

Abstract
Experimental vaccine adjuvants are being designed to target specific toll-like receptors (TLRs) alone or in combination, expressed by antigen presenting cells, notably dendritic cells (DCs). There is a need for high-content screening (HCS) platforms to explore how DC activation is affected by adjuvant combinations. Presented is a cell-based microarray approach, “immunoarray”, exposing DCs to a large number of adjuvant combinations. Microparticles encapsulating TLR ligands are printed onto arrays in a range of doses for each ligand, in all possible dose combinations. Dendritic cells are then co-localized with physisorbed microparticles on the immunoarray, adherent to isolated islands surrounded by a non-fouling background, and DC activation is quantified. Delivery of individual TLR ligands was capable of eliciting high levels of specific DC activation markers. For example, either TLR9 ligand, CpG, or TLR3 ligand, poly I:C, was capable of inducing among the highest 10% expression levels of CD86. In contrast, MHC-II expression in response to TLR4 agonist MPLA was among the highest, whereas either MPLA or poly I:C, was capable of producing among the highest levels of CCR7 expression, as well as inflammatory cytokine IL-12. However, in order to produce robust responses across all activation markers, adjuvant combinations were required, and combinations were more represented among the high responders. The immunoarray also enables investigation of interactions between adjuvants, and each TLR ligand suggested antagonism to other ligands, for various markers. Altogether, this work demonstrates feasibility of the immunoarray platform to screen microparticle-encapsulated adjuvant combinations for the development of improved and personalized vaccines.
Introduction
Modern experimental vaccines are being designed with an emphasis on specific, tailored formulations to elicit more precise and potent immunological responses.1 A critical component of a vaccine is the adjuvant, a molecule or compound that potentiates the specific type and magnitude of an immune response to co-formulated antigens.2–4 Various mechanisms underlying adjuvant activity have been uncovered, providing new strategies to optimize adjuvant formulations.5 Recognition of pathogen-associated molecular patterns (PAMPs) by the immune system is achieved via various pathogen-recognition receptors (PRRs), notably toll-like receptors (TLRs).6 TLRs act as sensors for different damage-associated molecular pattern (DAMP) or “danger” signals generated by TLR-agonists such as double stranded RNA, DNA, and glycolipids present on the surfaces of many pathogens.7 Microbial ligands bind to these receptors creating diverse immune responses that are the basis for multiple adjuvants currently in development.8–10
Numerous intracellular and surface bound TLRs have been identified on dendritic cells (DCs), which are cells of the innate immune system that act as the bridge for mounting an adaptive immune response against foreign antigens.7,11 Dendritic cells are the most efficient antigen presenting cells (APCs), capable of orchestrating lymphocyte function and directing the immune response toward either immunity or tolerance.12–15 Exploiting this potential, DCs have been manipulated both ex vivo and in vivo through controlled release schemes16 to treat a number of diseases such as cancer,17–19 infection20,21 and autoimmunity22 such as type-1 diabetes.23–25 A next step in DC modulation involves simultaneously providing combinations of multiple different TLR ligands, which is capable of inducing synergistic increases in antigen-specific immune responses.26 Targeting multiple TLRs may recapitulate in a well-controlled manner, classic adjuvants formulated from attenuated or killed viruses or bacteria, providing an opportunity to more precisely direct DC function.27,28 However, while this potential has been recognized, to date there is no systematic methodology to explore dose-dependent interplay of combined TLR stimulation in DCs. Such an approach would facilitate the development of combinatorial adjuvants for tailored immune responses.
One strategy to develop immunotherapeutic treatments involves using biomaterials as modulators of DCs.29–35 Polymeric, particle-based approaches have been developed that encapsulate combinations of antigen, adjuvant, chemokines, and other immunomodulating molecules for delivery to DCs via phagocytosis and controlled release.35–37 However, when considering combinatorial adjuvant approaches, the number of possible TLR ligand combinations is large, and identifying improved formulations using current immunological methods is challenging due to limitations in cell sourcing, as well as cost, labor, and time, in particular for personalized/precision medicine applications.38 To address this, we developed a cell-based microarray to screen combinatorial libraries of adjuvants formulated with biodegradable microparticles (MPs). The adjuvants monophosphoryl lipid A (MPLA, TLR4 ligand),39 CpG (TLR9 ligand),40 and polyinosinic:polycytidylic acid (poly I:C, TLR3 ligand)39 were encapsulated in poly(lactic-co-glycolic acid) (PLGA) MPs, printed on a DC-based microarray41 in 216 unique combinations, and cell surface markers of activation and cytokine production were quantified. Improved DC responses were indicated for particular dosing combinations of TLR ligands encapsulated in PLGA MPs. This immunoarray is hereby demonstrated to provide a robust, miniaturized platform to characterize tailored adjuvant formulations.
Experimental
Materials
A 50: 50 polymer composition of poly(d, l lactide-co-glycolide) (PLGA) with inherent viscosity 0.55–0.75 dL g−1 in hexafluoroisopropanol, HFIP (Lactel, AL, USA) was used to generate microparticles. Microparticles (MPs) were formed using a standard water-oil-water solvent evaporation technique. Briefly, the PLGA polymer was dissolved in methylene chloride at 20% concentration. To fabricate fluorescent particles, either rhodamine (RHOD) (100 μL of 5 mg mL−1 in PBS) (Sigma-Aldrich), fluorescein isothiocyanate (FITC) (100 μL of 5 mg mL−1 in PBS) (Sigma Aldrich), or 9-anthracenecarboxylic acid (ACA) (100 μL of 5 mg mL−1 in PBS) (Sigma Aldrich) were encapsulated. For adjuvant-loaded particle fabrication, LPS (1 mg mL−1 Fisher Scientific), CpG ODN (0.1 mg mL−1 InvivoGen), polyinosinic:-polycytidylic acid (poly I:C; 1 mg mL−1 Sigma-Aldrich), or monophosphoryl lipid A (1 mg mL−1 MPLA) was encapsulated in the primary water phase. The primary emulsion was generated by emulsifying 100 μL of adjuvants or dye with 1 mL of PLGA solution dissolved in methylene chloride (Fisher Scientific) using a tissue-miser homogenizer. The primary emulsion was then added to 10 mL of a 5% poly-vinyl alcohol (PVA) (MW ∼ 100 000 g mo−1) (Fisher Scientific) solution in PBS and homogenized to form the secondary emulsion. Afterward, the secondary emulsion was added to 100 mL of 0.5% PVA solution and the particles were agitated using a magnetic stirrer overnight to evaporate residual methylene chloride. The particles were subsequently washed, flash-frozen, and stored at −20 °C until used.
Dendritic cell culture
Dendritic cells were isolated from the bone marrow of 7 week old C57Bl6/j mice using a 10 day protocol.29 Briefly, bone marrow was isolated from the femur and tibia. Red blood cells were lysed by ACK lysing buffer (Whittaker) and the isolated precursor cells were incubated with DC-media consisting of 20 ng mL−1 of GM-CSF (R&D Systems), DMEM/F12 (1:1) with l-glutamine (Cellgro, Herndon, VA) and 10% fetal bovine serum (Bio-Whittaker), 1% sodium pyruvate (Lonza, Walkersville, MD) and 1% non-essential amino acid (Lonza, Walkersville, MD) for 2 days in a T-75 flask. The floating cells were collected after 48 h and re-seeded with fresh DC-media in a 6-well low-attachment plate (Corning Inc., NY) for 6 days. After 6 days of culture the cells were re-suspended in fresh media and seeded onto tissue-culture treated 6-well plates (Corning, Inc., NY) for 2 days. After 10 total days of culture DCs were lifted using 10 mM solution of Na2EDTA (Fisher Scientific) in PBS. Dendritic cells thus isolated were then tested via flow cytometry for immaturity (MHC-II + < 6% and CD86 + < 6%), purity (CD11c + > 90%) and viability (Trypan Blue > 99%).
Immunoarray fabrication
Microarrays were manufactured as described previously.41 Briefly, glass coverslips were cleaned in an oxygen plasma etcher (Terra Universal, Fullerton, CA). Arrays of (3-aminopropyl)trimethoxy-silane (NH2-terminated silane) (Sigma-Aldrich, St. Louis, MO) were printed on clean coverslips using a Calligrapher Miniarrayer printer (Bio-Rad, Hercules, CA) with a pin diameter of 400 μm. The silane printed coverslips were then coated with 200 Å of titanium (Ti; 99.995% pure) and 200 Å of gold (Au; 99.999% pure) (Williams Advanced Materials, Buffalo, NY). Following coating, gold-coated arrays were sonicated to remove gold from the amine spots, exposing NH2-terminated silane islands. The coverslips were incubated with 0.1 M, methyl-terminated alkanethiol (CH3(CH2)11SH) (Sigma) for 30 min. Substrates were incubated in 10% Pluronic® F-127 (BASF Corporation, USA) for 3 h to create a non-fouling surface around the adhesive amine islands (Fig. 1b). The immunoarrays were generated with 648 cell adhesive islands to provide 216 different conditions in triplicate. Fabricated MP solutions were printed onto cell adhesive islands using standard contact pin miniarraying (BioRad) equipment Following MP printing, the microarrays were desiccated for 30 min then stored at 4 °C. For adjuvant optimization experiments, particles encapsulated with CpG, poly I:C, or MPLA were used. Six dilutions of each MP formulation were generated and printed on the immunoarrays in all possible combinations, thus providing 216 different conditions. Dried MP arrays were rinsed and rehydrated for 30 minutes with PBS before seeding immature DCs. Following seeding, the arrays were rinsed with PBS to remove non-adherent cells. Fresh DC-media was added to the immunoarrays and adherent DCs were cultured for 24 h before performing immunocytochemistry.
