the principal effects on tyrosine phosphorylation modulated by the NF-κB–SOCS3 axis.
Finally, an important role for IL-1–mediated repression of SOCS3 in overriding RA-mediated inhibition of TH17 cell differentiation was directly established, as the inhibitory effect of RA was largely mitigated by specific deletion of Socs3 in developing TH17 cells. Thus, delivery of Cre to naïve CD4+ T cells from Socs3fl/fl mice via a Tat-Cre fusion protein led to a reduction in RA-induced Foxp3 expression and enhancement of IL-17 that was comparable in presence of IL-23 or IL-1β (Fig. 6a,b). Collectively, these data identify a SOCS3-dependent mechanism by which IL-1–induced NF-κB signaling regulates the amplitude and duration of STAT3 tyrosine phosphorylation induced by TH17-polarizing cytokines.
FIGURE 6. Deletion of SOCS3 abrogates the IL-1–dependent reversal of RA repression of TH17 development.
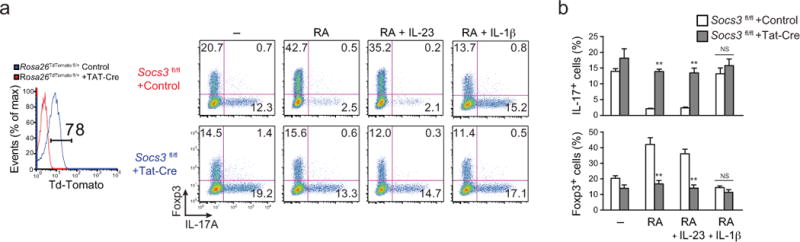
(a) Expression of Foxp3 and IL-17A (right composite panel) in naïve CD4+ T cells from Socs3fl/fl mice (control, red; Tat-Cre, blue) that were cultured under TH17 polarizing conditions with the indicated additions of RA, IL-1β and IL-23 with or without Tat-Cre peptide and Td-Tomato protein expression (left single plot) from Rosa26-floxed-STOP-TdTomato reporter mice (Rosa26TdTomato fl/+) treated with or without Tat-Cre peptide and cultured similarly under TH17 conditions and analyzed for Cre-mediated deletion efficiency. Numbers indicate the frequencies in each quadrant. (b) Frequencies of IL-17A– and Foxp3–positive cells from CD4+ T cells treated as in (a). Data are representative of one of four (deletion efficiency analysis with Rosa26TdTomato fl/+) or one of two (Socs3fl/fl with or without Tat-Cre) independent experiments (a) or are pooled from two independent experiments with 12 samples (n = 12) per group (b). Data are means and s.e.m. in b. NS = not significant, ** P < 0.01 (two-tailed unpaired t-test).
IL-1β modulates STAT3 vs. STAT5 binding to consensus sequences
In view of the dominant effect of IL-1 to override RA– and IL-2–induced repression of TH17 cell development, we reasoned that the actions of IL-1 signaling to enhance STAT3 phosphorylation might overcome pSTAT5 competition for binding to shared STAT-binding consensus sequences induced by RA-dependent IL-2 signaling15. Chromatin immunoprecipitation (ChIP) analyses were performed to quantitate the relative binding of pSTAT3 and pSTAT5 to consensus regulatory sites in the Il17a-Il17f and Foxp3 under various conditions of RA and IL-1β addition in TH17 polarized cells activated by IL-6 alone (STAT3) or IL-6 plus IL-2 (STAT5) (Fig. 7a,b). As a control for the STAT5-dependent effects of RA, TH17-polarized cells deficient for STAT5 (Stat5−/−) were included. We found that, with the exception of the intergenic enhancer 10 kb downstream of the Il17a transcription start site (Il17a+10), all STAT3-STAT5 binding sites in the Il17a-Il17f locus of TH17 cells treated with RA alone showed reciprocal, decreased STAT3 and increased STAT5 binding (Fig. 7a). The decrease in STAT3 binding caused by RA addition was reversed in STAT5-deficient cells, consistent with the actions of RA to enhance IL-2–dependent STAT5 signaling. Addition of IL-1β reversed both the RA-induced increase in binding of STAT5 and the decrease in binding of STAT3, consistent with the augmented STAT3 phosphorylation induced by IL-1 signaling (Fig. 5b–f). At the intergenic Il17a+10 site, both STAT3 and STAT5 binding were unaffected by addition of RA, although both were significantly affected by IL-1β, indicating that additional regulatory mechanisms might confer greater STAT3 binding at this cis-regulatory element, irrespective of IL-2–induced pSTAT5.
FIGURE 7. IL-1β reverses RA-mediated STAT5 binding in the Il17a-Il17f and Foxp3 gene loci.
