

Abstract
The observation of habitat‐specific phenotypes suggests the action of natural selection. The three‐spined stickleback (Gasterosteus aculeatus) has repeatedly colonized and adapted to diverse freshwater habitats across the northern hemisphere since the last glaciation, while giving rise to recurring phenotypes associated with specific habitats. Parapatric lake and river populations of sticklebacks harbour distinct parasite communities, a factor proposed to contribute to adaptive differentiation between these ecotypes. However, little is known about the transcriptional response to the distinct parasite pressure of those fish in a natural setting. Here, we sampled wild‐caught sticklebacks across four geographical locations from lake and river habitats differing in their parasite load. We compared gene expression profiles between lake and river populations using 77 whole‐transcriptome libraries from two immune‐relevant tissues, the head kidney and the spleen. Differential expression analyses revealed 139 genes with habitat‐specific expression patterns across the sampled population pairs. Among the 139 differentially expressed genes, eight are annotated with an immune function and 42 have been identified as differentially expressed in previous experimental studies in which fish have been immune challenged. Together, these findings reinforce the hypothesis that parasites contribute to adaptation of sticklebacks in lake and river habitats.
Keywords: habitat‐specific gene expression, immune genes, parasites, RNA‐Seq, three‐spined stickleback, transcriptomics
Introduction
The repeated occurrence of similar phenotypes associated with a distinct habitat is often attributed to the direct effect of natural selection (Elmer & Meyer 2011). Parallel phenotypic evolution among populations from geographically distant but ecologically similar habitats, referred to here as habitat‐specific phenotypes, is thought to reflect the advantages of those phenotypes in their respective habitat (Savolainen et al. 2013). Numerous examples have been documented including pharyngeal jaw and thick lips in cichlids (Albertson et al. 2005; Colombo et al. 2013), similar ecotype morphs of anolis lizards (Losos et al. 1998; Harmon et al. 2005), habitat‐specific pigmentation in isopods (Hargeby et al. 2004), repeated ecotypes with distinct shell sizes in the periwinkle snail (Butlin et al. 2014) and repeated differences of body depth and gill raker numbers between lake and stream sticklebacks (Berner et al. 2008; Kaeuffer et al. 2012; Lucek et al. 2014). Although phenotypic plasticity can contribute to such habitat‐specific phenotypes (Muschick et al. 2012; Machado‐Schiaffino et al. 2014; Moser et al. 2015), some of these traits have been shown to be genetically determined and under adaptive evolution (Hargeby et al. 2004; Albertson et al. 2005; Colombo et al. 2013). Adaptive genetic changes include those that result from polymorphisms that alter protein structures (Ffrench‐Constant et al. 1993; Hoekstra et al. 2006; Protas et al. 2006) as well as those that influence phenotypes via regulation of gene expression (Rebeiz et al. 2009; Chan et al. 2010). Gene expression has been associated with adaptive changes in morphological and physiological changes (Rebeiz et al. 2009; Manceau et al. 2011; Harrison et al. 2012) and is believed to contribute to adaptive divergence in natural populations (Pavey et al. 2010).
As gene expression bridges the underlying genotype to the ultimate morphological and physiological phenotypes, it can be considered as an extended molecular phenotype (Ranz & Machado 2006). Hence, it is interesting to evaluate whether or not gene expression patterns differ between contrasting habitats and if so whether they hold across geographically distant populations. Such habitat‐specific gene expression could arise due to several factors, such as genetically determined expression patterns among similar habitat types (ecotypes), as well plastic responses to extrinsic environmental conditions specific to a habitat. Aside from other mechanisms that might control regulation of transcription such as epigenetics, genetic studies have demonstrated variable degrees of heritability of gene expression and have for some phenotypes revealed the genetic basis underlying expression differences (Stamatoyannopoulos 2004; Gibson & Weir 2005; Gilad et al. 2008). There are examples of mutations affecting cis‐ and trans‐regulatory regions in the genome that silence or dramatically shift gene expression, including single nucleotide polymorphisms (SNPs) (Cheung & Spielman 2009; Fraser 2013), copy number variations (CNVs) (Haraksingh & Snyder 2013) and tandem repeats (Gemayel et al. 2010). Genomic changes in regulatory regions can alter the efficiency of transcription factors and thus affect expression of adjacent or remote genes. In sticklebacks, for example, frequent independent deletion events in the enhancer of Pitx1 suppress expression of the gene and result in repeated pelvic reduction in freshwater populations (Chan et al. 2010). Besides its heritable (genetic) component, gene expression is also a versatile phenotype that dynamically responds to changes in the environment (Gibson 2008) and holds the potential to facilitate plasticity to buffer against environmental changes (Franssen et al. 2011; Whitehead 2012; Morris et al. 2014). Despite the variability introduced by uncontrollable environmental factors, studies of gene expression in wild‐caught populations offer the opportunity to estimate the physiological responses of organisms in their environment, potentially providing insight into the role of gene expression variation in adaptation and acclimation to environmental stresses through genetic or plastic changes (Cheviron et al. 2008).
The repeated and independent postglacial colonization history of the three‐spined stickleback (Gasterosteus aculeatus) makes it a powerful study system to investigate habitat‐specific phenotypic evolution. Sticklebacks inhabit various marine and freshwater habitats across the northern hemisphere (MacKinnon & Rundle 2002), a distribution likely attributable to rapid adaptation from extensive standing genetic variation (Barrett & Schluter 2008; Eizaguirre et al. 2012a). Genetically diverged but geographically adjacent lake and river population pairs exhibit consistent morphological differentiation across multiple pairs, such as divergence for body depth and gill raker number (Berner et al. 2008; Kaeuffer et al. 2012; Lucek et al. 2014). These lake and river populations are also often referred to as ecotypes (Reusch et al. 2001). Many ecological factors differ between lake and river habitats, such as flow regime, temperature, food resource and predator communities, all contributing to the differentiation of lake and river stickleback ecotypes, for example in foraging traits (Berner et al. 2010) and antipredator traits (Lucek et al. 2014). Another important ecological difference between lakes and rivers is the locally distinct parasite communities (Kalbe et al. 2002; Eizaguirre et al. 2011; Karvonen et al. 2015). Besides harbouring different species of parasites between ecotypes, lake fish commonly have a higher parasite load than river fish comparing parapatric population pairs (Eizaguirre et al. 2011), and higher immuno‐competence (Scharsack et al. 2007). Lake fish also exhibit a higher diversity in the major histocompatibility complex (MHC) (Eizaguirre et al. 2011), believed to be a result of local adaptation (Eizaguirre et al. 2009, 2012b). Distinct immune expression patterns between lake and river individuals were detected upon multiple experimental parasite exposure of laboratory‐bred sticklebacks (Lenz et al. 2013). Altogether, these studies suggest that parasites play an important role in the differentiation of lake and river ecotypes by shaping the diversity and expression patterns of immune‐related genes. It is, however, not yet known whether the generality of these patterns holds in multiple lake‐river systems under natural conditions.
In this study, we performed an extensive transcriptomic survey using an RNAseq approach across four parapatric lake and river stickleback population pairs to investigate patterns of habitat‐specific gene expression. We used two major organs involved in immune response, the head kidney and the spleen. Differential expression analysis was performed between fish from lake and river habitats, and results were compared to the differentially expressed genes between laboratory‐bred individuals in controlled parasite infection experiments (Lenz et al. 2013; Haase et al. 2014). Our study describes gene expression differences in an ecological framework, highlighting habitat‐specific expression of genes that might be involved in adaptation.