Fig. 1.
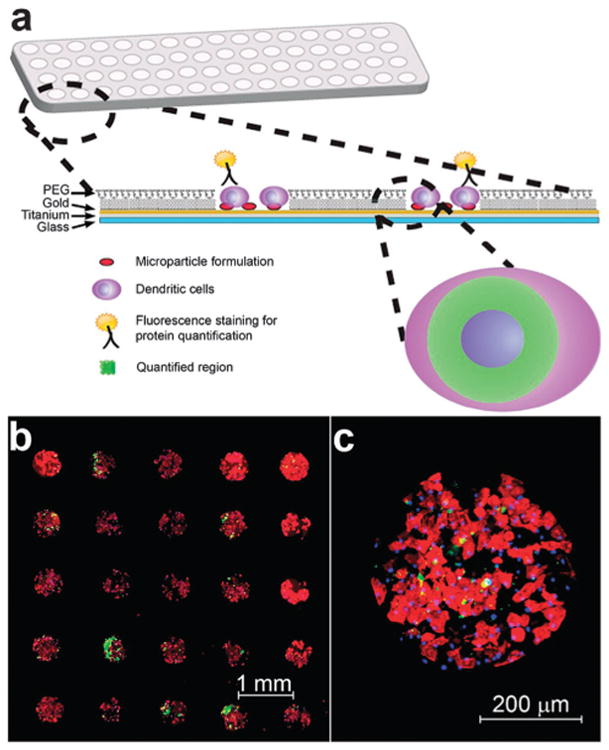
Schematic of immunoarray design and representative images of a previously described immunoarray of dendritic cells (DCs) cultured with co-localized microparticles.41 (a) PLGA microparticles encapsulated with adjuvants are printed on isolated microarray islands. Dendritic cells are cocultured with particles on the immunoarrays and characterized via immunocytochemistry. Arrays are imaged and quantified for expression of proteins that define DC activation and function. (b) Fluorescence microscopy mosaic image of a 5 × 5 section of a microarray seeded with murine bone-marrow derived dendritic cells illustrating fidelity of cell adhesion to isolated islands of adjuvant-encapsulated microparticles. Actin filaments stained using rhodamine phalloidin (red), FITC encapsulated particles demonstrating colocalization of microparticles (green) with DCs, and nuclear staining with Hoechst 34580 (blue). (c) Shown is a detail of a single island with DCs co-localized with microparticles.
Scanning electron microscope imaging
Microparticle morphology was characterized by scanning electron microscope (FEG-SEM JEOL JSM – 6335F, Major Analytical Instrumentation Center, University of Florida). Particles were dried for 16 h at room temperature, then coated with 5–10 nm thickness of gold and imaged at magnifications ranging from 30× to 60 000×. Dendritic cells cultured on the immunoarrays were fixed and imaged using scanning electron microscope by ICBR core electron microscopy facility at University of Florida, Gainesville.
Microparticle to dendritic cell ratio optimization
Prescribed numbers of particles can be printed by varying the concentration of MP solutions (in diH2O) in the source plate using the following experimentally determined relationship:
where: “source plate concentration” is the concentration of MP solutions in the wells of a 384-well plate from which the particles were lifted by the miniarrayer pin and printed on the immunoarray. Rhodamine encapsulated MPs (300, 600, 1500, 3000 and 9000 MPs per spot) were printed onto different islands. Dendritic cells were cultured on the vaccine array chip and cell number was quantified via nuclear staining (Hoechst 34580, Invitrogen) and fluorescent microscopy. The number of DCs was found to be 300 ± 33 per spot, thus providing MP: DC ratios of 1, 2, 5, 10 and 30. The DCs were then cultured on the vaccine array chip for 24 h upon which the cells were lifted and transferred to 96-well plates. A standard curve for rhodamineloaded MPs was generated and the number of particles internalized by DCs was quantified for each condition. Particle-printed arrays without DCs was used as a control to analyze the lifting of particles in the absence of DCs. It was observed that Trypsin/EDTA did not dislodge the physisorbed particles.
Immunocytochemistry
Following 24 h incubation on the immunoarrays, DCs were fixed with 4% paraformaldehyde (USB Corporation). Cells were then fluorescently stained either for surface markers CCR7 (Santa Cruz Biotechnology), CD86 (BD Biosciences), MHC-II (BD Biosciences), or intracellular cytokines IL-10 (BD Biosciences), or IL-12p40 (BD Biosciences) as previously reported.42 Intracellular staining for cytokines IL-10 and IL-12p40 was performed by first incubating seeded immunoarrays with monensin (0.7 μL mL−1) for the final 8 h of culture in order to block protein secretion. The arrays were then incubated with 4% paraformaldehyde followed by 0.01% triton X-100 (Fisher Scientific). Arrays were next washed and incubated with a blocking solution consisting of 1% goat serum along with 0.01% triton X-100 in PBS for 40 min to block the background before incubation with primary antibodies. Afterward, solutions of biotinylated secondary antibodies (Invitrogen) were incubated, followed by streptavidin-cross-linked alkaline phosphatase, and lastly the precipitating fluorescent substrate, ELF97 (Invitrogen). Nuclei were stained with Hoechst 34580 dye. Immunoarrays were then mounted and imaged.
Inter-spot cross-talk
In order to investigate cross-talk, or paracrine signaling between spots, center to center spot distances of 1000 or 1200 μm were investigated. Poly I:C loaded MPs were printed in dilutions to provide MP:DC ratio of 10, 5, 2.5,1 and 0 (no MPs). Randomized array configurations generated by MATLAB were used. Dendritic cells were cultured for 24 h and stained for intracellular IL-12p40. Production levels of IL-12p40 were quantified using Axiovision as described below.
Data acquisition – optical microscopy and image processing
Following immunocytochemistry, mounted immunoarrays were imaged using an Axiovert 200M Carl Zeiss inverted fluorescence microscope. The images were collected at 12-bit resolution (4096 different colors per pixel) with a filter sensitive for DAPI and a long pass filter (EX 365/12 EM 397) appropriate for the ELF 97 fluorescent precipitate. Image analysis was performed using AxioVision software. Briefly, nuclei were selected using DAPI staining, and a 4 μm rings distance of concentric circles were drawn around the nucleus (Fig. 1a). Next the mean relative fluorescence intensity of the area between the concentric circles were determined using the AxioVision software for each cell and utilized to determine the expression of different proteins. Cell nuclei were identified by DAPI staining and DC numbers per island were quantified by Carl Zeiss Axiovision automatic count object routine.
Statistical analyses
Overall statistical significance, least mean squares values and standard error were obtained using two-way ANOVA via Systat (Version 12, Systat Software, Inc., San Jose, CA), with independent variables being the individual experimental run identifier and the particle condition identifier. Pair-wise comparisons were made by Tukey's post-hoc analysis. A p-value of p < 0.05 was considered significant. Data was plotted using Sigmaplot (Version 10, Systat Software, Inc., San Jose, CA).
Results and discussion
Dendritic cell characterization on the immunoarray
The immunoarray developed here represents a high-content, cell-based array approach to screen combinatorial adjuvant microparticle formulations. PLGA MPs encapsulating three TLR ligands were printed onto solid substrates with discrete islands in hundreds of dosing combinations. A non-fouling background surrounding these cell-adhesive islands enabled dendritic cells (DCs) to be co-localized as isolated samples with unique particle/adjuvant formulations.41 Microparticles were printed on immunoarrays in a reproducible and quantitative manner, with minimal cross-contamination.41 Using these printing methods we could obtain homogenous deposition with as few as 16 ± 2 particles per spot. This miniaturized platform can decrease the use of reagents, time, and cost while providing the capability of screening greater than 30-fold more immunomodulatory molecules, based on the number of cells required, compared to traditional methods. Further, since the only complimentary equipment required is a fluorescent microscope, the immunoarray platform can easily be adopted into many biomedical laboratories.