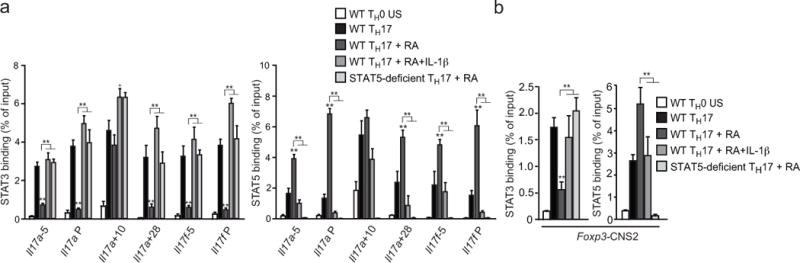
(a,b) Chromatin immunoprecipitation (ChIP) analysis of STAT3 and STAT5 binding in purified CD4+CD8− thymocytes isolated from wild-type (WT) or Stat5fl/fl.Cd4-cre mice and activated with plate-bound anti-CD3 and soluble anti-CD28 under TH0 or TH17 conditions (IL-6 +TGF-β, with or without the indicated additions of RA and IL-1β. On day 4, recovered cells were stimulated with IL-6 (pSTAT3) or IL-6+IL-2 (pSTAT5) and processed for ChIP-PCR to quantitate binding of STAT3 or STAT5 to the indicated sites within the Il17a-f (a) and Foxp3 (d) gene loci. Unstimulated TH0 cells were used as a negative control. Results are relative to input DNA. Data are representative of two independent experiments with 3 replicates per experiment (means and s.e.m. in a, b). *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
Competition between pSTAT5 and pSTAT3 binding at the CNS2 intronic enhancer of the Foxp3 gene has been proposed to regulate the heritable maintenance of Foxp3 expression—and thus the stability of the Treg cell developmental program29,30. We found that RA, by enhancing STAT5 binding, also restricts IL-6-mediated STAT3-binding at CNS2 in the Foxp3 locus during TH17 differentiation but is outcompeted by increased STAT3 binding promoted by IL-1. Consistent with this result, while IL-1 alone had no effect on repressing Foxp3 expression during iTreg cell development (Supplementary Fig. 8a), it significantly augmented the limited extinction of Foxp3 expression induced by IL-6 in secondary cultures of sorted Foxp3+ T cells derived from Il17fThy1.1Foxp3gfp dual reporter mice (Supplementary Fig. 8b). Indeed, even in the presence of exogenous RA, which promoted Foxp3 expression stability, greater than 50% of cells extinguished their expression of Foxp3, with a significant population converting into IL-17F-producing cells. Thus, IL-1 acts cooperatively with IL-6 to subvert the Foxp3–iTreg cell program in favor of TH17 development due, at least in in part, to its actions to alter the relative binding of pSTAT3 and pSTAT5 at the key CNS2 enhancer element in the Foxp3 locus.
DISCUSSION
The early developmental programs of iTreg and TH17 cells are intimately linked, reflected in the co-expression of lineage-specifying transcription factors Foxp3 and Rorγt downstream of TGF-β signaling5,6. The balance between antagonistic effects of RA and IL-2, which promote iTreg cell development while suppressing TH17 cell development, and IL-6 and IL-1, which promote TH17 development while suppressing iTreg cell development, is particularly critical in determining homeostatic versus host protective immunity to microbes in the intestines, where production of RA by resident DCs normally favors anti-microbial tolerance. Here, we show that IL-1 signaling overrides the effects of the RA–IL-2 axis in promoting iTreg cell differentiation to allow a host-protective TH17 response to an enteric pathogen, and we define a mechanism by which IL-1 signaling modulates the TH17–iTreg cell developmental balance. By enhancing the amplitude and duration of phospho-STAT3 activity induced by TH17-specifying cytokines (for example, IL-6 and IL-23), IL-1 alters the pSTAT3/pSTAT5 ratio in developing T cells to enhance TH17 cell development at the expense of iTreg cell development. Indeed, by favoring pSTAT3 binding over pSTAT5 binding at the CNS2 intronic enhancer in the Foxp3 locus, IL-1–dependent potentiation of STAT3 signaling directly subverts the Treg cell-stabilizing function of IL-2 promoted by RA, thereby contributing to plasticity in the Treg cell developmental program14,15,31,32. These findings extend the role of pro-inflammatory cytokines in overriding a dominant program of Treg cell development in the intestines to enable the recruitment of TH17-mediated host defense, and provide a mechanism by which IL-1 cooperates with the actions of TH17-specifying cytokines to promote TH17 cell development.
Although the contribution of IL-1 to TH17 differentiation is well established2,16, the mechanism of its action has been unclear. Here we identify a link between IL-1 signaling and repression of SOCS3 expression as a mechanism for specifically augmenting STAT3, but not STAT5, signaling. Recent studies have detailed the structural basis by which SOCS3 interacts with the gp130-Jak1 complex following ligand-mediated trans-phosphorylation of the cytoplasmic domain of IL-6–family receptors, explaining the specificity of SOCS3 for this family of receptors33,34—and presumably non-gp130 receptors such as the structurally related IL-23R26—but not the IL-2 receptor. The rapid induction of SOCS3 expression downstream of IL-6 (or IL-23) signaling provides a negative feedback loop that rapidly terminates IL-6 or IL-23 receptor signaling by inhibiting receptor-bound Jak1 or Jak2, respectively. Through repression of Socs3 expression, IL-1 indirectly sustains Jak-STAT interactions that potentiate pSTAT3 actions. Absent comparable effects on IL-2 receptor-induced pSTAT5, IL-1 signaling effectively shifts the ratio of pSTAT3/pSTAT5 in favor of pSTAT3, providing a competitive advantage for binding of pSTAT3 to shared DNA target sequences, as demonstrated in our ChIP studies.