Materials and methods
Sampling
Three‐spined sticklebacks were sampled in 2010 for genomic studies (Chain et al. 2014; Feulner et al. 2015), from which four parapatric lake–river population pairs were used in this study. These included two independent drainages from Germany: Großer Plöner See lake (G1_L) and Malenter Au river (G1_R), Westensee lake (G2_L) and Eider river (G2_R), one pair from Norway: Skogseidvatn lake (No_L) and Orraelva river (No_R), and one pair from Canada: Misty Lake (Ca_L) and Misty Stream (Ca_R) (See Table 1). All these lake‐river population pairs are significantly differentiated from each other, with a mean genome‐wide F ST ranging between 0.11 and 0.28 (for more detailed information about sampling sites and genetic differentiation between the populations, see Feulner et al. 2015). The two population pairs from Germany were sampled in May while the Norwegian and Canadian populations were sampled in September. About 20 individual fish per site were caught using dip nets or minnow traps and kept alive for a few hours in the water from where they were sampled until being euthanized using MS222 and dissection. For each population pair, the fish were treated identically after capture and lake fish and river fish were alternatively dissected. Fish standard length and weight were recorded and macroparasites screened following established procedures for three‐spined sticklebacks (Kalbe et al. 2002) (Table S1, Supporting information). Immediately after euthanasia, the whole head kidneys and spleens were dissected out and preserved in RNAlater (Sigma‐Aldrich) for later transcriptomic library preparation. These are the main immune organs in teleost fish and are commonly used for immunological studies (Press & Evensen 1999). Six individuals (three males and three females, except No_L with four males and two females) were selected for transcriptomic sequencing per sampling site. Fish selection was performed ignoring parasite screening results, but was nonrandom to ensure an equal sex distribution for each population and with a preference for larger fish to guaranty sufficient yield of RNA. Body weights of the selected fish suggest that all fish were older than 1 year (Table S1, Supporting information).
Table 1.
Summary of sample site information and number of individuals included in the transcriptomic analysis
| Population pair | Location | Habitat | Name | Head kidney | Spleen |
|---|---|---|---|---|---|
| G1 | Germany | Lake | Großer Ploener See (G1_L) | 6 | 6 |
| River | Malenter Au (G1_R) | 5 | 5 | ||
| G2 | Germany | Lake | Westensee (G2_L) | 6 | 5 |
| River | Eider (G2_R) | 6 | 6 | ||
| No | Norway | Lake | Skogseidvatnet (No_L) | 3 | 4 |
| River | Orraelva (No_R) | 4 | 4 | ||
| Ca | Canada | Lake | Misty Lake (Ca_L) | 5 | 3 |
| River | Misty Stream Inlet (Ca_R) | 6 | 3 |
RNA library preparation and sequencing
Total RNA (using the entire tissue dissected) was extracted from preserved samples using NucleoSpin® RNA (Mackerey‐Nagel) and reverse transcribed to cDNA using Omniscript RT kits (Qiagen). RNA was quantified with NanoDrop and Bioanalyzer, and ~1 μg of RNA in a concentration of 20 ng/μL was used for library construction. A few samples with poor RNA quality were excluded before constructing 77 libraries. Therefore, sample sizes per population vary between 3 and 6 individuals (Table 1). TruSeq RNA sample preparation kit (Illumina) was used for paired‐end library construction according to the manufacturer's instructions. Each sample was barcoded with a unique sequence index tag, and pools of 12 different barcoded samples were loaded in 8 lanes of a single flow cell of Illumina HiScanSQ machine.
Read filtering and mapping
Raw reads were quality filtered before read mapping in the following steps. All raw reads output to fastq files were 101 base pairs (bp) in length. Sequencing adaptors were removed using seqprep 0.4 (https://github.com/jstjohn/SeqPrep). prinseq 0.20.3 lite (Schmieder & Edwards 2011) was used to trim the read tails with a PHRED quality score below 20 as well as poly‐A tails longer than 10 bp. We kept read pairs for which both reads were longer than 60 bp after trimming. After filtering, read lengths varied from 60 to 101 bp, with about 60% of the reads exhibiting the initial 101‐bp length. Exact duplicates of both paired‐ends were removed with prinseq. The remaining quality‐filtered reads were aligned against the stickleback reference genome from ensembl version 68 (Flicek et al. 2012) using tophat2 v2.0.13 (Kim et al. 2013) with default settings. htseq 0.5.4p5 (Anders et al. 2014) was used to quantify read count for each gene using ensembl gene annotations (version 68) using default settings except for excluding reads with alignment quality below 5.
Gene expression analyses
Gene expression across all samples was evaluated with the bioconductor package edger 3.4.2 (Robinson et al. 2010). First, weakly expressed genes were filtered out when they had <1 read per million in half (38) of the 77 samples (Anders et al. 2013). All libraries were then simultaneously normalized with the trimmed mean of M‐value (TMM) method (Robinson & Oshlack 2010), implemented in the edger package. The TMM method computes the scaling factors as the weighted mean of log fold changes for the majority of genes between libraries, based on the assumption that the majority of genes are not differentially expressed. After applying the TMM method, most genes should have unified expression levels across individuals and the scaling factors for the libraries should be close to 1 (Dillies et al. 2012). Except for one head kidney library from G1_R with a scaling factor of 0.35, all other transcriptome libraries obtained scaling factors close to 1 (from 0.75 to 1.18, Table S2, Supporting information). The outlier library had fewer genes expressed compared to other libraries (12 769 vs. 15 735–17 341). This indicates a distinct expression profile likely dominated by technical artefacts, and therefore, this library was excluded from further analyses.
Next, the dispersion of the negative binomial distribution for the expression of each gene was estimated in edger. It represents the biological coefficient of variation of a gene's expression. This was used to evaluate the expression variance where a high dispersion value indicates high variance of gene expression pattern among samples. A principal component analysis (PCA) was then performed in r 3.0.1 (R Development Core Team 2008) using prcomp function based on log‐transformed normalized read counts of all 12 222 expressed genes (across both tissues and after filtering out weakly expressed genes as mentioned above) to assess differences in gene expression across libraries (Fig. 1).
Figure 1.
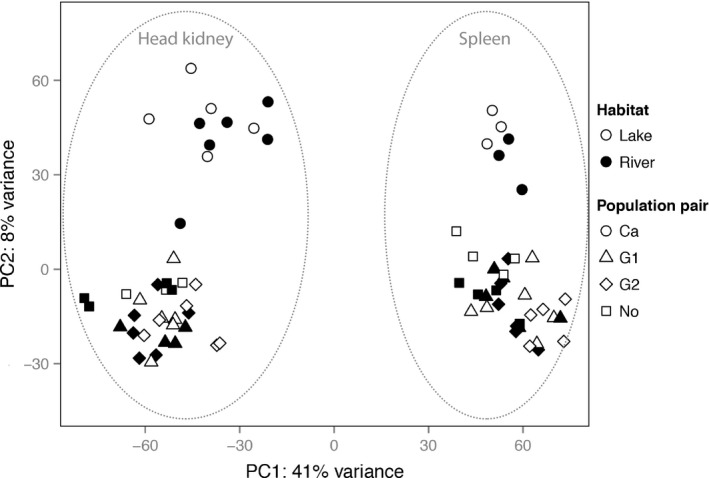
Principal component analysis (PCA) of gene expression profiles based on all genes after filtering out weakly expressed genes (See Methods). Head kidney samples and spleen samples are separated along the x‐axis, and the Canadian samples are separated along the y‐axis. PCA axes explain 41% (x‐axis) and 8% (y‐axis) of the total variation.
To identify habitat‐specific gene expression, that is the expression patterns that are similar within habitat types while significantly different between habitat types, we employed differential expression (DE) analyses that contrast lake and river fish from all four population pairs. On the basis of the PCA result (Fig. 1), DE analyses were performed separately for head kidney and spleen libraries in edger. Because the PCA results suggest that the Canadian populations are substantially diverged from the European populations, the DE analyses were also performed only among the three European population pairs (those results are presented in the Supplement only). Hence, four DE analyses were performed (comparing gene expression in the head kidney across all four population pairs, in spleen across all four population pairs, in head kidney across only the three European population pairs and in spleen across only the three European population pairs). Before conducting DE analyses, weakly expressed genes were filtered out to avoid bias in fold changes due to weak expression of some genes. Genes were filtered out from the DE analyses if they did not have at least 1 read per million in n of the samples, where n is the size of the smaller group (lake or river) in the DE comparisons (Anders et al. 2013). Libraries were renormalized within each comparison group with the TMM method in edger. A multi‐factor design was used in a negative binomial generalized linear model, which accounts for the variation attributed to different population pairs as well as for the variation associated to the sex of the individuals (Expression~Habitat type + Population pair + Sex). The gene‐wise dispersion was reestimated based on the generalized linear model within each comparison group. For each tissue, the distribution of dispersion values was left‐skewed with long tails, indicating that most genes had uniform expression, with a small proportion of genes having highly variable expression across individuals being compared (Fig. S1, Supporting information). We calculated the Pearson correlation of gene expression between all possible pairs of individuals within biological replicates (individuals of the same habitat, population pair and sex) using count data in r. The overall average correlation of gene expression across all pairwise comparisons was 0.86 (first quartile: 0.81 and third quartile: 0.95). Likelihood ratio tests for the contrast coefficient (lake vs. river) were performed, and P‐values were corrected for multiple testing using the Benjamini‐Hochberg method (Benjamini & Hochberg 1995). Genes with corrected P‐values smaller than 0.05 were categorized as differentially expressed genes (DE genes). In addition to performing all DE analyses in edger as described above, DE analyses were also performed with the default pipeline in the deseq2 package 1.0.19 (Love et al. 2014) giving similar results (Table S3, Supporting information).
