

Significance
We demonstrate that exposure to three environmental chemicals suggested to affect breast development—bisphenol A, mono-n-butyl phthalate, and polychlorinated biphenyl 153—at physiologically relevant doses results in unique responses and alterations in the proteome. This study provides insights into how the mammary epithelium changes in response to physiologically relevant exposures to xenobiotic chemicals. These changes could be correlated with increased risk of transformation or important changes in function.
Keywords: proteomics, mammary epithelium, environmental chemicals, organotypic culture, estrogen
Abstract
Common environmental contaminants such as bisphenols and phthalates and persistent contaminants such as polychlorinated biphenyls are thought to influence tissue homeostasis and carcinogenesis by acting as disrupters of endocrine function. In this study we investigated the direct effects of exposure to bisphenol A (BPA), mono-n-butyl phthalate (Pht), and polychlorinated biphenyl 153 (PCB153) on the proteome of primary organotypic cultures of the mouse mammary gland. At low-nanomolar doses each of these agents induced distinct effects on the proteomes of these cultures. Although BPA treatment produced effects that were similar to those induced by estradiol, there were some notable differences, including a reduction in the abundance of retinoblastoma-associated protein and increases in the Rho GTPases Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division cycle protein CDC42. Both Pht and PCB153 induced changes that were distinct from those induced by estrogen, including decreased levels of the transcriptional corepressor C-terminal binding protein 1. Interestingly, the three chemicals appeared to alter the abundance of distinct splice forms of many proteins as well as the abundance of several proteins that regulate RNA splicing. Our combined results indicate that the three classes of chemical have distinct effects on the proteome of normal mouse mammary cultures, some estrogen-like but most estrogen independent, that influence diverse biological processes including apoptosis, cell adhesion, and proliferation.
Breast cancers are caricatures of normal tissue development (1–3). The same underlying mechanisms that affect the cell processes needed for normal mammary development contribute to cancer progression. By affecting the distribution of interacting cells and communication among them through nontargeted effects, carcinogens can promote the outgrowth of altered cells with malignant potential. Exposure to environmental agents can affect mammary gland development and alter breast cancer risk. Epidemiologic studies have associated a number of environmental factors with increased breast cancer risk (4). Some of these factors may have low-level effects that are not genotoxic but alter immune responses, vascularity, or the microenvironment or that change susceptibility to carcinogens. A current weakness in models of cancer risk assessment is the lack of consideration of factors that influence the susceptibility of mixed cell populations to transformation. Indeed, ionizing radiation, the prototypical carcinogen, promotes changes in the biochemical properties in cultured mouse and human breast cells, in the stromal environment, and in intact mouse mammary gland (5–7).
Physiologically relevant 3D culture models can recapitulate crucial aspects of the dynamic and reciprocal signaling necessary for establishing and maintaining tissue-specific morphogenetic programs. The determination of the molecular mechanisms underlying mammary gland morphogenesis and tumorigenesis is facilitated by the use of model systems that are easier to manipulate and that facilitate higher throughput than in vivo genetics and pharmacogenomics. Organoid behavior in basement membrane gels (Matrigel) models terminal end bud invasion in normal development, whereas behavior in collagen gels approximates side-branching, epithelial-to-mesenchymal transition (EMT), and early neoplastic progression (8). Indeed, altered morphologies of breast cancer cells correlate with their gene-expression profiles (9). These cultures provide the opportunity to validate the effects of environmental stressors on human breast tissue, the critical next step for developing markers of exposure.
Our goal was to develop a model system for evaluating the impact of environmental stressors on breast tissues and elucidating the effects of low-level exposures. We investigated the perturbation of normal mammary development by environmental chemical stressors in mouse mammary tissue in 3D culture models. Using data obtained in the Breast Cancer and Environment Research Centers (10–13), we focused on xenobiotics that were detected in the serum and/or urine of girls undergoing puberty. We used mono-n-butyl phthalate (Pht), bis-phenol A (BPA), and polychlorinated biphenyl 153 (PCB) for our initial analysis. Recent studies have shown that treatment of mice with BPA during puberty leads to an increase in mammary stem cells and alters their functions (14). However, whether these effects are direct or indirect is not known. BPA, which produces dramatic changes in the appearance of human cells at doses ≥10 ng/mL, has been measured in the blood and tissues of pregnant women at 0.5–100 ng/mL (15). Di-n-butyl phthalate, an environmental contaminant with significant human exposure, is metabolized to the monoester mono-n-butyl phthalate (MBP), an active metabolite. Because a number of the compounds detected in serum are reported to be hormone disruptors, we used estradiol (E2) as a positive control. The experiments were designed to target the events in mammary epithelium during puberty, using this organotypic assay, which recapitulates many of the events in puberty (16) and is free from the systemic effects of these compounds. We hypothesized that exposure to environmental agents causes changes in the composition of mammary proteins that may lead to increased susceptibility to carcinogenesis.