Interestingly, IL-1 augmented IL-21–induced pSTAT3 similarly to that induced by IL-6 and IL-23. Although the IL-21 receptor is unlike other common γ chain cytokine receptors in primarily activating STAT3, not STAT5, as far as we are aware a direct role for SOCS3 in modulating IL-21 receptor signaling has not been defined. Further studies will be needed to determine whether the IL-21 receptor is unusual among common γ chain cytokine receptors in recruiting SOCS3, or whether other effects of IL-1 signaling contribute to the increased STAT3 phosphorylation induced by IL-21. It is notable that TGF-β has been reported to contribute to TH17 cell development through inhibition of IL-6– and IL-21–induced SOCS3 expression35, although another study found that TGF-β also increased STAT5 phosphorylation36, an effect not observed herein for IL-1 signaling. Thus, although both IL-1 and TGF-β might repress SOCS3 expression in concert with TH17-polarizing cytokines, albeit via fundamentally different signaling pathways, IL-1 would appear to preferentially alter pSTAT3/pSTAT5 ratios in favor of pSTAT3. In this regard, it is of interest that TH17 cell development can occur in the absence of TGF-β signaling—contingent on co-signaling of IL-1β with either IL-6 or IL-23 (ref. 37). It will therefore be of interest to determine whether this effect is mediated by IL-1–induced inhibition of SOCS3.
In addition to positioning SOCS3 for inhibition of the Jak catalytic domain, docking of the SH2 domain of SOCS3 to phosphotyrosine 759 (pY759) of the IL-6–activated gp130 cytoplasmic domain inhibits binding of the cytosolic tyrosine kinase, SHP2, which activates MAP and phosphatidylinositol-3-OH kinase signaling induced by IL-6, and presumably IL-23. Thus, while potentiating STAT3 phosphorylation, IL-1–mediated repression of SOCS3 might also potentiate STAT3-independent signals from the IL-6 receptor, the effects of which have not been explored. Moreover, deficiency of SOCS3 has been shown to increase STAT1 activation in myeloid cells38, raising the possibility that IL-1–induced suppression of SOCS3 might also impact the TH17–iTreg developmental balance via modulation of STAT1. It will be of interest to examine these issues in future studies.
A previous report found that IL-1 signaling increased expression of the transcription factors IRF4 and Rorγt, both of which are required for TH17 cell differentiation16. Expression of IRF4, which, unlike Rorγt42, is central to the development of other effector cells in addition to TH17 cells, appears to be regulated primarily by TCR signals and is STAT-independent. Hence, increased IRF expression is unlikely to be mediated by the STAT3-amplifying effects of IL-1 signaling described herein. However, through cooperative binding with the trans-factor BATF at consensus motifs in multiple TH17 lineage genes39,40, IRF4 contributes to chromatin accessibility at a number of sites that are also targeted by STAT3 (ref. 41), as well as sites targeted by both STAT3 and STAT5 (ref. 15). Thus, in addition to any STAT-independent effects that IL-1 signaling might have to enhance IRF4 expression, it can also indirectly modulate the effects of IRF4 actions by altering the balance of STAT binding at composite consensus sites in target genes. In contrast to IRF4, expression of Rorγt is STAT3-dependent43. Thus, the effects of IL-1 to augment the amplitude and duration of STAT3 tyrosine phosphorylation likely contribute to enhanced expression of Rorγt during TH17 cell differentiation, whether early, downstream of IL-6 signaling, or later, following expression of the IL-23 receptor.