Expression of cell surface markers and cytokine production upon interaction with particle formulations is quantified using standard immunocytochemistry and fluorescent microscopy (Fig. 1a). Cell attachment is tightly confined to the particle-coated silane islands as confirmed via actin (shown in red) and nuclei (shown in blue) staining of adherent DCs (Fig. 1b and c). Dendritic cell viability was assessed using 7-AAD on the immunoarrays and found to be 99% viable. Scanning electron microscope (SEM) micrographs revealed the interaction between DCs, microparticles on immunoarrays (Fig. 2).
Fig. 2.
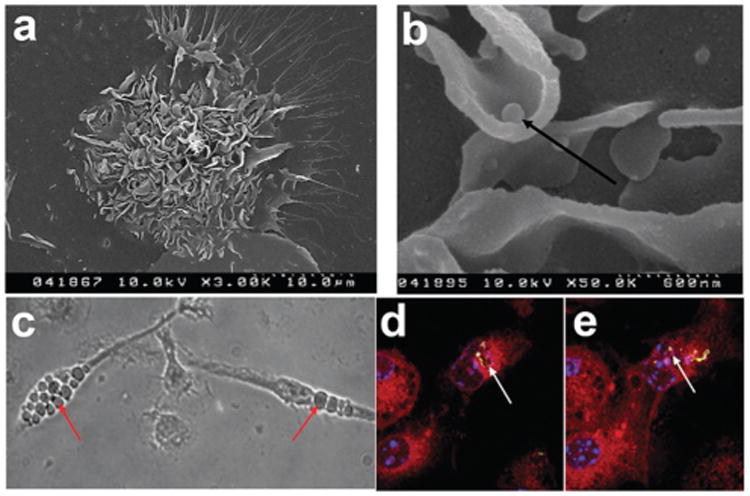
Characterization of microparticle phagocytosis by dendritic cells (DCs) on immunoarrays. (a) Scanning electron microscope micrograph of a DC on an island surrounded by particles (3000×). (b) Scanning electron microscope micrograph indicating DCs can lift physisorbed microparticles (arrow) from the array surface (50 000×). (c) Phase contrast image of DCs with phagocytosed particles localized in the cytosol. Particle internalization is highlighted (arrows). (d and e) Fluorescence confocal microscopy images of DCs phagocytosing microparticles. Confocal microscopy showing the co-localization of dye-loaded particles (arrows) with the cytosol (red). Multiple z-stacked images were acquired to confirm the internalization of the microparticles (data not shown). Actin filaments stained using rhodamine-conjugated phalloidin (red), FITC encapsulated particles demonstrating phagocytosis (yellow-green), and nuclear staining with Hoechst 34580 (blue).
Cell attachment was seen to occur with the formation of lamellipodia like structures, numerous dendritic processes and membrane ruffling (Fig. 2a). The ability to lift physisorbed MPs from the array surface was evident (Fig. 2b), with apparent internalization in the cytosol (Fig. 2c). Fluorescence confocal microscopy shows microparticles were localized internally, adjacent to the nucleus (Fig. 2d).
Particle uptake
In order to further characterize DC–MP interactions on the array, particle uptake was quantified (Fig. 3a). Immunoarrays were generated, varying the number of fluorescent dye-loaded particles per spot. DCs were then incubated on the immunoarrays for 24 h followed by nuclear staining. The time point of 24 h was chosen both for convenience as an incubation time for subsequent experiments, and because others have suggested that DC phagocytosis can saturate at this time.43 The percentage of particles phagocytosed plotted against the particle to DC ratio was found to exhibit a linear relationship. Approximately 90% of the physisorbed MPs on the array were taken up by DCs after 24 h incubation at a MP:DC ratio of 10 ± 2. In subsequent studies, this ratio was used to provide a high MP uptake level while limiting the number of non-phagocytosed particles.
Fig. 3.
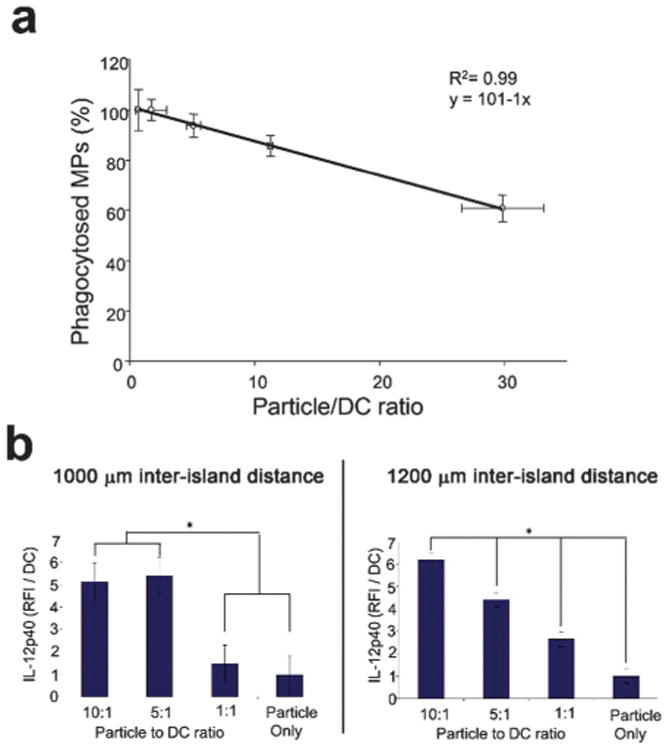
Quantification and optimization of microparticle (MP) to dendritic cell (DC) ratio and immunoarray island spacing. (a) Dendritic cells were cultured with rhodamine-encapsulated microparticles at varying ratios of MPs : DCs and the percentage of MPs phagocytosed was quantified. Dendritic cells were shown to phagocytose ∼90% of the printed particles within the first 24 h when the particle to DC ratio was maintained at 10: 1. A linear relationship was observed between the percent of particles phagocytosed and the MP:DC ratio. (b) Inter-island spacing was investigated to limit paracrine signaling. Poly I:C encapsulated MPs were printed with a range of MP:DC ratios. IL-12p40 production was quantified between immunoarrays printed in randomized configurations at either 1000 μm or 1200 μm center-to-center inter-island distances. Production of IL-12p40 at the 1000 μm spacing (b, left panel) does not provide the dose response fidelity seen with the 1200 μm spacing (b, right panel) suggesting negligible cross-talk between neighboring islands at the larger inter-island distance. (*: p < 0.05).
Island spacing optimization
A potential design constraint to miniaturized biological array platforms is the possibility of interactions between neighboring samples. Dendritic cells in particular have been shown to induce functional modulations in neighboring cells via paracrine signaling.30,44,45 While this phenomenon is generally accepted, the spatio-temporal modulations are not well characterized in culture. We explored the functional range of paracrine signaling between DCs by comparing distances in which a dose-dependent response was achievable (Fig. 3b). Doses were obtained by printing different numbers of poly I:C MPs to achieve a range of MP:DC ratios. Poly I:C is a potent inducer of IL-12 production in DCs.46 We therefore quantified IL-12p40, an active subunit of IL-12, for platform optimization studies. The 1000 μm spacing (Fig. 3b, left panel) does not provide the high fidelity dose response seen with the 1200 mm spacing (i.e., all doses are significant from each other), which suggests negligible cross-talk between neighboring islands at the larger inter-island distance (Fig. 3b, right panel). In subsequent studies, 1200 μm spacing was incorporated in the immunoarrays limit paracrine signaling between neighboring groups. The ability to elicit dose dependent cellular responses on the immunoarray is central to the platform validation. Having established island spacing, arrays were next fabricated with MPs loaded with a range of loading doses of LPS, an activating signal. Equal amounts of MPs were then printed on each island and DCs were cultured on the immunoarrays for 16 h before quantifying activation.
Combination adjuvant effects on dendritic cells
Increasing adjuvant quantity has been shown to increase DC activation45,47 until saturated levels are reached.26 The effect of varying combined adjuvant formulations on DC activation states is less explored but more intriguing, particularly in the pursuit of tunable immunotherapeutics. It is known that combining adjuvants may lead to differential activation of DCs, a requisite for successful induction of cellular and humoral responses.26,48,49 For instance, Th1 immune responses are stimulated through TLR3, TLR 4, TLR 7, TLR 8, and TLR 9 whereas TLR5 and TLR2 together with TLR1 or TLR6 supports a Th2 immune response.50–52 Utilizing the immunoarray platform facilitates investigation of the interplay between TLR signaling on DCs, a critical component to controlling the type and magnitude of antigen-specific immune responses. Microparticles encapsulated with MPLA, CpG, and poly I:C were printed on arrays consisting of 648 cell adhesive islands, testing six dilutions of each adjuvant in all possible combinations resulting in 216 unique combinations (i.e., 6̂3 = 216, and a replicate number of n = 3 on each array). DC activation was quantified via the markers CD86, MHC-II, IL-12p40, IL-10, and also CCR7 (chemokine receptor for lymphatic homing) (Fig. 5). A pro-inflammatory response is characterized by high expression of CD86, MHC-II, IL-12p40, CCR7 and low expression of IL-10. In all adjuvant combinations, it was observed that expression of MHC-II, CD86, CCR7, IL-12p40, and IL-10 were significantly different from the negative control condition of no-particles (Fig. 6a–e). This confirms that adjuvant loaded particles have a measurable effect on DC activation using this approach. Furthermore, it was observed that the condition consisting of CpG:MPLA:poly I:C at a ratio of 1c:2m:2p (c = CpG, m = MPLA, p = poly I:C) generated the highest pro-inflammatory state of DCs on the immunoarray, as indicated by high expression of MHC-II, CD86, IL-12, and CCR7 and low expression of IL-10 (Fig. 6f).