Functional analyses
Out of 20 787 stickleback genes, 13 568 are annotated with Gene Ontology [GO, (Ashburner et al. 2000)] terms in ensembl version 80. We complemented this with 13 044 gene annotations acquired from the Zebrafish Model Organism Database (ZFIN, Howe et al. 2013) genes associated with stickleback ensembl IDs, with annotation information from ftp://ftp.geneontology.org/pub/go/gene-associations/gene_association.zfin.gz. After merging all annotations, a total of 17 081 of 20 787 stickleback genes were annotated with GO terms. We tested for the enrichment of GO terms in our DE gene sets with the bioconductor package topgo (Alexa et al. 2006; Alexa & Rahnenfuhrer 2010), based on Fisher's exact tests. The gene pools against which we compared the DE gene sets were the genes having sufficient expression and entering the differential expression analyses (see gene expression analyses section above). Overrepresented GO terms were those with a multiple‐test corrected P‐value (Benjamini‐Hochberg's false discovery rate, FDR) smaller than 0.05. To infer the potential involvement of the habitat‐specific expressed genes in parasite defence in nature, we identified our DE genes that were also differential expressed in two previous laboratory‐controlled parasite exposure experiments (Lenz et al. 2013; Haase et al. 2014).
Results
Qualitative description of expression patterns
For each of the 77 transcriptome libraries, an average of 6.5 million read pairs of 101 bp were produced. After adapter cleaning, quality trimming, and duplicate‐ and length‐filtering, 92.78% of the reads remained for analyses (Table S2, Supporting information). On average, 88.10% of the quality‐filtered reads mapped to the reference genome and 2.71% of these mapped to multiple regions of the genome, which were subsequently excluded from further analyses. Out of a total of 22 456 genes annotated in the stickleback genome (ensembl version 68), an average of 16 397 (±944) genes were found expressed. The median number of reads mapping back to each expressed gene was 60 read pairs (first quartile to third quartile: 13–166). The principal component analysis (PCA) clearly separated the two tissue types along the first principal component, which accounted for 41% of the variance observed in the data set (Fig. 1). Within the same tissue type, the second principal component (variance explained: 8%) clearly separated European samples from the Canadian samples.
Differential expression (DE) analyses
After filtering out weakly expressed genes (see Methods), 12 105 genes expressed in head kidney and 12 451 expressed in spleen were contrasted between lake and river ecotypes across all four population pairs. A total of 139 genes showed significant differential expression after correction for multiple testing (Fig. 2). There were 73 DE genes in the head kidney, 74 DE genes in the spleen and 8 of these genes were shared between both tissues (Table S3, Supporting information). All 8 shared DE genes showed the same directional difference of expression between habitat types. A majority of the DE genes (75% in head kidney and 65% in spleen) showed higher expression in individuals from lakes than from rivers. Most of these same DE genes were identified using another commonly used software with default parameters (deseq2: 70 of 73 in the head kidney and 67 of 74 in the spleen, Table S3, Supporting information). Although the PCA analyses mentioned above suggested that the overall expression patterns of the European samples seemed distinct from the Canadian samples, a separate analysis of expression log fold changes between lake and river fish from the three European population pairs showed a strong positive correlation with that of all four population pairs together (linear regression, R 2 = 0.61, P < 0.001 for head kidney and R 2 = 0.82, P < 0.001 for spleen) and resulted in about half of the same DE genes (Table S4, Supporting information). The 5 DE genes with the smallest adjusted P‐value in the head kidney across all lake‐river comparisons include three genes that have higher expression in lake fish (leucine‐rich repeat containing 17, ryanodine receptor 3 and colony‐stimulating factor 1b) and two that have higher expression in river fish (cub and sushi multiple domains 3 and one uncharacterized protein‐coding gene ENSGACG00000000187). The five genes with smallest adjusted P‐values in the spleen include three that have higher expression in lake fish (solute carrier family 43, member 3b, actin binding LIM protein 1b and complement factor D) and two uncharacterized protein‐coding genes (ENSGACG00000000187 and ENSGACG00000012387) that have higher expression in river fish (see Table S3, Supporting information for all 139 DE genes identified).
Figure 2.
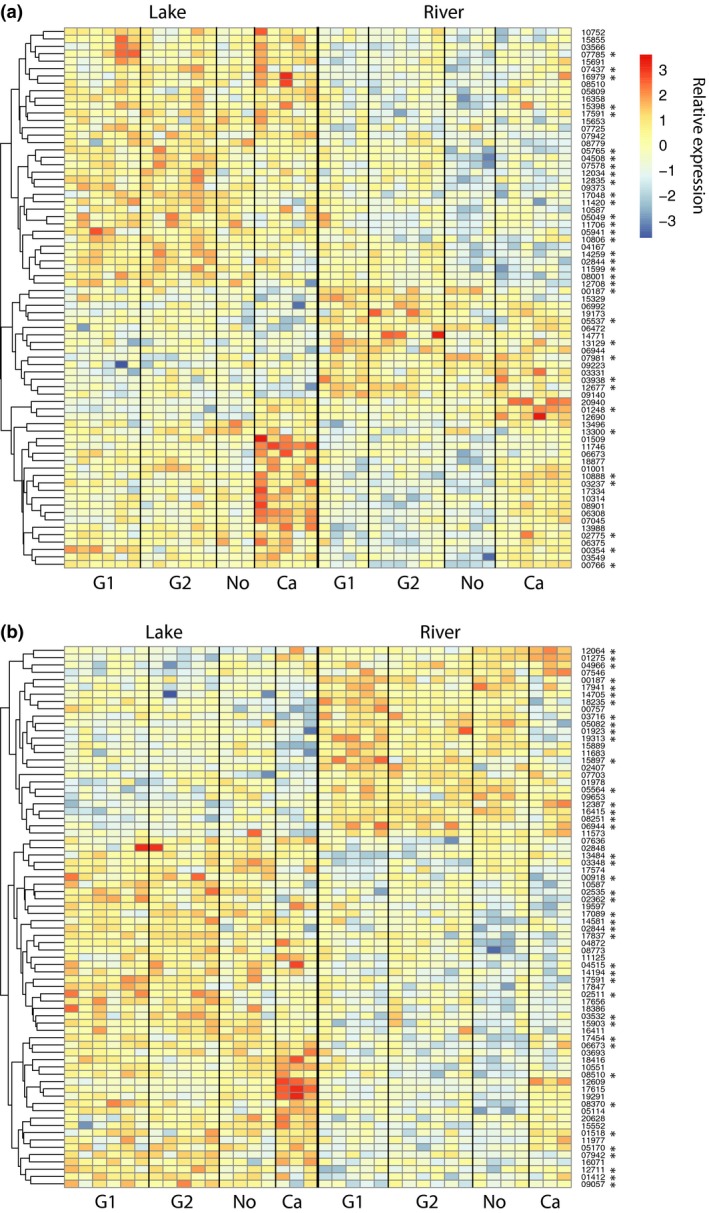
Heatmaps of DE gene expression profiles among all populations in (a) head kidney and (b) spleen. Each column represents one fish and each row represents one gene. Samples are organized by population affiliation as indicated at the bottom. Genes are clustered based on the similarities of the expression profiles between samples. The colour code corresponds to the relative expression intensity, which are the normalized read counts also scaled for each gene's expression intensity (median read count as 0), where red indicates higher expression and blue indicates lower expression. On the right side, the last five digits of the corresponding ensembl ID (ENSGACG00000000000) are shown. Asterisks indicate genes that were also identified in an analysis of the European populations only (Table S4, Supporting information).