iTreg cells differ from thymic (t)Treg cells in their tendency to lose Foxp3 expression during successive rounds of cell division. This reflects differences in the methylation status of CpG islands within CNS2 of the Foxp3 locus30,44,45. CNS2 is highly methylated in newly generated iTreg cells, whereas tTreg cells and mature iTreg cells have undergone demethylation of CNS2, which enables binding of Foxp3 and Runx1, among other factors, that confers stable expression of Foxp3 under limiting IL-2 conditions32. Although the mechanism by which CNS2 becomes demethylated during Treg cell maturation remains to be fully elucidated, it has recently been found that CNS is the major site of IL-2–induced STAT5 binding in the Foxp3 locus32, and, in contrast to other factors that bind CNS2, STAT5 can bind CNS2 irrespective of its methylation status36. Although unproven, this suggests that STAT5 might be an important factor in recruiting demethylation complexes to CNS2, in addition to its function in sustaining competency of CNS2 in immature iTreg cells as they divide32. Data presented herein favor a model wherein IL-1 signaling in intestinal T cells responding to enteropathogenic infection is important to override the RA–IL-2–STAT5 axis in favor of enhanced, sustained STAT3 signaling that inhibits STAT5 binding to Foxp3 CNS2 to silence expression of Foxp3 while promoting of expression Rorγt and other TH17 lineage genes that contain consensus sequences regulated by competitive antagonism of STAT5-STAT3 binding, thereby deviating nascent or immature iTreg cells towards TH17 cells development. These findings extend the role of pro-inflammatory cytokines in overriding a dominant program of Treg cell development in the intestines to enable the recruitment of TH17-mediated host defense, and provide a rationale for consideration of IL-1 blockade to re-establish homeostasis in the treatment of chronic inflammatory diseases mediated by the TH17 pathway.
METHODS
Mice
The following mouse strains used were purchased from Jackson Laboratory and/or bred at our facility: C57BL/6J (B6); B6.129P2-Tcrbtm1Mom/J (Tcrb−/−); B6.129S2-Il1btm1Dch (Il1b−/−); B6.Cg-Tg-Il17ftm1(Thy1)Weav (Il17f thy1.1); B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II); B6N.129(Cg)-Foxp3tm3Ayr/J (Foxp3gfp); B6.129S7-Il1r1tm1Imx/J (Il1r1−/−); B6.129S6-Stat5a-Stat5btm2Mam/Mmjax (Stat5fl/fl); B6.129S1-Relafl/fl (Relafl/fl); B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Rosa26TdTomato fl/+); and B6.129S4-Socs3tm1Ayos/J (Socs3fl/fl). B6.Cg-Tg(Cd4-Cre)1Cwi (Cd4-Cre) were purchased from Taconic and B6.CD45.1 from Frederick Cancer Center. All strains were on a B6 background. All intercrosses to generate additional strains like WT and IL-1R-deficient dual reporter mice (Il17f thy1.1.Foxp3gfp), Il17f thy1.1.CD45.1/CD45.2 and Stat5bfl/fl. Cd4-Cre mice were generated by crosses in our breeding facility. Animals were bred and maintained under specific pathogen-free condition in accordance with institutional animal care and use committee regulations.
C. rodentium infection and mAb treatments
C. rodentium strain DBS100 (ATCC), was used for all inoculations with the exception of experiments where whole body imaging was performed. For imaging experiments, the bioluminescent C. rodentium strain ICC180 (derived from DBS100) was used1. This strain harbors an constitutive luxCDABE operon encoding luciferase of nematode Photorhabdus luminescens. C. rodentium strain DBS100 was grown at 37 °C in LB broth and C. rodentium strain ICC180 was grown in LB containing kanamycin (100 μg/ml). Mice were inoculated with 2 × 109 cfu (high dose) in a total volume of 200 ml of PBS via gastric gavage.
For depletion of Il17f thy1.1 cells in vivo, mice were injected intraperitoneally (IP) with 400 μg anti-Thy1.1 (Univ. Alabama-Birmingham Epitope Recognition and Immunoreagent Core Facility, clone 19E1.2) on d3 and d7 post-infection. In all studies, age- and sex-matched mice were used (6–12 weeks of age).
Bioluminescence imaging
Mice were anesthetized with isoflurane and placed in a supine position in a custom built chamber for imaging with IVIS-100 system and Living Image Software (Xenogen, Inc.). Baseline images were collected prior to gavage with 2 × 109 cfu C. rodentium strain ICC180 and whole body images were taken at a binning of 4 over 1 for 3 min at the indicated times post infection with ICC180. Luminescence emitted from the same gate in individual mice was quantified as counts per second and pseudocolor images represent light intensity generated as a measure of colonization of the luminescent bacterial strain.