Fig. 5.
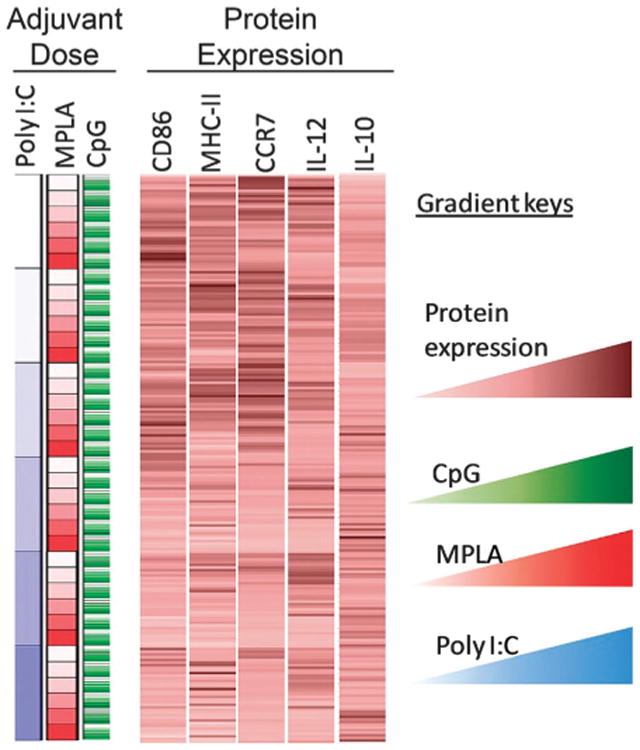
Protein expression and cytokine production from dendritic cells (DCs) upon exposure to different combinations of microparticle-encapsulated adjuvants. A color-coded map connecting external stimulation (in rows) to protein expression from DCs (columns). Columns display relative fluorescent intensity of each marker measured. Each row corresponds to a particular formulation of adjuvant combination. Average RFI per cell were measured across three array experiments per marker, averaged across experiments, and normalized across separate microarrays to each array's no particle control. Gradients are represented by lighter to darker color for CpG (green), MPLA (red), poly I:C (blue) and protein expression (brown).
Fig. 6.
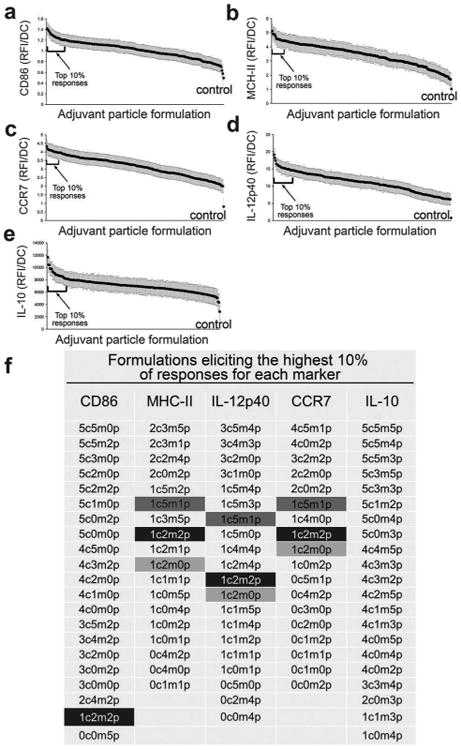
Combinations of microparticle-loaded adjuvants up-regulate dendritic cell activation markers to varying degrees. Microparticle formulations were organized left to right by the level of expression/production of markers from highest to lowest (mean ± standard error) for (a) CD86, (b) MHC-II (c) CCR7 (d) IL-12 (e) IL-10, where the x-axis represents each of the 216 unique formulations. Brackets identify formulations eliciting the highest 10% responses, and are listed in (f). Highlighted are adjuvant combinations with particularly high expression of more than one marker, with the condition consisting of CpG:MPLA:poly I:C at a ratio of 1c:2m:2p generating the highest pro-inflammatory state of DCs indicated by MHC-IIHiCD86HiIL-12HiCCR7HiIL-10Lo. Key: c – CpG; m – MPLA; p – poly I:C; 0 – no particle; 1–62 particles; 2 – 125 particles; 3 – 250 particles; 4 – 500 particles; 5 – 1000 particles.
Adjuvant interactions
To gain insight into the interactions between the three MP encapsulated adjuvants, analysis of the combinatorial effects was performed. The results are presented by arranging data in blocks of dosing categories along the x-axis (Fig. 7a – darker shades represent larger doses). Thus, this data is arranged in large (blue) blocks in order of increasing poly I:C dose; within this superset are smaller (red) blocks in order of increasing MPLA dose; and within this dataset is individual CpG doses (green) in increasing order. This hierarchical data organization was rearranged in all possible configurations to identify potential trends (data not shown).
Fig. 7.
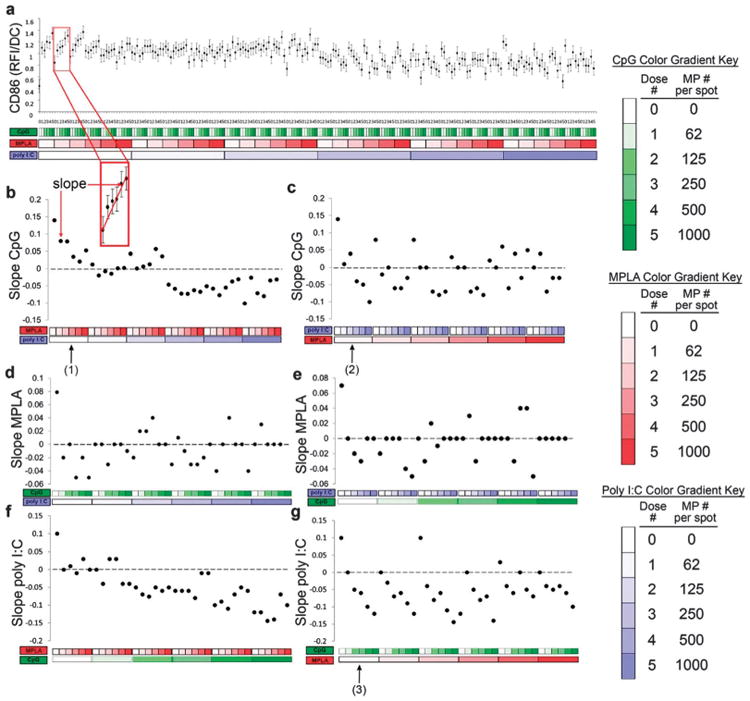
Analysis of dendritic cell (DC) CD86 expression (RFI) in response to combinatorial adjuvant-loaded microparticles (MPs). (a) CD86 expression (RFI) per DC is plotted against varying amounts of three MP-encapsulated adjuvants (CpG, MPLA and poly I:C; n = 9, with 3 replicates per array on 3 separate arrays). Data is arranged by grouping increasing particle numbers of CpG (green), MPLA (red), and lastly poly I:C (blue) with doses of 0, 62, 125, 250, 500 and 1000 particles per spot, represented by increased shading from lighter to darker colors. The x-axis represents different adjuvant concentrations, and the y-axis represents the expression of CD86 per DC, expressed in RFI. (b and c) Linear fits were calculated for each set of CpG dosing range (lowest to highest number of particles) for every combination of MPLA and poly I:C doses. An example showing the mapping of a single slope is illustrated in the inset. The slope of each linear fit was then calculated and plotted as a function of MPLA and poly I:C doses. Dotted lines represent a slope of zero. (b, arrow # 1) In the absence of poly I:C, the slope of CD86 expression demonstrates a positive response to CpG MPs, and introducing MPLA (red) dampens this effect as seen by the decreasing slope values plotted on the y-axis, with increasing MPLA dose. (b) Introducing poly I:C blocks the dampening effect MPLA has on CpG at lower doses and reverses the positive CD86 response to CpG, as evidenced by the negative slope values, at high poly I:C doses. (c, arrow # 2) In the absence of MPLA, poly I:C decreases CD86 responsiveness to CpG. (c) As MPLA is introduced, it mitigates some of the dampening effect of poly I:C on CpG dependent CD86 expression. (d and e) The adjuvant formulations were then reorganized for every combination dose of CpG and poly I:C. Linear fits were created for the slope of the MPLA response associated with the specific dose of CpG and poly I:C. (d) CD86 expression is modestly upregulated by the presence of MPLA alone (first data point). However, introduction of CpG and poly I:C appears to mitigate this response as most combination doses are near or equal to zero with no apparent trends present. (f and g) The adjuvant formulations were then reorganized for every combination dose of CpG and MPLA. Linear fits were created for the slope of the poly I:C response associated with the specific dose of CpG and MPLA. (f) Increasing CpG dose induces a negative responsiveness to poly I:C. This is further illustrated by reconfiguring the data (g, arrow # 3), showing that in the absence of MPLA, CpG decreases CD86 responsiveness to poly I:C.