Results and Discussion
Proteome Transformation After Xenobiotic Exposure.
We analyzed the effects of physiological levels of various xenobiotics, e.g., endocrine disruptors/estrogenic chemicals, on the proteome of primary organotypic cultures of mammary glands from 6- to 8-wk-old FVB/n mice. We compared relative protein abundances by quantitative mass spectrometry. Four biological replicates were exposed to 20 nM BPA, Pht, PCB153, or E2 for 6 d (Fig. 1A); exposure to 0.0002% DMSO was used as a vehicle control. Continuous exposure of organoids to BPA, MBP, and PCB altered developmental branching patterns, resulting in smaller structures with reduced numbers of invasive branches than seen in vehicle-treated controls (Fig. 1B). When invasive branches did occur in the PCB- and Pht-treated samples, they appeared to have altered morphology with thicker bases.
Fig. 1.

Analysis of proteome changes caused by environmental chemicals. (A) Schematic depicting the workflow for analysis of cell lines for the iTRAQ mass spectrometry experiment. Quadruplicate cultures of mouse mammary organoids cultured in Matrigel were treated with 20 nM BPA, Pht, PCB153, E2, or DMSO for 6 d. (B) Environmental chemicals cause morphological changes in organoid branching patterns. The organoids shown were treated with 10 nM of each chemical plus 0.0001% DMSO or with 0.0001% DMSO alone. (Scale bars, 100 μM.) (C) The number of proteins detected in each multiplex. FDRs were calculated using a target-decoy database containing the reversed sequences of all the proteins appended to the target database. The reported confidence scores reported by ProteinPilot software corresponding to 5% FDR are shown for each multiplex. (D) Venn diagram showing the number of proteins (5% FDR) with quantification values in each multiplex.
To define the proteome changes occurring after chemical exposure, we compared relative protein abundances by quantitative mass spectrometry. Tryptic peptides of the four independent mammary organoid cultures for each treatment were labeled with isobaric tags (iTRAQ 8plex reagents) and were analyzed by LC-MS (Fig. 1A). Samples were randomized to generate three multiplexes, and relative protein ratios were calculated from the reporter ions using ProteinPilot software (17). The total number of proteins detected at a 5% false-discovery rate (FDR) in each multiplex ranged from 4,292 to 5,081 (Fig. 1C), and 4,020 proteins had quantification values in all multiplexes (Fig. 1D and Dataset S1).
Treatment with each environmental chemical or E2 (Fig. 2A) resulted in distinct protein profiles. Analysis with the Limma package was used to define statistically significant differences compared with DMSO-treated control cultures. Approximately equal numbers of proteins were found in increased and decreased abundances in all treatments. However, treatment with E2 resulted in the greatest number of changes in abundance, with 7% of the proteins altered (Fig. 2 B and C). The other treatments changed approximately half that number of proteins, 2.8–4.2% of the total number of quantified species.
Fig. 2.

Distinct profiles of proteins with altered abundance in mammary epithelial cells upon treatment with environmental chemicals and estradiol at 10 nM. (A) Total number of proteins with altered abundance detected in each treatment group. Linear modeling was used to determine the significance of the relative expression of proteins with respect to the control cultures. (B) Number of proteins with increased (red) and decreased (blue) abundance in each treatment group. (C) Volcano plot showing significant P values (≤0.05) versus average normalized protein ratios (fold change ≥1.5) in each treatment compared with DMSO treatment. Dashed lines represent the applied thresholds. (D) Hierarchical clustering of 598 altered proteins with P ≤0.05 in at least one treatment. The heat map represents log2 fold changes vs. DMSO; blue, lower abundance; red, higher abundance.
Next we performed hierarchical clustering analysis, considering the relative abundance of altered proteins in the four treatment groups relative to control. The changes in the E2-treated cultures were distinct from the changes observed in the organoids treated with PCB or Pht (Fig. 2D and Dataset S2). However, the proteome responses to BPA, with some exceptions as discussed below, correlated well with the responses to E2. Moreover, the changes in protein abundance profiles of each of the environmental chemically treated cultures were distinct, suggesting that each chemical has a different mode of action in terms of its influence on branching morphogenesis.