Cell isolation, in vitro T cell differentiation, protein transduction and inhibitor treatment
Naïve CD4+ T cells from spleens or lymph nodes of 8–10-week-old mice were purified by flow cytometric sorting on a FACS Aria II instrument (BD Bioscience) by gating on the CD4+CD25−CD62LhiCD44lo fraction. Isolated CD4+ T cells were cultured in RPMI containing 10% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, non-essential amino acids, 50 μM β-mercaptoethanol, and 2 mM L-glutamine (R-10). For polyclonal plate-bound stimulation, sorted CD4+ T cells were stimulated with 5 μg of plate-bound anti-CD3 and 1 μg/ml of soluble anti-CD28. For polyclonal feeder-based stimulation, sorted CD4+ T cells were stimulated with 1 μg/ml of soluble anti-CD3 in presence of in presence of irradiated, T-cell depleted splenic or mesenteric lymph node feeder cells obtained from Il1b−/− mice. For antigen-specific stimulation, sorted naïve OTII TCR transgenic CD4+ T cells were activated with 5 μg/ml OVA peptide (OVAp) in presence of irradiated, T-cell depleted splenic feeder cells obtained from Il1b−/− mice. In all cases using feeder cells for TH17 polarization, T cells and feeder cells were added at a ratio of 1:1. All cultures were done in a volume of 200 μl in 96-well U-bottom plates. For TH17 or iTreg polarizations the following exogenous cytokines or antibodies were added at the indicated concentrations, unless stated otherwise: IL-6 (20 ng/ml; R&D Systems); TGF-β (2 ng/ml; R&D Systems); IL-21 (10 ng/ml; R&D Systems); IL-23 (5 ng/ml; R&D Systems); IL-1β (20 ng/ml; R&D Systems), hIL-2 (1U to 100 U; Roche), anti-IL-4 (10 μg/ml; clone XMG1.2); anti-IFN-γ (10 μg/ml; clone 11B11); anti-IL-2 (10 μg/ml; clone JES6-1A12, eBioscience). All-trans-retinoic acid (RA; Sigma) was added at various indicated concentrations (1 nM to 100 nM) and the retinoic acid antagonist LE 540 (Wako) was added at 1 μM concentration.
For purification of Foxp3+CD4+ T cells, Foxp3-GFP+ cells were sorted by flow cytometry and subjected to various conditions as indicated. For purification of CD4+ single-positive thymocytes, CD8+ cells were initially removed by magnetic depletion of CD8+ T cells by MACS (mouse CD8α MicroBeads, Miltenyi Biotec), and CD4+ single-positive cells were further purified by isolation of the flow-through by MACS (mouse CD4+ T Cell Isolation Kit II, Miltenyi Biotec).
In experiments where cells were treated with cell signaling inhibitors, recovered cells were washed and serum-starved in complete medium for 1 h prior to addition of the following reagents, used at the indicated concentrations: Janus kinase 2 protein (JAK2) inhibitor AG490 (50 μM), JNK-Inhibitor-SP600125 (1 μM), and NF-κB inhibitor PDTC (1 μM). With the exception of PDTC (Sigma), all inhibitors were purchased from Calbiochem.
In experiments where naïve CD4+ T cells were transduced by TAT-Cre protein, sorted naïve CD4+ T cells from Rosa26TdTomato fl/+reporter mice with a loxP-flanked STOP cassette were initially standardized by TAT-Cre transduction. Briefly cells were incubated in 1 ml of 100 μg/ml TAT-Cre peptide in serum free RPMI at 1:1 ratio for 15–20 min at 37 °C before washing and suspending into IL-7 (10 ng/ml) containing RPMI plus 10% FCS medium for 6, 12, 24 or 48 h, where maximum expression of the reporter was observed at 24 h. Subsequently sorted naïve CD4+ T cells from Ai14 td-Tomato reporter and Socs3fl/fl mice were treated in tandem with TAT-Cre or control peptide and cultured in IL-7-containing medium for 24 h followed by polarization under TH17 conditions, with the additional culture modifications indicated in figure legends.
Flow cytometry and phospho-flow analysis
The following mAbs were used for flow cytometric sorting: anti CD4-eFlour 780 (eBioscience, clone RM4-5); anti-CD25-FITC (BD Pharmingen, clone 7D4); anti-CD62L-PE (eBioscience, clone MEL-14); and anti-CD44-APC (eBioscience, clone IM-7). For flow cytometric analyses and intracellular staining, the following mAbs were used: anti CD3-FITC (eBioscience, clone 145-2C11) or anti-CD3-PerCP-Cy5.5 (BD Pharmingen, clone 145-2C11); anti-CD4-PeCy7 (eBioscience, clone RM4-5); anti-IL-17A-APC (eBioscience, clone 17B7); anti-CD25-PerCP-Cy5.5 (eBioscience, clone PC61.5), anti-CD45.1 Per-Cy5.5 (eBioscience, clone A20) and anti-RORγt-PE (eBioscience, clone AFKJS-9). In brief, differentiated TH17 cells were directly stained by anti-Thy1.1 (BD Pharmingen, clone OX-7) without secondary stimulation. Only in cases where intracellular IL-17A was detected, cells were stimulated with PMA (50 ng/ml; Sigma) and ionomycin (750 ng/ml; Calbiochem) for 4 h in the presence of Golgi Plug (BD Pharmingen). For detection of intracellular IL-17A and Foxp3 expression, cells were resuspended in Foxp3 staining Buffer (eBioscience). In all cases, LIVE/DEAD Fixable Near-IR Dead Cell Stain (Invitrogen) was included prior to surface staining to exclude dead cells in flow-cytometric analyses.