Linear regressions were generated for every adjuvant dosing range in the presence of a fixed amount of the other two adjuvants for all possible configurations in order to elucidate the effect of each adjuvant on surface expression of DC activation marker, costimulatory molecule CD86 (Fig. 7b–g).
The slope of each linear fit was calculated, where a positive slope indicates increasing surface expression and a negative slope indicates decreasing expression when exposed to the adjuvant named on the y-axis, and when in combination with fixed amount of the other adjuvants. Using this method, it is thus possible to interpret interaction effects between two or three adjuvants used in combination by comparing how the dose responses are altered upon introduction of additional adjuvants.
Several dominant interactions emerged from the linear regression analysis of the CD86 data. First, exposure to CpG MPs alone displayed the greatest capacity to upregulate expression of CD86 in DCs (Fig. 7b, d and f leftmost data points). Both the addition of MPLA or poly I:C dampens responsiveness to CpG, but while higher dose MPLA can block the response to CpG, higher dose poly I:C can actually reverse it. Additionally, while MPLA had a modest effect on CD86 expression by itself, in combination with CpG or poly I:C this effect was mitigated (Fig. 7d and e). Lastly, the addition of MPLA or CpG dampens responsiveness to poly I:C, and higher CpG doses can reverse the poly I:C response, as previously reported.53
Expression of MHC class II molecules in response to MP-encapsulated adjuvants was quantified (Fig. 8). MHC-II is a critical component of DC activation, allowing presentation of foreign peptides to antigen-specific T-cells.54 Each MP encapsulated adjuvant individually increased MHC-II expression, with similar effects (Fig. 8b, d and f leftmost points). The dose dependent response to CpG with the addition of either MPLA or poly I:C decreased MHC-II surface expression (Fig. 8b and c). On the other hand, dose dependent responses to MPLA were non-linearly modified by the addition of CpG or poly I:C (Fig. 8d and e). Lastly, dose dependent responses to poly I:C with the addition of either MPLA or CpG decreased MHC-II expression (Fig. 8f and g).
Fig. 8.
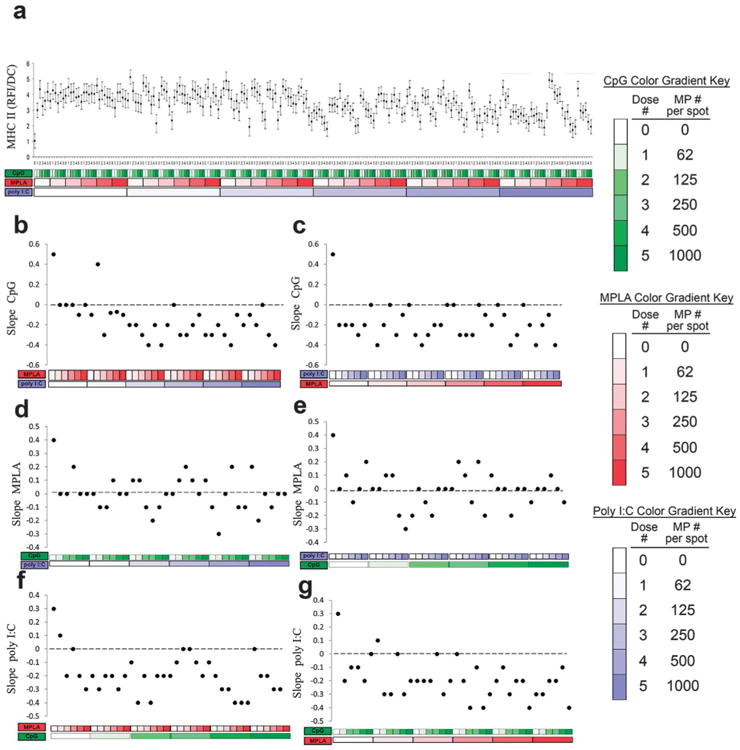
Analysis of dendritic cell (DC) MHC-II expression (RFI) in response to combinatorial adjuvant-loaded microparticles (MPs). (a) MHC-II expression (RFI) per cell is plotted against varying amounts of three MP-encapsulated adjuvants (CpG, MPLA and poly I:C; n = 9, with 3 replicates per array on 3 separate arrays). Data is arranged by grouping increasing particle numbers of CpG (green), MPLA (red), and lastly poly I:C (blue) with doses of 0, 62, 125, 250, 500 and 1000 particles per spot, represented by increased shading from lighter to darker colors. (b and c) Linear fits were calculated for each set of CpG dosing range (lowest to highest number of particles) for every combination of MPLA (red) and poly I:C (blue) doses. The slope of each linear fit was then calculated and plotted as a function of MPLA and poly I:C doses. Dotted lines represent a slope of zero. (d and e) The adjuvant formulations were then reorganized for every combination dose of CpG and poly I:C. Linear fits were created for the slope of the MPLA response associated with the specific dose of CpG and poly I:C. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and poly I:C (blue) doses. Dotted lines represent a slope of zero. (f and g) The adjuvant formulations were then reorganized for every combination dose of CpG and MPLA. Linear fits were created for the slope of the poly I:C response associated with the specific dose of CpG and MPLA. Linear fits were calculated for each set of poly I:C dosing range. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and MPLA (red) doses. Dotted lines represent a slope of zero.
Secretion of the IL-12, a dominant Th1-promoting cytokine,55 was also investigated (Fig. 9). Each adjuvant individually upregulated IL-12 production, with MPLA providing a somewhat larger effect (Fig. 9b, d and f leftmost points). Introduction of either MPLA or poly I:C was capable of reversing the dose dependent response to CpG (Fig. 9b and c). In contrast, the addition of either CpG or poly I:C had a tendency to maintain positive dose dependent responses to MPLA (Fig. 9d and e). Lastly, introduction of either MPLA or CpG was capable of reversing the dose dependent responses to poly I:C (Fig. 9f and g). Overall, these observations suggest significant interactions between MPLA (TLR4) and either poly I:C (TLR3) or CpG (TLR9) on DC production of IL-12p40, which corroborates previous findings.27,39,56
Fig. 9.
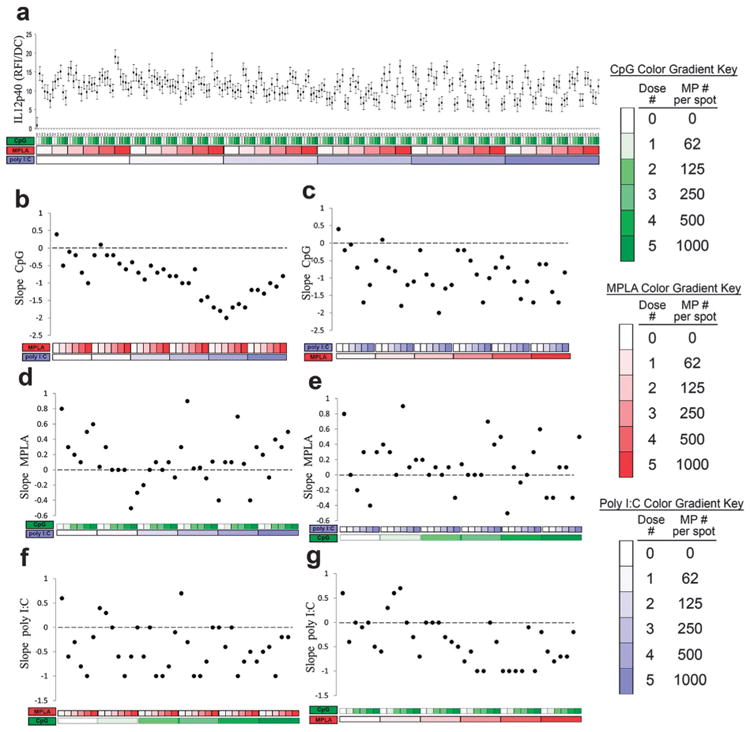
Analysis of dendritic cell (DC) IL-12 production in response to combinatorial adjuvant-loaded microparticles (MPs). (a) IL-12 production per cell is plotted against varying amounts of three MP-encapsulated adjuvants (CpG, MPLA and poly I:C; n = 9, with 3 replicates per array on 3 separate arrays). Data is arranged by grouping increasing particle numbers of CpG (green), MPLA (red), and lastly poly I:C (blue) with doses of 0, 62, 125, 250, 500 and 1000 particles per spot, represented by increased shading from lighter to darker colors. (b and c) Linear fits were calculated for each set of CpG dosing range (lowest to highest number of particles) for every combination of MPLA (red) and poly I:C (blue) doses. The slope of each linear fit was then calculated and plotted as a function of MPLA and poly I:C doses. Dotted lines represent a slope of zero. (d and e) The adjuvant formulations were then reorganized for every combination dose of CpG and poly I:C. Linear fits were created for the slope of the MPLA response associated with the specific dose of CpG and poly I:C. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and poly I:C (blue) doses. Dotted lines represent a slope of zero. (f and g) The adjuvant formulations were then reorganized for every combination dose of CpG and MPLA. Linear fits were created for the slope of the poly I:C response associated with the specific dose of CpG and MPLA. Linear fits were calculated for each set of poly I:C dosing range. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and MPLA (red) doses. Dotted lines represent a slope of zero.