Functional analyses of DE genes
GO annotations from ensembl and the ZFIN database were available for 105 of the 139 DE genes (Table S3, Supporting information). The DE genes in head kidney had no significant GO term enrichment, while the DE genes in spleen were enriched for collagen (GO:0005581, with 3 of 18 genes annotated with this term in the gene pool), extracellular region (GO:0005576, with 8 of 265 genes) and extracellular matrix part (GO:0044420, with 3 of 20 genes). Applying a less stringent cut‐off for DE genes (FDR < 0.10) to test for enrichment of GO terms (FDR < 0.05), only extracellular region (GO:0005576) remained significant in the spleen, with no additional terms found in both tissues. The top 50 GO terms from the enrichment analyses of original DE gene sets (FDR < 0.05) are provided in the Dryad database (see Data Accessibility Section). To specifically investigate the differential expression of immune genes in the sampled immune‐related tissues, a list of 1126 stickleback genes with putative immune functions was acquired from a previous study (Haase et al. 2014). Among the DE genes between lake and river fish, three of the 73 DE genes in the head kidney and five of the 74 DE genes in the spleen are putatively immune genes (Table 2). These included macrophage receptors, an interferon regulatory factor and a gene annotated with the functions of antigen processing and presentation and immune response.
Table 2.
Differentially expressed genes between all lake and river populations with putative immune functions
| Gene ID | Gene name | GO term (biological process) | Tissue | Log fold‐changea | FDR |
|---|---|---|---|---|---|
| ENSGACG00000001509 |
Marco
Macrophage receptor with collagenous structure |
Scavenger receptor activity (molecular function) | Head kidney | 0.73 | 0.0053 |
| ENSGACG00000016979 |
CMKLR1 (2 of 2)
chemokine‐like receptor 1 |
G‐protein coupled receptor signalling pathway | Head kidney | 0.77 | 0.0070 |
| ENSGACG00000015855 |
RAB27A
Member RAS oncogene family |
Nucleocytoplasmic transport Small GTPase mediated signal transduction Signal transduction Intracellular protein transport |
Head kidney | 0.56 | 0.026 |
| ENSGACG00000010551 |
Mst1ra
Macrophage stimulating 1 receptor a |
Protein phosphorylation | Spleen | 0.89 | 0.0030 |
| ENSGACG00000012609 |
LGALS1 (2 of 3)
Lectin, galactoside binding, soluble, 1 |
Carbohydrate binding (molecular function) | Spleen | 0.73 | 0.0038 |
| ENSGACG00000004966 |
IRF4 (2 of 2)
Interferon regulatory factor 4b |
Regulation of transcription, DNA‐templated | Spleen | −0.59 | 0.028 |
| ENSGACG00000019291 |
irak3
Interleukin‐1 receptor‐associated kinase 3 |
Signal transduction Protein phosphorylation |
Spleen | 0.42 | 0.048 |
| ENSGACG00000001978 |
Antigen processing and presentation Immune response |
Spleen | −1.44 | 0.048 |
Positive values represent higher expression in lake fish than in river fish and vice versa.
While our analysis only detected very few immune function genes showing differential gene expression, the parasite survey of our sampled fish showed that lake fish harbour higher parasite loads than river fish (Table S1, Supporting information). This has already been demonstrated previously using a larger sample size (Fig. 1 in Feulner et al. 2015). To further investigate the role of parasite infection and potential resistance in driving differential gene expression between lake and river habitats, we compared our results with two laboratory‐controlled parasite exposure experiments that assessed gene expression in sticklebacks from the same German populations as used in our study. Lenz et al. (2013) described the transcriptional responses of laboratory‐bred lake and river sticklebacks under either controlled or parasite‐challenged conditions. That study used three parasites that are found in the natural environment of those fish: Diplostomum pseudospathaceum, Anguillicola crassus and Camallanus lacustris. These parasites were also found in our sampled fish (see discussion and Table S1, Supporting information). Of 166 DE genes between twice parasite‐exposed lake and river fish (Lenz et al. 2013), 51 and 73 genes showed the same directional differences of expression between habitat types in our study among all lake‐river population pairs, in the head kidney and in the spleen, respectively. Some of the differences between the two studies are likely due to that the majority of DE genes in Lenz et al. 2013 were highly expressed in river fish as they are exposed to equal dosage of parasites compared to lake fish, while in our study the majority of DE genes were highly expressed in lake fish as the river fish were exposed to less parasites in nature. Nevertheless, among those genes with same directional differences, one gene methyltransferase like 13 (mettl13) was also identified significantly differentially expressed in our study (Table 3, also see Discussion for more details). In addition, 10 of the 1057 DE genes between control and parasite‐challenged fish (Lenz et al. 2013) overlapped with our set of DE genes (four in the head kidney and six in the spleen). In another recent parasite infection study, laboratory‐bred lake sticklebacks (from the G1_L population) were challenged with the trematode D. pseudospathaceum (Haase et al. 2014), and DE was assessed in the head kidney and in the gill. Of 1060 DE genes between control and challenged fish in the head kidney (Haase et al. 2014), six overlapped with the DE genes from our study (all in the spleen). Of 1415 DE genes in the gill (Haase et al. 2014), 25 overlapped with our set of DE genes (12 in the head kidney and 14 in the spleen, including 1 in both tissues, Table 3).
Table 3.
Differentially expressed genes between lake and river populations also found as differentially expressed in previous parasite infection studies
| Gene ID | Gene name | Comparisons in Lenz et al. 2013, a | Tissue in Lenz et al. 2013 | Log fold‐change in Lenz et al. 2013, b | Tissue in this study | Log fold‐change in this studyc | FDR in this study |
|---|---|---|---|---|---|---|---|
| ENSGACG00000011746 |
fyco1a
FYVE and coiled‐coil domain containing 1a |
Control vs. infected | Head kidney | −3.21 | Head kidney | 0.71 | 0.0096 |
| ENSGACG00000010806 |
sox7
SRY‐box containing gene 7 |
Control vs. infected | Head kidney | −2.58 | Head kidney | 0.74 | 0.038 |
| ENSGACG00000013129 | MRPL49 (2 of 2) mitochondrial ribosomal protein L49 | Control vs. infected | Head kidney | −2.69 | Head kidney | −0.91 | 0.017 |
| ENSGACG00000015653 |
lmo1
LIM domain only 1 |
Control vs. infected | Head kidney | −1.20 | Head kidney | 1.13 | 0.033 |
| ENSGACG00000014705 |
mettl13
Methyltransferase‐like 13 |
Lake vs. River in 2nd infection; control vs. infected | Head kidney | −4.4 and −2.69 | Spleen | −0.63 | 0.028 |
| ENSGACG00000011977 |
ppdpfa
Pancreatic progenitor cell differentiation and proliferation factor a |
Control vs. infected | Head kidney | 1.03 | Spleen | 3.17 | 0.011 |
| ENSGACG00000001923 |
n6amt2
N‐6 adenine‐specific DNA methyltransferase 2 |
Control vs. infected | Head kidney | 1.56 | Spleen | −0.88 | 0.025 |
| ENSGACG00000004515 |
Cfd
Complement factor D (adipsin) |
Control vs. infected | Head kidney | −1.48 | Spleen | 1.16 | 0.00065 |
| ENSGACG00000012609 | LGALS1 (2 of 3) lectin, galactoside binding, soluble, 1 | Control vs. infected | Head kidney | −5.09 | Spleen | 0.73 | 0.0038 |
| ENSGACG00000011683 |
slc5a6b
Solute carrier family 5, member 6 |
Control vs. infected | Head kidney | −2.46 | Spleen | −0.45 | 0.045 |
| Gene ID | Gene name | Comparisons in Haase et al. 2014, a | Tissue in Haase et al. 2014 | Log fold‐change in Haase et al. 2014, b | Tissue in this study | Log fold‐change in this studyc | FDR |
|---|---|---|---|---|---|---|---|
| ENSGACG00000003716 | CASQ2 (1 of 2) calsequestrin 2 | Control vs. clone XII | Head kidney | 2.8 | Spleen | −0.97 | 0.0080 |
| ENSGACG00000017615 |
Smox
Spermine oxidase |
Control vs. clone I, control vs. XII and control vs. clone mix | Head kidney | 4.79, 5.81 and 5.11 | Spleen | 0.65 | 0.011 |
| ENSGACG00000011977 |
ppdpfa
Pancreatic progenitor cell differentiation and proliferation factor a |
Control vs. clone I | Head kidney | −4.08 | Spleen | 3.17 | 0.011 |
| ENSGACG00000004966 | IRF4 (2 of 2) interferon regulatory factor 4b | Control vs. clone I, control vs. XII and control vs. clone mix | Head kidney | 1.41, 2.26 and 1.79 | Spleen | −0.59 | 0.028 |
| ENSGACG00000020628 |
angptl5
Angiopoietin‐like 5 |
Control vs. clone mix | Head kidney | 2.83 | Spleen | 0.71 | 0.028 |
| ENSGACG00000017656 | SVIL (2 of 2) supervillin | Control vs. clone mix | Head kidney | 2.96 | Spleen | 0.59 | 0.040 |
| ENSGACG00000008510 |
apnl
Actinoporin‐like protein |
Control vs. clone mix | Gill | −1.20 | Head kidney and spleen | 0.88 and 1.05 | 0.0093 and 0.0030 |
For the DE genes Haase et al. 2014 identified in gill, only the overlapped DE genes we identified in both head kidney and spleen are shown.