Functional Classes of Proteins and Pathways Changed by Exposure to Environmental Chemicals.
To determine whether a subset of proteins altered by exposure could sensitize mammary epithelium to transformation, differentiation, and other premalignant processes, we took an approach that considers the direction and type of all signals in a pathway and identified the biological processes characteristic of the proteins altered by each chemical (18, 19). The Elim pruning method, which iteratively removes the genes mapped to a significant gene ontology (GO) term from more general (higher level) GO terms was used to overcome the limitation of errors introduced by considering genes multiple times (20). A modest number of biological processes (29/385) were affected by more than one chemical exposure (Fig. 3A). Only activation of the complement cascade, lectin pathway, involved in the regulation of immune processes, was common to all four treatments (Fig. 3B and Dataset S3). BPA, E2, and PCB all exerted effects on the proteins that regulate glycolysis. Protein autophosphorylation, which affects regulatory mechanisms and can promote proliferation and transformation (21), was perturbed in E2- and PCB-treated cultures. A comparison of BPA- vs. E2-treated cultures revealed alterations in extracellular matrix organization, glycolytic process, cell migration, and activation of Ral GTPase activity. Recently, RalA activity was shown to promote the invasion of breast cancer cells in vitro (22). All 10 of the biological processes affected by both Pht and PCB were defined by a single protein per process. In the main, however, these analyses revealed that, in concordance with the slight overlap of differentially expressed proteins, the majority of altered biological processes were chemical-specific.
Fig. 3.

Functional classes of proteins changed after exposure. (A and B) Altered biological processes (A) and molecular functions (B) from each treatment. P values were corrected using the Elim method. (C and D) The overlap of GO terms found in more than one condition. The intensity of the shading indicates the significance (P value) of the pathway. All black and gray boxes have a P value of ≤0.05; white boxes have a P value of ≥0.05 and are not significantly altered; n = 4 in each group.
Next, we performed GO analysis of the molecular functions (Fig. 3C). As expected from the morphological changes observed, structural molecule activity was altered in all conditions (Fig. 3D and Dataset S4). DNA-binding functions were affected by the environmental chemicals but not by E2. The chromatin-binding Toll-like receptor (Tlr) ligand Hmgb1 was reduced in all three environmental chemical treatments. Tlr-2 signaling is an important contributor to the maintenance of the mammary epithelial repopulating unit and is required for normal ductal outgrowth (23). Two overlaps in affected function between BPA- and E2-treated organoids were observed: heparin binding and integrin binding.
To identify the potential functional significance of responses to these agents, we performed an analysis of pathways affected by the different agents. We used an impact analysis approach that applies overrepresentation of differentially abundant proteins in a given pathway, i.e., enrichment analysis, plus perturbation of that pathway, which takes into consideration the direction of all signals in a pathway and the position, role, and type of every protein (24). This analysis showed that proteins changing in abundance with treatment across multiple conditions were involved in pathways related to growth and proliferation, metabolism, development, cell cycle, and protein synthesis and processing (Dataset S5). Cultures treated with BPA and E2 showed proteome alterations affecting four pathways in common: glycolysis/glucogenesis, carbon metabolism in cancer, ECM-receptor interaction, and biosynthesis of amino acids. Steroid hormone biosynthesis was affected in PCB-treated (P = 0.025) and Pht-treated (P = 0.043) cultures. Protein processing in endoplasmic reticulum was affected in all treatments but was significant only in BPA (P = 0.039). ER protein-processing proteins that were altered were Pdia4, Cair, P4hb, and Prkcsh, all of which were up-regulated, and Rad23a, which was down-regulated.
Chemical-Specific Proteome Changes.
BPA.
Responses in BPA-treated cultures were distinct from those seen with E2 treatment. Focal adhesion assembly (P = 0.006), substrate adhesion-dependent cell spreading/cell migration (P = 0.014), epithelium morphogenesis (P = 0.017), PI3 kinase (P = 0.008), DNA replication (P = 0.009), and nuclear division (P = 0.019) were altered. At the pathway level, cellular proliferation pathways such as pancreatic cancer (P = 0.039) and viral carcinogenesis (P = 0.047) were uniquely impacted by BPA. This treatment resulted in the down-regulation of the tumor-suppressor retinoblastoma 1 (Rb1), which is lost in a majority of human cancers. In the mammary gland, Rb appears to function redundantly with retinoblastoma-like-1 (p107) and -2 (p130). Neither p107 nor p130 was detected in our analysis. Deletion of Rb1 alone is not sufficient to perturb mammary development (25). However, transgenic expression of an inhibitory fragment of the SV40 large T-antigen that inactivates all three proteins without inactivating p53 is sufficient to induce a premalignant state of enhanced proliferation and apoptosis in mice (26).