For phosphor-flow analysis, sorted naïve CD4+ T cells were differentiated with plate-bound anti-CD3 and soluble anti-CD28 under TH17 polarizing conditions alone or in presence of RA or RA plus IL-1β. After 4 days, cells were briefly rested in neutral media before stimulating with IL-6 alone (50 ng/ml, R&D Systems) or with IL-2 (500 U/ml) for 20 min. The cells were then washed and suspended in pre-warmed PBS before fixing with 4% paraformaldehyde for 10 min in a 37 °C incubator. The cells were washed again and resuspended in ice-cold phospho-wash buffer before adding chilled 100% methanol in drops and keeping incubating for 30 min followed by 2 washes in PBS containing 0.05% Tween. Before incubation in anti-pSTAT3 (anti-STAT3 PE, pY694, BD Pharmingen) or anti-pSTAT5 (anti-STAT5 AlexaFluor APC, pY694, BD Pharmingen) Ab (1 h), cells were pre-incubated in PBS-Tween containing 3% goat serum and 1 μl Fc block for 15 min. Samples were acquired on an LSRII instrument (BD Biosciences), and data were analyzed with FlowJo software (Tree Star).
Isolation and in staining of lamina propria lymphocytes
For isolation of lamina propria lymphocytes (LPLs), the large intestine was removed, cleared of luminal contents and fat, cut into small pieces and washed in chilled HBSS without Ca2+ or Mg2+. Minced tissue pieces were incubated in presence of EDTA for 30 min and vortexed thoroughly to remove epithelial cells, then incubated in RPMI containing collagenase IV (1 mg/ml, Sigma-Aldrich), dispase (0.5 mg/ml, Gibco, Invitrogen) and DNaseI (0.25 mg/ml, Sigma-Aldrich). LP lymphocytes were collected by centrifugation. In all experiments using Il17fthy1.1Foxp3gfp reporter mice, unmanipulated LPLs were analyzed after surface staining with anti-CD3, anti-CD4, anti-CD25, anti-CD45.1, anti-TCR-β and anti-Thy1.1. For analyses using Il1r1+/+Il17fthy1.1 (CD45.1/CD45.2) and Il1r1−/−Il17fthy1.1 (CD45.2) recipient Tcrb−/− mice, unmanipulated LPLs were analyzed after surface staining with anti-TCRβ, anti-CD25, anti-CD45.1 and anti-CD90.1 and subsequently fixed in fixation buffer (Foxp3-staining buffer, eBioscience) before staining intracellularly with anti-Foxp3 mAb.
Adoptive transfers and in vivo LE135 treatment
For co-transfer of CD4+ T cells from congenically marked Il1r1+/+Il17fthy1.1 (CD45.1/CD45.2) and Il1r1−/−Il17fthy1.1 (CD45.2) reporter mice to Tcrb−/− mice, CD4+ T cells were isolated directly ex vivo by MACS-based depletion of PE-labeled antibodies against CD8α, CD11b, CD11c, CD19, CD45R (B220), CD49b (DX5), CD105, MHC-class II, γδ TCR and Ter-119 captured by anti-PE magnetic microbeads (Miltenyi Biotec). For co-transfers of WT and IL-1R1-deficient Foxp3+ T cells, CD4+Foxp3+GFP+IL-17F.Thy1.1− T cells were sorted by flow cytometry from Citrobacter-infected donor mice. In all cases, recovered T cells were transferred by IP injection at indicated doses and time-points.
For in vivo treatment of Il1r1−/− reporter mice with retinoic acid inhibitor LE135 (Torcis Bioscience), 100 μg of LE135 dissolved in DMSO (10 mg/ml) was gavaged in a volume of 200 μl (DMSO:PBS=1:20) from d3 through d7 PI and analyzed at d8 PI by flow cytometry or on d10 by histopathology. Control mice was gavaged on the same schedule with vehicle alone.
ELISA
For determination of cytokine concentrations in vivo, supernatants from differentiated T cells were collected on 3 day of culture and analyzed by capture ELISA. For determination of cytokine concentration ex vivo, colonic tissues from uninfected or C. rodentium-infected mice were collected at the indicated time points post infection, finely minced and cultured in 24-well plate for 24 h in R-10 medium supplemented with 10 μg/ml gentamicin. Supernatants were collected after centrifugation and analyzed by capture ELISA. IL-2, IFN-γ, IL-17A and IL-22 ELISAs were done using mouse quantikine ELISA kits per manufacturer’s recommendations (R&D Systems).
Immunoblot
B6 CD4+ T cells were cultured under TH17 polarizing conditions for 5 d in presence of anti-CD3 and irradiated, T cell-depleted splenocytes. Following Ficoll separation to isolate viable cells, T cells were rested in neutral media and restimulated with IL-6 (20 ng/ml), IL-1β (20 ng/mL) or IL-21 (20 ng/ml) for the indicated times. Cell lysates were prepared in lysis buffer (RIPA buffer; 50 mM Tris-HCl pH?, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing a protease & phosphatase inhibitor mixture (Pierce). Protein was quantified by Bradford Assay before equivalent amounts were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). Primary antibodies were: anti-STAT3 (Cell Signaling #9132), anti-phospho-tyrosine(705)-STAT3 (Cell signaling Technology #9145), or anti-phospho-serine(727)-STAT3 (Cell signaling Technology #9134). HRP-cojugated Donkey anti-rabbit or HRP-conjuaged anti-mouse antibody (Affinity Bioreagents) were used to detect target protein by ECL detection kit (GE Healthcare or Pierce SuperSignal Dura).