Comparisons where the genes were previously identified as differentially expressed are indicated. In Lenz et al. 2013; DE gene sets between control naïve fish from lake and from river, between twice exposed fish from lake and from river (2nd infection), and between infected fish and control fish were compared to DE gene sets in this study. In Haase et al. 2014, DE gene sets between control fish and infected fish with different parasite clones were compared.
In lake‐river comparisons, positive log fold‐change values represent higher expression in lake fish and vice versa. In control‐infection comparisons, positive values represent upregulation with infection compared to control.
Positive log fold‐change values represent higher expression in lake fish and vice versa.
Discussion
Habitat‐specific expression
This study investigated transcriptional profiles of three‐spined sticklebacks from contrasting lake and river habitats across a wide geographical scale. Physical and ecological differences between lake and river habitats, consisting of differences in flow regime, vegetation, food resources and parasite communities among others, can influence individual fitness, behaviour, life history, morphology and physiology. Studies contrasting lake and river sticklebacks have mainly focused on their morphology (Berner et al. 2010; Lucek et al. 2014) and genomic variation (Deagle et al. 2012; Roesti et al. 2012; Chain et al. 2014; Feulner et al. 2015). Here, we evaluated how lake and river ecotypes differ in gene expression profiles in their natural environments. We have identified habitat‐specific gene expression patterns, that is differential expression between habitats across four lake‐river pairs, three from European locations and one from Canada. For differentially expressed genes, fish from the same habitat have a similar expression, which is distinct from the expression in fish from the contrasting habitat. These habitat‐specific expression patterns suggest that a part of the transcriptome (about 1%) is shaped by the global environmental contrast across all lake‐river pairs, although a larger fraction may be affected by local habitat differences within a given population pair or expressed in other tissues or during a different season or ontogenetic stage. These findings add to the growing discussion of parallelism at the regulatory level between contrasting ecotypes and morphs (Derome et al. 2006; Pavey et al. 2011; Manousaki et al. 2013).
Plasticity and heritability of gene expression
A combination of evolutionary mechanisms could be shaping the habitat‐specific expression patterns observed in this study. Freshwater sticklebacks likely possess the innate ability to regulate certain genes in acclimating to the different conditions in lakes and rivers (Stutz et al. 2015). This plasticity could result in habitat‐specific expression patterns. Alternatively, differential expression across habitats might also reveal adaptive genetic differences between lake and river fish. These alternative explanations for habitat‐specific patterns are by no means mutually exclusive and may both contribute to shape the gene expression profiles of lake and river sticklebacks. Setting our study into the context of previous findings, we further evaluated these explanations. Using the same individuals from this study (as well as additional individuals), recent genomic studies have shown little evidence for sequence‐based habitat‐specific patterns using genome scan approaches with single nucleotide polymorphisms (SNPs; Feulner et al. 2015) and with copy number variations (Chain et al. 2014). Hence, from a genomic perspective, despite significant differentiation between lake and river sticklebacks at a regional scale and across a wider continental scale (Deagle et al. 2012; Roesti et al. 2012; Feulner et al. 2015), there is little evidence for parallel genetic differentiation between lake and river sticklebacks across the distribution area of the fish. In other words, genetic differences between freshwater ecotypes of sticklebacks are for the large part not shared across population pairs, whereas here we identified several genes with habitat‐specific gene expression. This discrepancy is consistent with the observation that phenotypes are similar among lake‐river populations while the genetic basis is different (Deagle et al. 2012; Kaeuffer et al. 2012; Feulner et al. 2015). Gene expression, which bridges the underlying genetic basis and the ultimate phenotypes, might contribute to the understanding of the discrepancy between phenotypes and genotypes. Habitat‐specific expression patterns could be controlled by various trans‐regulatory elements from different genomic sources in different populations. Another explanation is that pathways regulating expression might be triggered at different steps in signalling cascades and therefore leave distinct signatures in the genomes of different populations (Pritchard et al. 2010). Based on controlled laboratory studies, there is evidence that expression differences in sticklebacks can be largely heritable (Leder et al. 2014). In addition, a laboratory‐controlled experiment in which laboratory‐bred G1_L and G1_R sticklebacks exhibited different transcriptional responses to parasite exposure suggested that the genetic background plays an important role in differential gene expression between fish ecotypes (Lenz et al. 2013). It is interesting that this differentiation between lake and river fish was most pronounced in their adaptive immune response (triggered upon 2nd exposure) to parasites, most likely resembling the differences we are observing in nature, where the fish are very likely to have multiple encounters with parasites. In the light of these studies, adaptive genetic differences between lake and river sticklebacks appear to be a likely explanation for habitat‐specific expression patterns. However, a reciprocal transplant experiment suggested that environmentally induced plasticity strongly affects the expression of some carefully selected immune genes (Stutz et al. 2015). Hence, plasticity in gene expression might have also shaped the habitat‐specific expression pattern of some of the genes identified in this study.
Immunological relevance of DE genes
Large‐scale observational studies such as the current one are complementary to experimental studies in general, and here to the stickleback system in particular. Previous studies on sticklebacks in German lake‐river systems highlighted that lake fish harbour higher parasite loads than river fish in terms of intensity and species diversity (Kalbe et al. 2002; Eizaguirre et al. 2011, 2012b). This trend of contrasting parasite loads was further confirmed across a wide geographic range including all populations used in our study (Feulner et al. 2015). Experiments have established that lake and river sticklebacks have differences in immune‐competence due to habitat‐specific adaptation to the distinct parasite communities (Scharsack et al. 2007). It was further investigated that genetic differences in MHC genotypes between lake and river fish provide a basis for parasite‐mediated local adaptation (Eizaguirre et al. 2011, 2012a) following the idea that parasite resistance could represent a magic trait involved in speciation (Eizaguirre et al. 2009). As the differences in parasite pressure between niches could be a force driving divergent adaptation in lake and river sticklebacks, we surveyed gene expression in immune tissues with a specific focus on genes involved in immune functions. Across the 139 candidate genes, we found three putative immune genes in the head kidney and five in the spleen with habitat‐specific expression patterns (Table 2). We found that genes with an immune function were not overrepresented, which indicates that under natural conditions, other factors besides parasites and immunity also contribute to the differentiation between ecotypes. The overrepresented GO terms from these habitat‐specific expressed genes suggest the gene products are often extracellular components, such as collagen‐structured proteins. Given the generic GO terms, their contribution to habitat‐specific adaptation is open to speculation. Nevertheless, a detailed examination of the DE genes showing most significant expression differences (with smallest adjusted P‐values) between lakes and rivers revealed some associations with immune‐related functions. One of the genes that is highly expressed in lake fish and differentially expressed in both the head kidney and in the spleen is colony‐stimulating factor 1b (csf1b), which is involved in macrophage production and differentiation (Stanley et al. 1976). Another DE gene in the head kidney which is highly expressed in lake fish, leucine‐rich repeat containing 17 (lrrc17), regulates osteoclasts in mice cells (Kim et al. 2009). The repeated domain of this gene is involved in a variety of protein–protein interactions, including binding to pathogen‐associated molecular patterns and surface receptors and thus has been studied in pathogen–host interactions (Kedzierski et al. 2004). Some DE genes with putative immune functions are in contrast more highly expressed in river fish. For example, an uncharacterized protein‐coding gene (ENSGACG00000000187) is differentially expressed in both head kidney and spleen, and its sequence is homologous to NOD‐like receptor family CARD domain containing 3 (NLRC3). NLRC3 is a negative regulator of innate immune signalling (Zhang et al. 2014), which inhibits the activity of T cells (Conti et al. 2005) and Toll‐like receptor (Schneider et al. 2012). Another DE gene that is highly expressed in river populations in the head kidney is cub and sushi multiple domains 3 (csmd3), reported to be associated with periodontal pathogen colonization in human (Divaris et al. 2012). The putative immune‐related function of these candidate habitat‐specific genes is consistent with the hypothesis that parasites act as important selective agents driving differentiation between river and lake sticklebacks (Wegner et al. 2003; Scharsack et al. 2007; Eizaguirre et al. 2011, 2012b; Feulner et al. 2015).