Production of the anti-inflammatory cytokine IL-10 was quantified (Fig. 10). IL-10 was also upregulated by each adjuvant individually, as expected,57,58 with similar responses across adjuvants (Fig. 10b, d and f leftmost points). Positive dose dependent responses to CpG were maintained for nearly all combinations including MPLA and/or poly I:C (Fig. 10b and c). In contrast, the addition of either CpG or poly I:C dampened and reversed the dose dependent response to MPLA (Fig. 10d and e). Positive dose dependent responses to poly I:C were also maintained for nearly all combinations including MPLA and/or CpG (Fig. 10b and c).
Fig. 10.
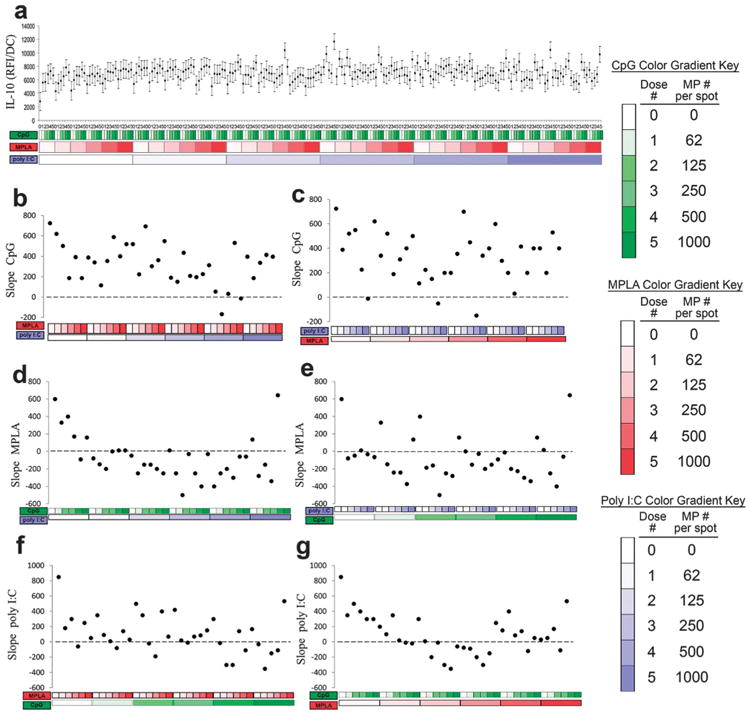
Analysis of dendritic cell (DC) IL-10 production in response to combinatorial adjuvant-loaded microparticles (MPs). (a) IL-10 production per cell is plotted against varying amounts of three MP-encapsulated adjuvants (CpG, MPLA and poly I:C; n = 9, with 3 replicates per array on 3 separate arrays). Data is arranged by grouping increasing particle numbers of CpG (green), MPLA (red), and lastly poly I:C (blue) with doses of 0, 62, 125, 250, 500 and 1000 particles per spot, represented by increased shading from lighter to darker colors. (b and c) Linear fits were calculated for each set of CpG dosing range (lowest to highest number of particles) for every combination of MPLA (red) and poly I:C (blue) doses. The slope of each linear fit was then calculated and plotted as a function of MPLA and poly I:C doses. Dotted lines represent a slope of zero. (d and e) The adjuvant formulations were then reorganized for every combination dose of CpG and poly I:C. Linear fits were created for the slope of the MPLA response associated with the specific dose of CpG and poly I:C. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and poly I:C (blue) doses. Dotted lines represent a slope of zero. (f and g) The adjuvant formulations were then reorganized for every combination dose of CpG and MPLA. Linear fits were created for the slope of the poly I:C response associated with the specific dose of CpG and MPLA. Linear fits were calculated for each set of poly I:C dosing range. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and MPLA (red) doses. Dotted lines represent a slope of zero.
In addition to cytokine signaling, the chemokine receptor CCR7, important for DC lymph node homing,40,59 was quantified (Fig. 11). Expression of CCR7 was upregulated by each adjuvant alone (Fig. 11b, d and f leftmost points). However, the addition of either MPLA and/or poly I:C reversed the dose dependent response to CpG (Fig. 11b and c). In contrast, the dose dependent responses to MPLA were largely muted by the addition of either CpG or poly I:C (Fig. 11d and e). Lastly, the addition of either MPLA and/or CpG reversed the dose dependent response to poly I:C (Fig. 11f and g).
Fig. 11.
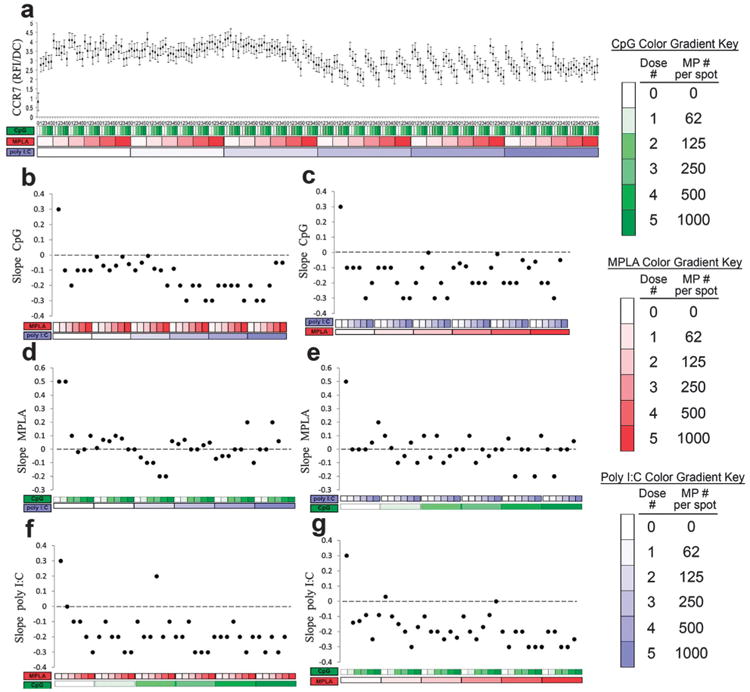
Analysis of dendritic cell (DC) CCR7 expression in response to combinatorial adjuvant-loaded microparticles (MPs). (a) CCR7 expression per cell is plotted against varying amounts of three MP-encapsulated adjuvants (CpG, MPLA and poly I:C; n = 9, with 3 replicates per array on 3 separate arrays). Data is arranged by grouping increasing particle numbers of CpG (green), MPLA (red), and lastly poly I:C (blue) with doses of 0, 62, 125, 250, 500 and 1000 particles per spot, represented by increased shading from lighter to darker colors. (b and c) Linear fits were calculated for each set of CpG dosing range (lowest to highest number of particles) for every combination of MPLA (red) and poly I:C (blue) doses. The slope of each linear fit was then calculated and plotted as a function of MPLA and poly I:C doses. Dotted lines represent a slope of zero. (d and e) The adjuvant formulations were then reorganized for every combination dose of CpG and poly I:C. Linear fits were created for the slope of the MPLA response associated with the specific dose of CpG and poly I:C. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and poly I:C (blue) doses. Dotted lines represent a slope of zero. (f and g) The adjuvant formulations were then reorganized for every combination dose of CpG and MPLA. Linear fits were created for the slope of the poly I:C response associated with the specific dose of CpG and MPLA. Linear fits were calculated for each set of poly I:C dosing range. The slope of each linear fit was then calculated and plotted as a function of CpG (green) and MPLA (red) doses. Dotted lines represent a slope of zero.