The Rho GTPases cell division cycle protein CDC42 (Cdc42) and Ras-related C3 botulinum toxin substrate 1 (Rac1), which regulate mammary epithelial invasion into diverse stromal environments (16, 27, 28), were up-regulated by BPA exposure. Similarly BPA previously has been found to regulate Rac1/Cdc42 expression positively in hippocampal neurons (29). Overexpression of Cdc42 in the developing mammary gland results in increased side branching and abnormal terminal end bud morphology through increased MEC contractility and motility rather than through effects on proliferation (30). Up-regulation of Cdc42 activity may play a key role in tamoxifen resistance (31). Rac1 is up-regulated in malignant but not in benign breast cancers (32). Rac1 activity can regulate estrogen receptor transcriptional activity positively in human breast cancer cell lines (33). In contrast to BPA, estrogen has been reported to regulate Rac1 expression negatively in monocytes (34) and vascular smooth muscle cells (35), although in the present study E2 treatment did not alter Rac1 expression significantly. We speculate that through its positive regulation of Rac1 levels BPA may function as an estrogen sensitizer by antagonizing a negative feedback loop of estrogen on Rac1 levels.
PCB.
PCB-treated cells showed changes in multiple proteins involved in defense response, cell differentiation (P = 8.6e-4), proliferation (P = 0.007), vesicle-mediated transport (P = 0.029), and cellular response to oxygen-containing compounds (chemicals) (P = 0.043). In BPA-treated cultures, functions were GTP-dependent protein binding, cholesterol binding, and carbohydrate binding. Among the molecular functions that were the most altered by PCB treatment were Notch binding (Aak1 and Ncor2, both decreased in abundance), protein kinase activity and protein tyrosine phosphatase activity, DNA binding, and histone deacetylase binding.
The most significant pathways affected by PCB treatment were Notch signaling (P = 0.009), steroid hormone biosynthesis (P = 0.025), and mRNA surveillance (P = 0.035). The Notch signaling pathway has been implicated in both the development and progression of breast cancer. Specifically, the levels of the phosphoprotein C-terminal–binding protein 1 (CTBP1) were increased. CTBP1 is a transcriptional repressor with roles in cellular proliferation and apoptosis (36, 37). It down-regulates the transcription of Brca1 and E-cadherin in human breast cancer cells (38). However, we did not observe significant differences in E-cadherin protein levels in control and PCB-treated organoids. Brca1 was not detected. Nuclear receptor corepressor 2 (NCOR2), a member of the thyroid hormone- and retinoic acid receptor-associated corepressor family, was down-regulated. Aberrant expression of this protein is associated with certain cancers, including breast (39, 40). The mRNA surveillance pathway is a quality-control mechanism that detects and degrades abnormal mRNAs (41, 42). The nonsense-mediated decay mRNA surveillance pathway down-regulates aberrant E-cadherin expression in gastric cancer cells (43).
Phthalate.
Phthalates are considered endocrine disruptors because of their complex effects on several hormonal systems, including estrogen (44). Of the three chemicals tested, Pht treatment induced the proteome response that was the most distinct from that of E2 (Fig. 2D). The top pathways affected included apoptosis (P = 5.3e-4), proliferation of mammary gland epithelial cells (P = 0.003), proliferation of mesenchymal cells (P = 0.005), tissue regeneration, (P = 0.007), and ubiquitin-protein transferase activity (P = 0.003).
In Pht-treated cultures, the most dysregulated functions were glutathione peroxidase activity, protein serine/threonine phosphatase activity, and protein 2A phosphatase (PP2A) regulatory activity. The PP2A holoenzyme contains three subunits: scaffold subunit A, regulatory subunit B, and catalytic subunit C. Protein phosphatase 2A activator, regulatory subunit B (PPP2R4), which was found in higher levels (1.8-fold, P = 0.004), regulates estrogen receptor alpha (ERα) expression by modulating mRNA stability and is involved in the phosphatidylinositol-3 kinase (PI3K)/serine/threonine-protein kinase (Akt) signaling pathway (45, 46). Immunoglobulin-binding protein 1, a regulatory protein of the catalytic subunit of PP2A that alters activity and substrate specificity (47), was down-regulated (−1.6-fold, P = 0.003). The scaffold (PPP2R1A) (1.5-fold, P = 0.037) and β-catalytic (twofold, P = 004) subunits were up-regulated. PPP2R4/PP2A regulates ERα expression by modulating ER mRNA stability; hence, it has been considered a potential therapeutic target for breast cancer (45). PPP2R4 is involved in the PI3K/Akt signaling pathway that modulates the interaction between BRCA1 and ER-α (46). Mutations of PPP2R4 contribute to multiple cancer types, including breast (48). PPP2R4 inhibits telomerase activity in human breast cancer cells (49) and is inhibited by tamoxifen. Finally, ubiquitin–protein transferase activity was affected, with the up-regulation of ubiquitin-like modifier activating enzyme 1 (Uba1), ubiquitin-conjugating enzyme (Ube) E2 D3, Ube2n, Ube2l3, and kelch-like protein 42 observed.