To investigate how differences in parasite load between lake and river populations may be reflected in gene expression in the wild, we compared the set of DE genes with the DE gene sets identified in two previous parasite infection experiments performed on G1 stickleback populations. Despite using different conditions, sequencing technologies and bioinformatic analyses to identify DE genes, this exercise provides information on immune‐related functions of DE genes given their putative role in parasite defence based on experimental studies. The two laboratory‐controlled parasite exposure experiments that we compared our results with used three‐spined sticklebacks subjected to infection with parasites that are found in their natural environment: the three parasites Diplostomum pseudospathaceum, A. crassus and C. lacustris in a study by Lenz et al. (2013), and D. pseudospathaceum in a separate study by Haase et al. (2014). An independent parasite survey performed on our own transcriptome‐sequenced fish (Table S1, Supporting information) showed that lake fish have a significantly higher abundance of Diplostomum sp. than river fish (negative binomial GLM, z = −4.87, P < 0.001, see Fig. S2, Supporting information), whereas A. crassus did not show a habitat‐specific pattern (binomial GLM, z = −0.075, P = 0.94) and the lake‐specific parasite C. lacustris (Eizaguirre et al. 2011) was only found in one G1_L fish in our samples. Lenz et al. (2013) assessed gene expression in the head kidney following parasite infection carried out with one of the European population pairs (G1_L and G1_R) used in our study. Among the DE genes found in that study, methyltransferase like 13 (mettl13) was expressed at lower levels in the parasite‐challenged fish compared to controls, and in lake vs. river individuals after a 2nd parasite infection. In our study, this same gene was also differentially expressed with lower expression in the lake populations in the spleen. These results suggest that mettl13 expression is downregulated when the fish are challenged with more parasites, for example in lakes vs. rivers. mettl13 is therefore an interesting candidate for mediating a differential expression between lake and river sticklebacks shaped by the contrasting parasite environment. These comparisons to experimental studies demonstrate another way of inferring functional insights of candidate genes, which goes beyond functional annotations based on sequence similarity with model organisms. These transcriptomic results are in line with the hypothesis that parasite‐mediated selection contributes to lake and river population differentiation; however, it does not act alone but in interaction with other factors under natural conditions.
Limits of the study
Even though we have been able to gain insight into the role of gene expression in population differentiation, various factors confound the analysis of wild‐caught animals. For instance, temporal variation in expression, genetic background differences and stochastic environmental fluctuations introduce variation at the transcriptomic level (Harrison et al. 2012; Lenz 2015). Because our samples are derived from different regions and have been caught at different times of the year, geographical and seasonal factors influenced the observed expression patterns. An important biotic aspect with respect to this study is that fish accumulate parasites from spring to autumn, and their immune system responds differently to early and to late parasite infections (Rohlenová et al. 2011). Furthermore, our study focused on macroparasites, but we acknowledge that there are more pathogens and factors in the natural environment that affect fitness, physiology and immune response. For example, it was found that gut microbiota composition in lake sticklebacks might contribute to shape the genetic polymorphism of MHC class IIb genes (Bolnick et al. 2014), a known genetic basis that vary between fish populations (e.g. Eizaguirre et al. 2011). Hence, microparasites most likely also impact the gene expression of the fish in their natural environments.
In addition, factors like temperature and light condition can vary substantially across geographical regions and seasons. Environmental factors cannot be controlled for sampling on large geographical scale and add noise to the data, reducing the ability to detect habitat‐specific patterns. However, for each location, parapatric lake and river fish were processed at the same time and alternatively dissected, minimizing the variation between lake and river fish within sampling locations. Despite analysing wild‐caught individuals, the majority of our samples showed reasonable correlations between replicated individuals (same habitat, population and sex), resulting in an average Pearson correlation of 0.86. Moreover, including multiple lake‐river contrasts can help to overcome some of the variance among wild‐caught samples, as it is unlikely that environmental fluctuations would produce habitat‐specific expression patterns across multiple individuals and populations by chance. Therefore, our results are conservative estimates of habitat‐specific gene expression across the replicated systems.
Having a single population pair from Canada might also affect some results. As the Canadian populations were rather distinct from the other populations, we also conducted DE analyses only on the three European population pairs for a comparison. However, differential expression between lake and river in the two data sets (with and without the Canadian population pair) was significantly positively correlated and about half of the DE genes are found in both data sets (Table S4, Supporting information). Therefore, including one geographically distant population pair from Canada allows identifying habitat‐specific patterns on a more global scale. It provides an opportunity to examine which genes show consistent habitat‐specific expression patterns in fish across continents, forming a subset of the DE genes from all four population pairs (asterisks in Fig. 2).
As we studied the transcriptomic profiles of wild‐caught fish, a large number of replication in terms of individuals and populations is required to accommodate environmental variations. This results into trading off sample size and sequencing depth. The Encyclopedia of DNA Elements (ENCODE) consortium recommends 30 million pair‐end reads of length >30 nucleotides, in which 20–25 million reads are mappable to the genome for evaluating transcriptional profiles. In our study, the sequencing depths are generally 5× lower than the recommendation, limiting our ability to detect genes with low expression. When we used a more stringent cut‐off to filter out weakly expressed genes (at least two reads per million in half of the samples), 10 715 genes (compared to 12 183 with the original cut‐off) in the head kidney and 11 012 genes (compared to 12 503) in the spleen passed the filtering step. A total of 36 of 73 DE genes in the head kidney and 58 of 74 DE genes in the spleen remained with the higher cut‐off, suggesting at least half of the detected DE results are robust against the low sequencing depth.
Conclusions and prospects
Despite some intrinsic shortcomings, studying gene expression in wild‐caught animals provides a view on differential expression responses caused by both genetic and environmental factors. Our study provides additional evidence that environmental differences, which contrast lakes and rivers and among those the distinct parasite community, shape differential gene expression patterns in sticklebacks. We utilize results of previous laboratory‐controlled experiments to explain the patterns we detected in the wild. This comparison suggests that among other factors the distinct parasite community is most likely an important explanatory factor causing expression differences between habitats. Our results add to previous laboratory results by examining the expression patterns of candidate genes under natural conditions. Those genes identified both here and in previous laboratory studies deserve special attention in potential follow‐up studies.
M.M. funded the project. F.J.J.C., C.E. and M.K. organized and contributed to the sample collections and dissections. F.J.J.C., I.E.S., M.S. and P.G.D.F. prepared the RNA samples. Y.H. performed quality assessment of the sequencing data, transcriptome mapping and processed data for analysis. Y.H., F.J.J.C., M.P. and P.G.D.F. designed the differential expression analyses, and all authors contributed to discussions on research design and results interpretations. Y.H. drafted the manuscript together with F.J.J.C. and P.G.D.F. All authors revised the manuscript.