DC activation via TLR activation can occur via MyD88 dependent or MyD88 independent pathways, with simultaneous activation potentially eliciting synergistic effects on DC activation.56,60 For instance, activation of TLR2/6, TLR9 (MyD88 dependent) along with TLR3 (MyD88 independent) in DCs has generated strong adaptive responses against HIV infection in mice,61 highlighting the potential of signaling through non-overlapping pathways. In addition to TLRs, nucleotide-binding domain (NOD), leucine rich repeat containing family [Nod-like receptors (NLRs)], and the retinoic-acid inducible protein 1 [RIG-I-like receptors (RLRs)] can interact synergistically62 or antagonistically.63 Ultimately, the role of simultaneous activation of variable signaling pathways has been recognized, and vaccines are being developed that utilize multiple ligands to induce robust immune responses.62
Identifying adjuvant combinations that synergize to promote enhanced immune responses is central to the goal of developing more potent and precise vaccines. To that end, we sought to explore the interactions of TLR agonists CpG, MPLA, and poly I:C in their ability to either induce synergistic or antagonistic DC activation states. When examining surface expression of activation markers (MHC-II, CD86, CCR7) separately, there was a tendency for inhibitory interactions between adjuvants. Similar antagonistic interactions have been shown due to simultaneous activation of TLRs on DCs. For example, inhibitory interactions between TLR4 and TLR2, TLR4 and Dectin-1, TLR2 and TLR9, and TLR3 and TLR2 have been observed.64 On the other hand, cytokine production (IL-12, IL-10) maintained more positive dose dependent responses overall. Previous studies show augmentation of IL-12 production when MyD88 dependent (CpG) and MyD88 independent (poly I:C) pathways were activated simulta-neously56 which supports our findings. However this effect was not observed for surface expression of CD86, corroborating the results found here. Notably, in order to produce robust responses across all activation markers, adjuvant combinations were required, and combinations were more represented among the high responders (Fig. 6f). While mechanisms governing interaction effects between adjuvants were not investigated, considerations may include aspects such as targeting the ligands to subcellular locations of each TLR, the competition of phagocytosis of differently formulated microparticles, the interplay between downstream signaling pathways,56,65 and the dynamics of adjuvant-induced cytokine secretion.66,67
The task of resolving the interplay of TLR networks is arduous, necessitating the development of new tools, such as the immunoarrays here, which facilitate such discovery. A previous approach employed microfluidics to investigate combinatorial adjuvant signaling on DC cytokine production.68 They found MPLA to synergize with multiple TLR agonists in the production of IL-6, IL-12, and TNF-a with the strongest DC activation achieved by MPLA together with Gardiquimodt™ or CpG-B. While encouraging, this approach is limited to screening soluble adjuvants and quantifying cytokine secretion. In contrast, the immunoarray developed here is capable of evaluating cell surface activation markers in addition to cytokine secretion, thereby providing a more comprehensive assessment of DC activation. Further, our approach utilizes microparticle encapsulated adjuvants which have the advantage of providing vaccine stability, controlled release, and preferential targeting to DCs. While different, both approaches underscore the utility of miniaturized platforms to aid in vaccine development through major cost reductions and parallel analyses.
Conclusions
A significant barrier to high-content investigation of new drug candidates is the limited availability of compounds and relevant cells.69 Thus, technologies that aim to reduce compounds and cells required for screening greatly expedite identification of leads. Polymeric, particle-based approaches encapsulating adjuvants can provide delivery to DCs via phagocytosis and controlled release.35–37 A recently described parallel particle production method describes rapid fabrication of hundreds of unique MP formulations.70 Integrating this method with the immunoarray platform may present a robust strategy to generate and quantify large libraries of factor-loaded MPs requiring limited cells and reagents. With thirteen TLR ligands identified to date, the task of resolving the combinatorial effects is challenging using standard techniques. Thus, a high-content approach mapping these interactions is enabling. This work demonstrates feasibility of a MP-based DC immunoarray platform to screen adjuvant combinations for the development of improved and personalized vaccines.
Fig. 4.
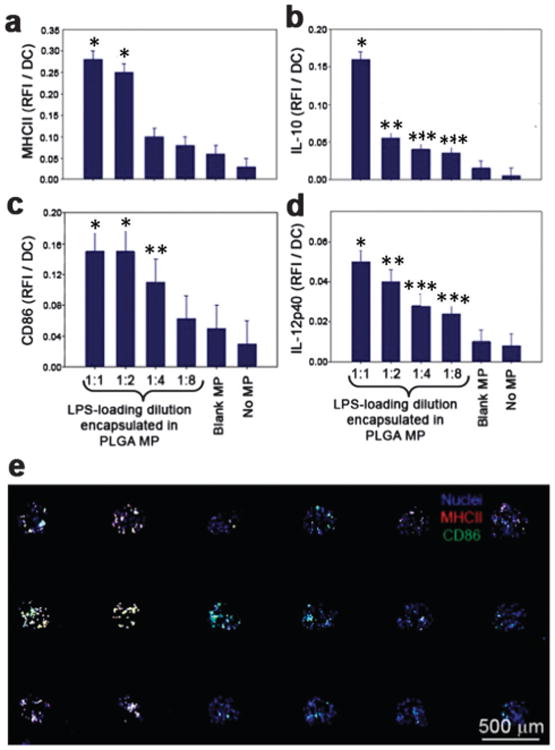
Dose-dependent dendritic cell activation on immunoarrays. Microparticles were fabricated with a range of LPS loading doses and printed in equal numbers onto the immunoarrays. DCs were then cultured on the platform for 16 h before fixation and immunocytochemistry was performed. (a) MHC-II expression was upregulated 6-fold at the highest dose of LPS compared to the unloaded MP control. * significant from 1:4,1:8, blank MP and no MP. (b) IL-10 production was shown to increase 10-fold for the highest LPS dose compared to unloaded MPs. * significant from all other groups; ** significant from 1:1, 1:8, blank MP and no MP; *** significant from 1:1, blank MP and no MP (c) expression of the co-stimulatory signal CD86 revealed a dose-dependent response to LPS that reached a maximum at the second highest dose, a 3-fold increase compared to the unloaded control. * significant from 1:4, 1:8, blank MP and no MP; ** significant from blank MP and no MP. (d) Pro-inflammatory cytokine IL-12 showed increased production for each dose of LPS. * significant from 1:4, 1:8, blank MP and no MP; ** significant from 1:8, blank MP and no MP; *** significant from 1:1, blank MP and no MP. No differences were found between the unloaded blank MPs and the no MP control for all markers. (e) A representative mosaic image of a 3 × 6 immunoarray showing the expression of MHC-II and CD86 by DCs after exposure to LPS loaded PLGA MPs. Dendritic cell nuclei are shown as blue.
Acknowledgments
Research reported in this publication was supported by the following grants: NSF DGE-0802270 (graduate research fellowship to MRC); R21 AI094360, a Juvenile Diabetes Research Foundation Innovative Award, R01 DK091658, and R01 DK098589 (to BGK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Notes and references
- 1.Amorij JP, Kersten GF, Saluja V, Tonnis WF, Hinrichs WL, Slütter B, Bal SM, Bouwstra JA, Huckriede A, Jiskoot W. J Controlled Release. 2012;161:363–376. doi: 10.1016/j.jconrel.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 2.Lindblad EB, Elhay MJ, Silva R, Appelberg R, Andersen P. Infect Immun. 1997;65:623–629. doi: 10.1128/iai.65.2.623-629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chedid L, Audibert F, Lefrancier P, Choay J, Lederer E. Proc Natl Acad Sci U S A. 1976;73:2472–2475. doi: 10.1073/pnas.73.7.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wack A, Rappuoli R. Curr Opin Immunol. 2005;17:411–418. doi: 10.1016/j.coi.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janeway CA, Medzhitov R. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Barton GM, Kagan JC. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pålsson-McDermott EM, O'Neill LA. Biochem Soc Trans. 2007;35:1437–1444. doi: 10.1042/BST0351437. [DOI] [PubMed] [Google Scholar]
- 9.Pashine A, Valiante NM, Ulmer JB. Nat Med. 2005;11:S63–68. doi: 10.1038/nm1210. [DOI] [PubMed] [Google Scholar]
- 10.Ishii KJ, Akira S. J Clin Immunol. 2007;27:363–371. doi: 10.1007/s10875-007-9087-x. [DOI] [PubMed] [Google Scholar]
- 11.Blasius AL, Beutler B. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Morel PA, Vasquez AC, Feili-Hariri M. J Leukocyte Biol. 1999;66:276–280. doi: 10.1002/jlb.66.2.276. [DOI] [PubMed] [Google Scholar]
- 13.Miretti MM, Beck S. Hum Genomics. 2006;2:244–251. doi: 10.1186/1479-7364-2-4-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medzhitov R, Janeway CA. Semin Immunol. 1998;10:351–353. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- 15.Matzinger P. Ann N Y Acad Sci. 2002;961:341–342. doi: 10.1111/j.1749-6632.2002.tb03118.x. [DOI] [PubMed] [Google Scholar]