At the pathway level, treatment with Pht affected ubiquitin-mediated proteolysis (P = 0.008), an important cellular mechanism for targeting abnormal or short-lived proteins for degradation. Protein ubiquitination is a multistep enzymatic process that regulates the stability, function, and/or localization of the modified proteins. Uba1, found in lower abundance in Pht-treated cultures, catalyzes the first step in ubiquitin conjugation to mark cellular proteins for degradation. This protein is required for the DNA damage response, and mutations in the catalytic domain result in cell-cycle arrest and decreased DNA synthesis (50, 51). Similar to Uba1, the ubiquitin-conjugating enzymes E2L3, E2D3, and E2N were decreased in abundance. Studies in mice suggest that E2N, also known as “Ubc13,” plays a role in DNA repair (52). The E2L3 enzyme participates in c-Fos degradation and NF-κB maturation in vitro and potentially could affect the pathways involved in cellular proliferation (53). We identified several genes classified in the Huntington’s disease pathway, including Bax (BCL2-associated X protein), glutathione peroxidase 1 (Gpx1), and Huntingtin. Bax (increased 2.5-fold) accelerates programmed cell death (54), functions as an apoptotic activator, and is regulated by p53 (55). Multiple studies have implicated Gpx1 (increased 1.8-fold) in breast cancer development and progression (56–58).
In summary, these data suggest that the primary actions of the chemicals, whose direct effects we analyzed in primary mammary organoid cultures, were not the result of hormone disruption. Many of the GO terms mapped to proteins with common ancestry, i.e., general, terms. Thus, the processes and functions that lead to proliferation and differentiation were affected, with diverse downstream consequences at the pathway level. Indeed, in all conditions we observed decreases in proliferation and stemness that were concordant with a phenotype that is more susceptible to transformation.
Alternative Splicing.
In addition to changes in protein abundance, alterations in isoform expression constitute proteome alterations that are dynamically regulated during development and tumorigenesis (59). For example, alternative splicing of epidermal growth factor receptor results in the expression of a constitutively active isoform that is selectively expressed in prostate and ovarian cancers but not in the normal tissues from which they arise. We hypothesized that if exposure to environmental chemicals altered organoid development, the mechanisms might include their effects on the expression of particular protein isoforms (60). To investigate changes in splicing across the proteome, we took advantage of the copious sequence data produced by the MS/MS-based proteomic approach used in this study.
Isoform-specific quantification was accomplished by calculating protein ratios using spectra from peptides that were distinct to each isoform. We observed 72 differentially expressed proteins translated from alternative splice variants in a treatment-dependent manner (Dataset S6). A subset of the protein variants with potential cancer relevance is shown in Table 1. The isoforms of the NADH-binding transcriptional repressor CtBP1 are important regulators of tumor suppressors (61) that are required for high-fidelity replication of DNA during mitosis (38, 62) and play a role in breast cancer chemoresistance (63). Isoform 2 of CtBP1 was down-regulated by PCB and, to a lesser extent, by Pht in organoids treated with these chemicals. This down-regulation is consistent with the antiproliferative effect of these chemicals. An antagonist of the oncogenic Notch receptor signaling pathway, Numb, was up-regulated in response to Pht treatment. In the breast, loss of Numb expression is associated with triple-negative breast cancers (64). In mammary cancers that retain Numb expression, its function as a negative regulator of Notch signaling is impaired (65). Elevation of Numb protein in renal epithelial cells promotes a migratory program characterized by EMT (66). Consistent with the induction of a migratory phenotype, Pht treatment also down-regulated Focad-1, a focal adhesion protein that is a suppressor of glial tumor invasion (67), and α-catenin, which is involved in actin assembly at focal adhesions, linking E-cadherin/β-catenin to the actin cytoskeleton. Loss of α-catenin in the hair follicle is sufficient to promote skin cancer (68). The ubiquitin ligase cullin-4B (CUL4B) was decreased in BPA- and E2-treated cultures. Mutations in this protein were identified in BRCA+ family members with breast cancer and were absent in unaffected family members (69). CUL4B is a component of Wnt-induced targeting that regulates cell-cycle progression (70).