Data accessibility
The raw reads of RNA‐Seq data (fastq format) and mapping files (bam format) are available through the European Nucleotide Archive (study Accession no.: PRJEB8677, URL: http://www.ebi.ac.uk/ena/data/view/PRJEB8677). htseq read counts, edger results and topGO results are archived in Dryad (doi:10.5061/dryad.hq50s). Morphological and parasite data are included in Table S1 (Supporting information).
Supporting information
Fig. S1 Distribution of dispersion values in (a) head kidney samples and (b) spleen samples.
Fig. S2 Counts of Diplostomum sp. across stickleback populations.
Table S1 Morphological data and parasite loads of sequenced fish
Table S2 Summary of library statistics in sequence quality filtering, sequence mapping and library scaling factor.
Table S3 Differentially expressed genes between lake and river across four population pairs.
Table S4 Differentially expressed genes between lake and river in European populations.
Acknowledgements
We thank Derk Wachsmuth for computational assistance and MPI technicians, especially Anja Hasselmeyer for help with laboratory work. We thank Anika Witten for help with RNA‐Seq library preparation and sequencing. We thank Ludovic Mallet for discussions on the study. We thank the International Max Planck Research School for Evolutionary Biology for research support. We also acknowledge the work and generosity of sample collectors including Andrew Hendry, Renaud Kaeuffer, Shahin Muttalib, Caroline Leblond, Noémie Erin, Per Jakobsen, Tom Klepaker and Hendrik Schultz. We thank Sean Rogers and five reviewers for their insightful and constructive comments on a previous version of this manuscript.
Reference
- Albertson RC, Streelman JT, Kocher TD, Yelick PC (2005) Integration and evolution of the cichlid mandible: the molecular basis of alternate feeding strategies. Proceedings of the National Academy of Sciences of the United States of America, 102, 16287–16292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenfuhrer J (2010) topGO: Enrichment analysis for Gene Ontology. R package version 2.18.0.
- Alexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics, 22, 1600–1607. [DOI] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y et al (2013) Count‐based differential expression analysis of RNA sequencing data using R and Bioconductor. Nature Protocols, 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2014) HTSeq – A Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA et al (2000) Gene Ontology: tool for the unification of biology. Nature Genetics, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RD, Schluter D (2008) Adaptation from standing genetic variation. Trends in Ecology & Evolution, 23, 38–44. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological), 57, 289–300. [Google Scholar]
- Berner D, Adams DC, Grandchamp AC, Hendry AP (2008) Natural selection drives patterns of lake‐stream divergence in stickleback foraging morphology. Journal of Evolutionary Biology, 21, 1653–1665. [DOI] [PubMed] [Google Scholar]
- Berner D, Roesti M, Hendry AP, Salzburger W (2010) Constraints on speciation suggested by comparing lake‐stream stickleback divergence across two continents. Molecular Ecology, 19, 4963–4978. [DOI] [PubMed] [Google Scholar]
- Bolnick DI, Snowberg LK, Caporaso JG et al (2014) Major Histocompatibility Complex class IIb polymorphism influences gut microbiota composition and diversity. Molecular Ecology, 23, 4831–4845. [DOI] [PubMed] [Google Scholar]
- Butlin RK, Saura M, Charrier G et al (2014) Parallel evolution of local adaptation and reproductive isolation in the face of gene flow. Evolution, 68, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain FJ, Feulner PG, Panchal M et al (2014) Extensive copy‐number variation of young genes across stickleback populations. PLoS Genetics, 10, e1004830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YF, Marks ME, Jones FC et al (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science, 327, 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS (2009) Genetics of human gene expression: mapping DNA variants that influence gene expression. Nature Reviews Genetics, 10, 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheviron ZA, Whitehead A, Brumfield RT (2008) Transcriptomic variation and plasticity in rufous‐collared sparrows (Zonotrichia capensis) along an altitudinal gradient. Molecular Ecology, 17, 4556–4569. [DOI] [PubMed] [Google Scholar]
- Colombo M, Diepeveen ET, Muschick M et al (2013) The ecological and genetic basis of convergent thick‐lipped phenotypes in cichlid fishes. Molecular Ecology, 22, 670–684. [DOI] [PubMed] [Google Scholar]
- Conti BJ, Davis BK, Zhang J et al (2005) CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. The Journal of Biological Chemistry, 280, 18375–18385. [DOI] [PubMed] [Google Scholar]
- Deagle BE, Jones FC, Chan YF et al (2012) Population genomics of parallel phenotypic evolution in stickleback across stream‐lake ecological transitions. Proceedings of the Royal Society London Series B: Biological Sciences, 279, 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derome N, Duchesne P, Bernatchez L (2006) Parallelism in gene transcription among sympatric lake whitefish (Coregonus clupeaformis Mitchill) ecotypes. Molecular Ecology, 15, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Dillies MA, Rau A, Aubert J et al (2012) A comprehensive evaluation of normalization methods for Illumina high‐throughput RNA sequencing data analysis. Briefings in Bioinformatics, 14, 671–683. [DOI] [PubMed] [Google Scholar]
- Divaris K, Monda KL, North KE et al (2012) Genome‐wide association study of periodontal pathogen colonization. Journal of Dental Research, 91, 21s–28s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Traulsen A, Milinski M (2009) Speciation accelerated and stabilized by pleiotropic major histocompatibility complex immunogenes. Ecology Letters, 12, 5–12. [DOI] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Sommerfeld RD et al (2011) Parasite diversity, patterns of MHC II variation and olfactory based mate choice in diverging three‐spined stickleback ecotypes. Evolutionary Ecology, 25, 605–622. [Google Scholar]
- Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012a) Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nature Communications, 3, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012b) Divergent selection on locally adapted major histocompatibility complex immune genes experimentally proven in the field. Ecology Letters, 15, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer KR, Meyer A (2011) Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends in Ecology & Evolution, 26, 298–306. [DOI] [PubMed] [Google Scholar]
- Feulner PG, Chain FJ, Panchal M et al (2015) Genomics of divergence along a continuum of parapatric population differentiation. PLoS Genetics, 11, e1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ffrench‐Constant RH, Rocheleau TA, Steichen JC, Chalmers AE (1993) A point mutation in a Drosophila GABA receptor confers insecticide resistance. Nature, 363, 449–451. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D et al (2012) Ensembl 2012. Nucleic Acids Research, 40, D84–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen SU, Gu J, Bergmann N et al (2011) Transcriptomic resilience to global warming in the seagrass Zostera marina, a marine foundation species. Proceedings of the National Academy of Sciences of the United States of America, 108, 19276–19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB (2013) Gene expression drives local adaptation in humans. Genome Research, 23, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annual Review of Genetics, 44, 445–477. [DOI] [PubMed] [Google Scholar]
- Gibson G (2008) The environmental contribution to gene expression profiles. Nature Reviews Genetics, 9, 575–581. [DOI] [PubMed] [Google Scholar]
- Gibson G, Weir B (2005) The quantitative genetics of transcription. Trends in Genetics, 21, 616–623. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Pritchard JK (2008) Revealing the architecture of gene regulation: the promise of eQTL studies. Trends in Genetics, 24, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase D, Rieger JK, Witten A et al (2014) Specific gene expression responses to parasite genotypes reveal redundancy of innate immunity in vertebrates. PLoS ONE, 9, e108001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraksingh RR, Snyder MP (2013) Impacts of variation in the human genome on gene regulation. Journal of Molecular Biology, 425, 3970–3977. [DOI] [PubMed] [Google Scholar]