- 16.Lewis JS, Roy K, Keselowsky BG. MRS Bull. 2014;39:25–34. doi: 10.1557/mrs.2013.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller M, Reichardt W, Koerner J, Groettrup M. J Controlled Release. 2012;162:159–166. doi: 10.1016/j.jconrel.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Adv Drug Delivery Rev. 2011;63:943–955. doi: 10.1016/j.addr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 19.Morse MA, Deng Y, Coleman D, Hull S, Kitrell-Fisher E, Nair S, Schlom J, Ryback ME, Lyerly HK. Clin Cancer Res. 1999;5:1331–1338. [PubMed] [Google Scholar]
- 20.Lin CY, Lin SJ, Yang YC, Wang DY, Cheng HF, Yeh MK. Hum Vaccines Immunother. 2015;11:650–656. doi: 10.1080/21645515.2015.1009345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou XF, Liu B, Yu XH, Zha X, Zhang XZ, Chen Y, Wang XY, Jin YH, Wu YG, Shan YM, Liu JQ, Kong W, Shen JC. J Controlled Release. 2007;121:200–207. doi: 10.1016/j.jconrel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keselowsky BG, Xia CQ, Clare-Salzler M. Hum Vaccines. 2011;7:37–44. doi: 10.4161/hv.7.1.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo J, Clare-Salzler MJ. Autoimmun Rev. 2006;5:419–423. doi: 10.1016/j.autrev.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. Clin Immunol. 2015;160:90–102. doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG. Sci Rep. 2015;5:13155. doi: 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García-Sastre A, Compans R, Pulendran B. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coussens LM, Zitvogel L, Palucka AK. Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acharya AP, Dolgova NV, Clare-Salzler MJ, Keselowsky BG. Biomaterials. 2008;29:4736–4750. doi: 10.1016/j.biomaterials.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 30.Acharya AP, Dolgova NV, Xia CQ, Clare-Salzler MJ, Keselowsky BG. Acta Biomater. 2011;7:180–192. doi: 10.1016/j.actbio.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 31.Phillips BE, Giannoukakis N, Trucco M. Pediatr Endocrinol Rev. 2008;5:873–879. [PubMed] [Google Scholar]
- 32.Steinman RM. Immunity. 2008;29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Lu L, Thomson AW. Transplantation. 2002;73:S19–22. doi: 10.1097/00007890-200201151-00008. [DOI] [PubMed] [Google Scholar]
- 34.Buonerba C, Ferro M, Di Lorenzo G. Expert Rev Anticancer Ther. 2011;11:25–28. doi: 10.1586/era.10.180. [DOI] [PubMed] [Google Scholar]
- 35.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Nat Mater. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy ST, Swartz MA, Hubbell JA. Trends Immunol. 2006;27:573–579. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Singh A, Qin H, Fernandez I, Wei JS, Lin J, Kwak LW, Roy K. J Controlled Release. 2011;155:184–192. doi: 10.1016/j.jconrel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Wistuba II, Gelovani JG, Jacoby JJ, Davis SE, Herbst RS. Nat Rev Clin Oncol. 2011;8:135–141. doi: 10.1038/nrclinonc.2011.2. [DOI] [PubMed] [Google Scholar]
- 39.Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, Groettrup M. Vaccine. 2008;26:1626–1637. doi: 10.1016/j.vaccine.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 40.Elamanchili P, Lutsiak CM, Hamdy S, Diwan M, Samuel J. J Immunother. 2007;30:378–395. doi: 10.1097/CJI.0b013e31802cf3e3. [DOI] [PubMed] [Google Scholar]
- 41.Acharya AP, Clare-Salzler MJ, Keselowsky BG. Biomaterials. 2009;30:4168–4177. doi: 10.1016/j.biomaterials.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 42.Lewis JS, Dolgova NV, Chancellor TJ, Acharya AP, Karpiak JV, Lele TP, Keselowsky BG. Biomaterials. 2013;34:9063–9070. doi: 10.1016/j.biomaterials.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutsiak ME, Robinson DR, Coester C, Kwon GS, Samuel J. Pharm Res. 2002;19:1480–1487. doi: 10.1023/a:1020452531828. [DOI] [PubMed] [Google Scholar]
- 44.Rand U, Rinas M, Schwerk J, Nöhren G, Linnes M, Kröger A, Flossdorf M, Kály-Kullai K, Hauser H, Höfer T, Köster M. Mol Syst Biol. 2012;8:584. doi: 10.1038/msb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acharya AP, Dolgova NV, Moore NM, Xia CQ, Clare-Salzler MJ, Becker ML, Gallant ND, Keselowsky BG. Biomaterials. 2010;31:7444–7454. doi: 10.1016/j.biomaterials.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 46.Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, Puccetti P. Immunity. 1998;9:315–323. doi: 10.1016/s1074-7613(00)80614-7. [DOI] [PubMed] [Google Scholar]
- 47.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O'Hagan DT, Pétrilli V, Tschopp J, O'Neill LA, Lavelle EC. Proc Natl Acad Sci U S A. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salvador A, Igartua M, Hernández RM, Pedraz JL. Vaccine. 2012;30:589–596. doi: 10.1016/j.vaccine.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 49.Aranda F, Llopiz D, Díaz-Valdés N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martínez M, Durantez M, Mansilla C, Prieto J, Lasarte JJ, Borrás-Cuesta F, Sarobe P. Cancer Res. 2011;71:3214–3224. doi: 10.1158/0008-5472.CAN-10-3259. [DOI] [PubMed] [Google Scholar]
- 50.Gnjatic S, Sawhney NB, Bhardwaj N. Cancer J. 2010;16:382–391. doi: 10.1097/PPO.0b013e3181eaca65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pulendran B. Immunol Res. 2004;29:187–196. doi: 10.1385/IR:29:1-3:187. [DOI] [PubMed] [Google Scholar]
- 52.Barton GM, Medzhitov R. Curr Opin Immunol. 2002;14:380–383. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 53.Skold AE, Hasan M, Vargas L, Saidi H, Bosquet N, Le Grand R, Smith CIE, Spetz AL. Blood. 2012;120:768–777. doi: 10.1182/blood-2011-12-397778. [DOI] [PubMed] [Google Scholar]
- 54.Chow A, Toomre D, Garrett W, Mellman I. Nature. 2002;418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 55.Steinman RM, Banchereau J. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 56.Zhu Q, Egelston C, Vivekanandhan A, Uematsu S, Akira S, Klinman DM, Belyakov IM, Berzofsky JA. Proc Natl Acad Sci U S A. 2008;105:16260–16265. doi: 10.1073/pnas.0805325105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ngoi SM, Tovey MG, Vella AT. J Immunol. 2008;181:7670–7680. doi: 10.4049/jimmunol.181.11.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruchaud-Sparagano MH, Mills R, Scott J, Simpson AJ. Immunol Cell Biol. 2014;92:799–809. doi: 10.1038/icb.2014.55. [DOI] [PubMed] [Google Scholar]
- 59.Scandella E, Men Y, Gillessen S, Förster R, Groettrup M. Blood. 2002;100:1354–1361. doi: 10.1182/blood-2001-11-0017. [DOI] [PubMed] [Google Scholar]
- 60.Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 61.Zhu Q, Egelston C, Gagnon S, Sui Y, Belyakov IM, Klinman DM, Berzofsky JA. J Clin Invest. 2010;120:607–616. doi: 10.1172/JCI39293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duthie MS, Windish HP, Fox CB, Reed SG. Immunol Rev. 2011;239:178–196. doi: 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Negishi H, Yanai H, Nakajima A, Koshiba R, Atarashi K, Matsuda A, Matsuki K, Miki S, Doi T, Aderem A, Nishio J, Smale ST, Honda K, Taniguchi T. Nat Immunol. 2012;13:659–666. doi: 10.1038/ni.2307. [DOI] [PubMed] [Google Scholar]
- 64.Timmermans K, Plantinga TS, Kox M, Vaneker M, Scheffer GJ, Adema GJ, Joosten LAB, Netea MG. Clin Vaccine Immunol. 2013;20:427–432. doi: 10.1128/CVI.00703-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EEM, Trinchieri G, Caux C, Garrone P. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Re F, Strominger JL. J Immunol. 2004;173:7548–7555. doi: 10.4049/jimmunol.173.12.7548. [DOI] [PubMed] [Google Scholar]
- 67.Willems F, Marchant A, Delville JP, Gérard C, Delvaux A, Velu T, de Boer M, Goldman M. Eur J Immunol. 1994;24:1007–1009. doi: 10.1002/eji.1830240435. [DOI] [PubMed] [Google Scholar]
- 68.Garcia-Cordero JL, Nembrini C, Stano A, Hubbell JA, Maerkl SJ. Integr Biol. 2013;5:650–658. doi: 10.1039/c3ib20263a. [DOI] [PubMed] [Google Scholar]
- 69.Carstens MR, Fisher RC, Acharya AP, Butterworth EA, Scott E, Huang EH, Keselowsky BG. Proc Natl Acad Sci U S A. 2015;112:8732–8737. doi: 10.1073/pnas.1505374112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Acharya AP, Lewis JS, Keselowsky BG. Biomaterials. 2013;34:3422–3430. doi: 10.1016/j.biomaterials.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]