Table 1.
Splice variants and mRNA-processing proteins altered by chemical treatment
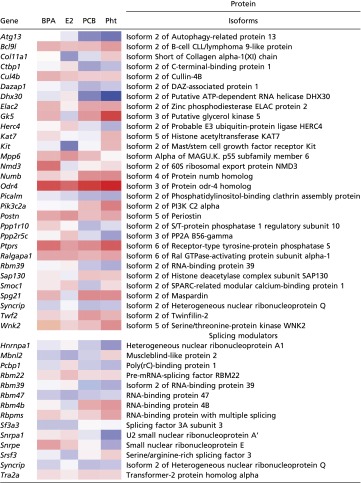 |
The table lists altered proteins with P ≤ 0.05 in at least one treatment. The heat map represents log2 fold changes vs. DMSO; blue, lower abundance; red, higher abundance.
Several mRNA-processing proteins, including splicing regulators and modulators, many of which function in cell growth and differentiation in cancer, also were detected in altered abundance. We found altered expression of the regulators serine/arginine-rich splicing factor 3 (SRSF3), splicing factor 3A3 (SF3A3), heterogeneous nuclear ribonuclear proteins Q and A1, small nuclear ribonuclear proteins A′ and E, regulation of nuclear pre-mRNA domain-containing protein 2 (RPRD2), RNA-binding proteins 22, 4b, 39, 47, and MS, and the RNA helicase DHX30 (Table 1). Perturbation of the splicing machinery also can activate the p53 pathway (71, 72).
The observation of differentially expressed proteins translated from alternative splice variants, many of which function in cell growth and differentiation in cancer (8, 73), is intriguing. Alternative splicing significantly increases functional gene diversity, and aberrant splicing contributes to tumor progression and resistance to cancer therapy (59). Dysregulation of splicing, which plays critical roles in numerous cancers at multiple points in disease progression, has been estimated to occur in 50% of tumors (71, 74, 75). We previously have observed that alternative splicing of p120-catenin (76) and Rac1b contribute to fundamental changes such as EMT that occur during tumorigenesis (77).
Conclusions
In summary, although multiple proteins showed similar changes, treatment with each chemical resulted in a distinct protein-expression profile. Pathway analysis showed that the proteins changing in abundance with treatment across conditions were involved in pathways related to growth and proliferation, development, cell cycle, and protein synthesis. We also identified more than 30 protein isoforms, indicating that environmental chemicals affect splicing. Our results with these prototypical chemicals suggest suppression of proliferation and differentiation in the setting of altered expression of protein isoforms.
In this study we examined the effects of physiological levels of xenobiotic agents on relative protein abundances in mouse mammary organoids. The dramatic changes in organoid morphology that we observed with nanomolar concentrations of these agents suggested the suppression of cell proliferation and/or differentiation, a surprising result. In support of this idea, Biro et al. reported in their puberty studies (10–12, 78) that breast development is accelerated in girls with a high body mass index, but the highest blood and urine levels of environmental chemicals were associated with delayed breast development. In an independent study, we found that differentiation and cell-cycle genes are suppressed in the stem-like cells that disseminate from primary patient-derived xenograft tumors (79). We exploited the mechanisms of morphogenesis identified in normal mouse epithelium that have informed our understanding of tumor growth and invasion in human breast cancer. The results of the global proteome profiling and quantification study presented here pointed to the important finding that, at low doses, each chemical exposure had unique effects on diverse molecular functions. Although BPA elicited the proteome response most similar to the response to E2, there were some important differences, notably in the up-regulation of Rac1 and Cdc42 by BPA. Pht and PCB treatment elicited a proteome response consistent with a migratory, antiproliferative phenotype.
A caveat of mammary organoid studies is the heterogeneity of the cell types present. Organoids have epithelial cells as well as some stromal fibroblasts and inflammatory cells. It is possible that the chemicals alter the balance of mammary cell types (luminal, basal, and less differentiated cells) in the organoids (8, 80, 81). As a result, proteins with altered expression may contribute to disrupted breast development or facilitate the induction of breast cancer. Ultimately, the information about the protein-level effects of environmental stressors we obtained by using a model system will be validated in pubertal mice and in mice orthotopically transplanted with human breast epithelium.