- Hargeby A, Johansson J, Ahnesjo J (2004) Habitat‐specific pigmentation in a freshwater isopod: adaptive evolution over a small spatiotemporal scale. Evolution, 58, 81–94. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Kolbe JJ, Cheverud JM, Losos JB (2005) Convergence and the multidimensional niche. Evolution, 59, 409–421. [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Mank JE (2012) The evolution of gene expression and the transcriptome‐phenotype relationship. Seminars in Cell and Developmental Biology, 23, 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP (2006) A single amino acid mutation contributes to adaptive beach mouse color pattern. Science, 313, 101–104. [DOI] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF et al (2013) The zebrafish reference genome sequence and its relationship to the human genome. Nature, 496, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer R, Peichel CL, Bolnick DI, Hendry AP (2012) Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback. Evolution, 66, 402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbe M, Wegner KM, Reusch TBH (2002) Dispersion patterns of parasites in 0+ year three‐spined sticklebacks: a cross population comparison. Journal of Fish Biology, 60, 1529–1542. [Google Scholar]
- Karvonen A, Lucek K, Marques DA, Seehausen O (2015) Divergent macroparasite infections in parapatric Swiss lake‐stream pairs of threespine stickleback (Gasterosteus aculeatus). PLoS ONE, 10, e0130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski L, Montgomery J, Curtis J, Handman E (2004) Leucine‐rich repeats in host‐pathogen interactions. Archivum Immunologiae et Therapia Experimentalis, 52, 104–112. [PubMed] [Google Scholar]
- Kim T, Kim K, Lee SH et al (2009) Identification of LRRc17 as a negative regulator of receptor activator of NF‐κB Ligand (RANKL)‐induced osteoclast differentiation. Journal of Biological Chemistry, 284, 15308–15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C et al (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder EH, McCairns RJ, Leinonen T et al (2014) The evolution and adaptive potential of transcriptional variation in sticklebacks‐signatures of selection and widespread heritability. Molecular Biology and Evolution, 32, 674–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz TL (2015) Transcription in space ‐ environmental vs. genetic effects on differential immune gene expression. Molecular Ecology, 24, 4583–4585. [DOI] [PubMed] [Google Scholar]
- Lenz TL, Eizaguirre C, Rotter B, Kalbe M, Milinski M (2013) Exploring local immunological adaptation of two stickleback ecotypes by experimental infection and transcriptome‐wide digital gene expression analysis. Molecular Ecology, 22, 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB, Jackman TR, Larson A, Queiroz K, Rodriguez‐Schettino L (1998) Contingency and determinism in replicated adaptive radiations of island lizards. Science, 279, 2115–2118. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐Seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucek K, Lemoine M, Seehausen O (2014) Contemporary ecotypic divergence during a recent range expansion was facilitated by adaptive introgression. Journal of Evolutionary Biology, 27, 2233–2248. [DOI] [PubMed] [Google Scholar]
- Machado‐Schiaffino G, Henning F, Meyer A (2014) Species‐specific differences in adaptive phenotypic plasticity in an ecologically relevant trophic trait: hypertrophic lips in Midas cichlid fishes. Evolution, 68, 2086–2091. [DOI] [PubMed] [Google Scholar]
- MacKinnon JS, Rundle HD (2002) Speciation in nature: the threespine stickleback model systems. Trends in Ecology & Evolution, 17, 480–488. [Google Scholar]
- Manceau M, Domingues VS, Mallarino R, Hoekstra HE (2011) The developmental role of Agouti in color pattern evolution. Science, 331, 1062–1065. [DOI] [PubMed] [Google Scholar]
- Manousaki T, Hull PM, Kusche H et al (2013) Parsing parallel evolution: ecological divergence and differential gene expression in the adaptive radiations of thick‐lipped Midas cichlid fishes from Nicaragua. Molecular Ecology, 22, 650–669. [DOI] [PubMed] [Google Scholar]
- Morris MR, Richard R, Leder EH et al (2014) Gene expression plasticity evolves in response to colonization of freshwater lakes in threespine stickleback. Molecular Ecology, 23, 3226–3240. [DOI] [PubMed] [Google Scholar]
- Moser D, Kueng B, Berner D (2015) Lake‐stream divergence in stickleback life history: a plastic response to trophic niche differentiation? Evolutionary Biology, 42, 328–338. [Google Scholar]
- Muschick M, Indermaur A, Salzburger W (2012) Convergent evolution within an adaptive radiation of cichlid fishes. Current Biology, 22, 2362–2368. [DOI] [PubMed] [Google Scholar]
- Pavey SA, Collin H, Nosil P, Rogers SM (2010) The role of gene expression in ecological speciation. Annals of the New York Academy of Sciences, 1206, 110–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey SA, Sutherland BJ, Leong J et al (2011) Ecological transcriptomics of lake‐type and riverine sockeye salmon (Oncorhynchus nerka). BMC Ecology, 11, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press C, Evensen O (1999) The morphology of the immune system in teleost fishes. Fish & Shellfish Immunology, 9, 309–318. [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G (2010) The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Current Biology, 20, R208–R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protas ME, Hersey C, Kochanek D et al (2006) Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nature Genetics, 38, 107–111. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2008) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3‐900051‐07‐0, URL http://www.R-project.org [Google Scholar]
- Ranz JM, Machado CA (2006) Uncovering evolutionary patterns of gene expression using microarrays. Trends in Ecology & Evolution, 21, 29–37. [DOI] [PubMed] [Google Scholar]
- Rebeiz M, Pool JE, Kassner VA, Aquadro CF, Carroll SB (2009) Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science, 326, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch TB, Wegner KM, Kalbe M (2001) Rapid genetic divergence in postglacial populations of threespine stickleback (Gasterosteus aculeatus): the role of habitat type, drainage and geographical proximity. Molecular Ecology, 10, 2435–2445. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biology, 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesti M, Hendry AP, Salzburger W, Berner D (2012) Genome divergence during evolutionary diversification as revealed in replicate lake‐stream stickleback population pairs. Molecular Ecology, 21, 2852–2862. [DOI] [PubMed] [Google Scholar]
- Rohlenová K, Morand S, Hyršl P et al (2011) Are fish immune systems really affected by parasites? an immunoecological study of common carp (Cyprinus carpio). Parasit Vectors, 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen O, Lascoux M, Merila J (2013) Ecological genomics of local adaptation. Nature Reviews Genetics, 14, 807–820. [DOI] [PubMed] [Google Scholar]
- Scharsack JP, Kalbe M, Harrod C, Rauch G (2007) Habitat‐specific adaptation of immune responses of stickleback (Gasterosteus aculeatus) lake and river ecotypes. Proceedings of the Royal Society London Series B: Biological Sciences, 274, 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Zimmermann AG, Roberts RA et al (2012) The innate immune sensor NLRC3 attenuates Toll‐like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF‐kappaB. Nature Immunology, 13, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA (2004) The genomics of gene expression. Genomics, 84, 449–457. [DOI] [PubMed] [Google Scholar]
- Stanley ER, Cifone M, Heard PM, Defendi V (1976) Factors regulating macrophage production and growth: identity of colony‐stimulating factor and macrophage growth factor. Journal of Experimental Medicine, 143, 631–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz WE, Schmerer M, Coates JL, Bolnick DI (2015) Among‐lake reciprocal transplants induce convergent expression of immune genes in threespine stickleback. Molecular Ecology, 24, 4629–4646. [DOI] [PubMed] [Google Scholar]
- Wegner KM, Reusch TB, Kalbe M (2003) Multiple parasites are driving major histocompatibility complex polymorphism in the wild. Journal of Evolutionary Biology, 16, 224–232. [DOI] [PubMed] [Google Scholar]
- Whitehead A (2012) Comparative genomics in ecological physiology: toward a more nuanced understanding of acclimation and adaptation. The Journal of Experimental Biology, 215, 884–891. [DOI] [PubMed] [Google Scholar]
- Zhang L, Mo J, Swanson KV et al (2014) NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity, 40, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Distribution of dispersion values in (a) head kidney samples and (b) spleen samples.
Fig. S2 Counts of Diplostomum sp. across stickleback populations.
Table S1 Morphological data and parasite loads of sequenced fish
Table S2 Summary of library statistics in sequence quality filtering, sequence mapping and library scaling factor.
Table S3 Differentially expressed genes between lake and river across four population pairs.
Table S4 Differentially expressed genes between lake and river in European populations.
Data Availability Statement
The raw reads of RNA‐Seq data (fastq format) and mapping files (bam format) are available through the European Nucleotide Archive (study Accession no.: PRJEB8677, URL: http://www.ebi.ac.uk/ena/data/view/PRJEB8677). htseq read counts, edger results and topGO results are archived in Dryad (doi:10.5061/dryad.hq50s). Morphological and parasite data are included in Table S1 (Supporting information).