Real-time PCR
Total RNA was isolated using RNeasy Isolation Kit per manufacturer’s instructions (Qiagen) and treated with DNAse (Qiagen). cDNA synthesis was performed with Superscript III first-strand synthesis system (Invitrogen) and real-time PCR was performed on a Bio-Rad iCycler with primer pairs specific for cDNAs of Il17f, Il1r1, Il1r2, IlrAcP, Rara, Rarb, Rarc, Rxra, Rxrb, Rxrc, Rorc and 18S transcripts with iQTM SYBR Green Supermix (Bio-rad). The primer sets were:
Il17f: for 5′-CGCCATTCAGCAAGAAATCC-3′, rev 5′-CTCCAACCTGAAGGAATTAGAACAG-3′;
Il1r1: for 5′-CCGGGCACGCCCAGGAGAA -3′, rev 5′- TCCAGCGACAGCAGAGGCAC -3′;
Il1r2: for, 5′-TCGGAGAAGCCCACAGTCCA -3′, rev, 5′-GGAGTGAGGTGCCAAGGGGC -3′;
IlrAcP: for, 5′- TTGCCACCCCAGATCTATTC -3′, rev 5′-CCAGACCTCATTGTGGGAGT -3′;
Raraα: for, 5′- TCCGACGAAGCATCCAGAA -3′; rev 5′- GGTTCCGGGTCACCTTGTT -3′;
Rarβ: for, 5′- CAGTGAGCTGGCCACCAAGT -3′; rev, 5′- GCGATGGTCAGACCTGTGAA -3′;
Rarγ: for, 5′- GTCAGCTCCTGTGAAGGCTGC -3′; rev, 5′- GTTTCGATCGTTCCTTACAG -3′;
Rxrα: for, 5′- TAGTCGCAGACATGGACACC -3′; rev, 5′- GTTGGAGAGTTGAGGGACGA -3′;
Rxrβ: for, 5′- GCACAGAAACTCAGCCCATT -3′; rev, 5′- CATCCTCATGTCACGCATTT -3′;
Rxrγ: for, 5′- GCCTGGGATTGGAAATATGA -3′; rev, 5′- ACACCGTAGTGCTTCCCTGA -3′;
Rorc: for, 5′-CAGCCAACATGTGGAAAAGCT-3′; rev, 5′-GGGAAGGCGGCTTGGA-3′
Cd25: for, 5′- TACAAGAACGGCACCATCCTAA -3′; rev, 5′-TTGCTGCTCCAGGAGTTTCC -3′
Cd122: for, 5′- GTCCATGCCAAGTCGAACCT-3′; rev, 5′-GGATGCCTGCCTCACAAGAG -3′ Socs3 for, 5′-AGTGCAGAGTAGTGACTAAACATTACAAGA-3′; rev, 5′-AGCAGGCGAGTGTAGAGTCAGAGT-3′
18SrRNA: for: 5′- GCCGCTAGAGGTGAAATTCTTG -3′; rev, 5′- CATTCTTGGCAAATGCTTTCG -3′.
Reactions were run in duplicate and samples were normalized to 18S as a fold-induction over controls unless otherwise stated.
Chromatin immunoprecipitation (ChIP) assay
CD4+ single-positive thymocytes from WT or Stat5fl/fl.Cd4-Cre mice were polarized for 3-4 d under TH0 (no cytokine) or TH17 polarizing conditions (IL-6 + TGF-β), with or without RA or RA + IL-1β, followed by 20 min stimulation in CO2 incubator by addition of IL-6 (50 ng/ml, R&D Systems) or IL-2 (500 U/ml). The cells were cross-linked with 1% (vol/vol) formaldehyde for 5 min, quenched with 125 mM glycine and then suspended in HALT protease-inhibitor containing PBS. Chromatin Immunoprecipitation (ChIP) was performed with ExactaChIP Kit from R&D systems in accordance with the manufacturer’s instruction. Briefly, cells were suspended in lysis buffer containing protease-inhibitor and immunoprecipitated overnight (4 °C) with biotinylated anti-STAT3 (R&D systems) or anti-STAT5 (R&D systems). Bound DNA was collected using agarose-streptavidin beads and purified with QIAprep Spin Miniprep Kit (Qiagen). Immunoprecipitated DNA was quantitated by real-time PCR with iQTM SYBR Green Supermix (Bio-rad) on a Bio-Rad iCycler using following primer pairs specific for the indicated elements in the Il17a-Il17f locus2 and Foxp3 locus.