Materials and Methods
Chemicals.
FGF-2, Pht, PCB153, BPA, and 17-β-estradiol and, unless otherwise described, all other chemicals were purchased from Sigma.
Organoid Cultures.
Experiments were approved by the University of California, San Francisco Institutional Animal Use and Care Committee. Primary mammary gland organoids were prepared according published procedures (16, 82). Mammary glands from 6- to 8-wk-old FVB/n mice were removed aseptically, minced with scalpel blades, and digested with 0.1% collagenase/trypsin for 30 min at 37 °C. The digested cells were centrifuged at 1,500 rpm for 10 min using a Beckman Allegra X-12R centrifuge with an SX4750 rotor, supernatant containing fat tissue was discarded, and cell pellets were resuspended in 40 U/mL DNase I. Organoid clusters were separated from single-cell organoids by differential centrifugation. Aggregates were resuspended and embedded in growth factor-reduced Matrigel at a 1 aggregate/μL concentration. The Matrigel pellets were overlaid with DMEM/F12 supplemented with insulin-selenium-transferrin, penicillin-streptomycin, and 5 nM FGF-2, and compounds were dissolved in 0.0001% DMSO or in DMSO alone. Quadruplicate cultures of mouse mammary organoids were treated with 20 nM BPA, Pht, PCB153, E2, or DMSO for 6 d. Organoids in 50-μL Matrigel drops were photographed by bright-field microscopy after 6 d of growth at 37 °C, 5% CO2.
Sample Preparation.
Four biological replicate samples from each treatment were prepared for proteomic analyses. For each sample, organoids from 0.5 mL of Matrigel droplets were isolated by dissolution of Matrigel in 2.5 mM EDTA in PBS at 4 °C. After three washes with the PBS-EDTA solution, organoids were solubilized in 8 M urea containing Complete protease inhibitor mixture (Roche) and were clarified by centrifugation for 10 min at 16,500 × g. Fifty micrograms of protein extract from mammary organoids were trypsin digested and labeled with iTRAQ 8plex reagent (Sigma-Aldrich) as previously described (Fig. 1A) (83).
LC-MS Analysis.
Peptides from the three multiplexes were first separated offline into 20 fractions using alkaline pH reversed-phase HPLC (84). Peptides from each fraction were separated using a nanoLC Ultra 2D Plus system (SCIEX) interfaced with a 5600 Triple TOF mass spectrometer (SCIEX). The peptides were initially loaded onto a guard column (300 μm i.d. × 5 mm, 5-μm particle size, 100-Å pore size; Acclaim PepMap300 C18; Thermo-Fisher) and were washed with the aqueous loading solvent that consisted of 2% solvent B [98% acetonitrile (ACN)/0.1% formic acid (FA)] in solvent A (2% ACN/0.1% FA), flow rate 12 μL/min for 3 min. Then the peptides were separated on a C18 Acclaim PepMap100 column (75 μm i.d. × 150 mm, 3-mm particle size, 100-Å pore size; Thermo Fisher Scientific) heated at 40 °C with a column oven. Peptides were eluted at a flow rate of 300 nL/min with a gradient of 2–35% solvent B for 90 min. In positive ion mode, MS scans from m/z 400–1,600 were acquired followed by MS/MS scans of the 20 most abundant ions with an exclusion time of 15 s.
Bioinformatics and Statistics.
Proteins were identified and quantified using ProteinPilot v. 4.5 software (17) and the UniProt v. 201302 human database with isoform sequences. iTRAQ ratios were calculated using the pooled DMSO control as the denominator, and bias correction and background subtraction were applied. Protein ratios were calculated using only ratios from the peptide spectra that were distinct to each protein or protein form, thus eliminating any masking of changes in expression caused by the sharing of peptides among proteins. A target-decoy database containing the reversed sequences of all the proteins appended to the target database was used to calculate peptide and protein FDR rates (85). The limma method was utilized to estimate the differences in expression between treated and control cultures (86). Proteins were identified as differentially expressed if the corresponding q values were less than 0.05, based on the Benjamini–Hochberg method, and if the absolute log expression was less than 0.06. Hierarchical clustering was performed using MultiExperiment Viewer using Euclidean distance and average linkage clustering (87). The significantly impacted pathways, biological processes, and molecular interactions were analyzed using Advaita Bioinformatic iPathwayGuide software (www.advaitabio.com/ipathwayguide.html) in the context of pathways obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Release 73.0+/03–16) (88, 89) and the gene ontology from the Gene Ontology Consortium database (2014-Sep19) (90). The Elim pruning method, which iteratively removes the genes mapped to a significant GO term from more general (higher level) GO terms, was used to overcome the limitation of errors introduced by considering genes multiple times (20).