Il17a-5: for, 5′- CAGGTATTATTCTCAGGGCTTTGG -3′; rev 5′- TGGCAATGGTGTCTTTTCTTTG -3′
Il17aP: for, 5′- CACCTCACACGAGGCACAAG -3′; rev 5′- ATGTTTGCGCGTCCTGATC -3′
Il17a+10: for, 5′- GGATTAAGGGCACACGTGTTG -3′; rev 5′- TTTCCCCACTCTGTCTTTCCA -3′
Il17a+28: for, 5′- TCATCGGCTCCCACACAGA -3′; rev 5′- GGCAGTACCGAAGCTGTTTCA -3′
Il17fP: for, 5′- CCCACAAAGCAACACTCTTGTC -3′; rev 5′- ACTGCATGACCCGAAAGCA -3′
Il17f-5: for, 5′- GCATCGCATCTTTCAAACCA -3′; rev 5′- TTAGGATAAGCGCCCAGTGAAT -3′
Foxp3 CNS2 for, 5′-GTTGCCGATGAAGCCCAAT-3′; rev 5′-ATCTGGGCCCTGTTGTCACA-3′
Data are expressed as relative values normalized to the input DNA samples (percentage of input).
Histolopathological evaluation
Tissue samples obtained from proximal, middle and distal portions of large intestines were fixed in 10% neutral buffered formalin, embedded in paraffin to prepare 5 μm sections and stained with H&E. The tissue sections were examined and scored to evaluate tissue pathology, as previously described3. In all scoring, the identity of specimens was concealed from the pathologist.
Statistical analysis
For statistical analyses, P-values were calculated by paired or unpaired Student’s t-test, unless stated otherwise in figure legends. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank H. Qin (University of Alabama at Birmingham), L. Xu (University of Pennsylvania) and members of the Weaver laboratory for their helpful advice and comments. We are grateful to G. Frankel and S. Wiles (Imperial College, London) for provision of the bioluminescent C. rodentium strain and T. DeSilva (University of Alabama at Birmingham) for provision of Rosa26TdTomato fl/+ mice. We also thank B.J. Parsons for expert technical assistance, G. Gaskins for editorial assistance, and the UAB Small Animal Imaging Facility, UAB Center for AIDS Research Flow Cytometry Core, and UAB Epitope Recognition and Immunoreagent Core Facility for imaging studies, flow cytometric sorting, and antibody preparations, respectively. This work was supported by grants from the NIH (PO1DK71176 and R01DK093015 to C.T.W. and R.D.H., and R01AI047833 to W.S.P.) and the Crohn’s and Colitis Foundation of America (C.T.W, R.B. and D.B.O.).
Footnotes
AUTHOR CONTRIBUTIONS
R.B., S.K.W., R.D.H. and C.T.W designed the studies. R.B., S.K.W., S.B. and R.D.H. performed the experiments. C.L.Z. advised on design and execution of figures. T.R.S. performed pathological analyses. E.N.B. assisted in the design and interpretation of studies involving SOCS3. W.S.P. provided TAT-Cre peptide and provided guidance on its use in naïve CD4+ T cells. R.B., S.K.W. and C.T.W. wrote and edited the manuscript.
References
- 1.Basu R, Hatton RD, Weaver CT. The TH17 family: flexibility follows function. Immunol Rev. 2013;252:89–103. doi: 10.1111/imr.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Mangan PR, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 4.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 5.Zhou L, et al. TGF-beta-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Littman DR, Rudensky AY. TH17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 7.Vicente-Suarez I, et al. Unique lamina propria stromal cells imprint the functional phenotype of mucosal dendritic cells. Mucosal Immunol. 2014;8:141–151. doi: 10.1038/mi.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 10.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 14.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Yang XP, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atarashi K, et al. ATP drives lamina propria TH17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishigame H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Basu R, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 23.Hirota K, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452:356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang XP, et al. Dual function of interleukin-1beta for the regulation of interleukin-6-induced suppressor of cytokine signaling 3 expression. J Biol Chem. 2004;279:45279–45289. doi: 10.1074/jbc.M313072200. [DOI] [PubMed] [Google Scholar]
- 28.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 29.Laurence A, et al. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity. 2012;37:209–222. doi: 10.1016/j.immuni.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng Y, et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Y, et al. Control of the inheritance of regulatory T cell Identity by a cis element in the Foxp3 locus. Cell. 2014;158:749–763. doi: 10.1016/j.cell.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol. 2014;26:13–19. doi: 10.1016/j.smim.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kershaw NJ, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–476. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin H, et al. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogawa C, et al. TGF-β-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J Immunol. 2014;192:475–483. doi: 10.4049/jimmunol.1301892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghoreschi K, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lang R, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 39.Glasmacher E, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li P, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543–546. doi: 10.1038/nature11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciofani M, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 43.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 44.Miyao T, et al. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Ohkura N, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785–799. doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 46.Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun. 2006;74:5391–5396. doi: 10.1128/IAI.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang XP, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bleich A, et al. Refined histopathologic scoring system improves power to detect colitis QTL in mice. Mamm Genome. 2004;15:865–871. doi: 10.1007/s00335-004-2392-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.