Supplementary Material
Acknowledgments
This work was supported by Grants 17UB-8705 and 21UB-8011 from the California Breast Cancer Research Program and by Grant ES019458 from the National Institute of Environmental Health Sciences and the National Cancer Institute. Mass spectrometry analysis was performed in the University of California, San Francisco Sandler-Moore Mass Spectrometry Core Facility, which receives support from the Sandler Family Foundation and the Gordon and Betty Moore Foundation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1600645113/-/DCSupplemental.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74(7):365–381. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Committee on Breast Cancer and the Environment . The Scientific Evidence, Research Methodology, and Future Directions. National Academy; Washington, DC: 2012. [Google Scholar]
- 5.Andarawewa KL, et al. Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 2007;67(18):8662–8670. doi: 10.1158/0008-5472.CAN-07-1294. [DOI] [PubMed] [Google Scholar]
- 6.Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment - tumorigenesis and therapy. Nat Rev Cancer. 2005;5(11):867–875. doi: 10.1038/nrc1735. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen DH, et al. Radiation acts on the microenvironment to affect breast carcinogenesis by distinct mechanisms that decrease cancer latency and affect tumor type. Cancer Cell. 2011;19(5):640–651. doi: 10.1016/j.ccr.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen-Ngoc KV, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci USA. 2012;109(39):E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1(1):84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biro FM, et al. Onset of breast development in a longitudinal cohort. Pediatrics. 2013;132(6):1019–1027. doi: 10.1542/peds.2012-3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mervish N, et al. BCERP Dietary predictors of urinary environmental biomarkers in young girls, BCERP, 2004-7. Environ Res. 2014;133:12–19. doi: 10.1016/j.envres.2014.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolff MS, et al. Breast Cancer and Environment Research Program Phthalate exposure and pubertal development in a longitudinal study of US girls. Hum Reprod. 2014;29(7):1558–1566. doi: 10.1093/humrep/deu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biro FM, et al. Pubertal assessment method and baseline characteristics in a mixed longitudinal study of girls. Pediatrics. 2010;126(3):e583–e590. doi: 10.1542/peds.2009-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, et al. Pubertal bisphenol A exposure alters murine mammary stem cell function leading to early neoplasia in regenerated glands. Cancer Prev Res (Phila) 2014;7(4):445–455. doi: 10.1158/1940-6207.CAPR-13-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schönfelder G, et al. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110(11):A703–A707. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14(4):570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shilov IV, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6(9):1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Draghici S, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17(10):1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donato M, et al. Analysis and correction of crosstalk effects in pathway analysis. Genome Res. 2013;23(11):1885–1893. doi: 10.1101/gr.153551.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22(13):1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 21.Hartman Z, Zhao H, Agazie YM. HER2 stabilizes EGFR and itself by altering autophosphorylation patterns in a manner that overcomes regulatory mechanisms and promotes proliferative and transformation signaling. Oncogene. 2013;32(35):4169–4180. doi: 10.1038/onc.2012.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, et al. RILP suppresses invasion of breast cancer cells by modulating the activity of RalA through interaction with RalGDS. Cell Death Dis. 2015;6:e1923. doi: 10.1038/cddis.2015.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheeren FA, et al. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol. 2014;16(12):1238–1248. doi: 10.1038/ncb3058. [DOI] [PubMed] [Google Scholar]
- 24.Tarca AL, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson GW, Wagner KU, Hennighausen L. Functional mammary gland development and oncogene-induced tumor formation are not affected by the absence of the retinoblastoma gene. Oncogene. 2001;20(48):7115–7119. doi: 10.1038/sj.onc.1204888. [DOI] [PubMed] [Google Scholar]
- 26.Simin K, et al. pRb inactivation in mammary cells reveals common mechanisms for tumor initiation and progression in divergent epithelia. PLoS Biol. 2004;2(2):E22. doi: 10.1371/journal.pbio.0020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu W, Nelson CM. PI3K regulates branch initiation and extension of cultured mammary epithelia via Akt and Rac1 respectively. Dev Biol. 2013;379(2):235–245. doi: 10.1016/j.ydbio.2013.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390(6660):632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, et al. Bisphenol A promotes dendritic morphogenesis of hippocampal neurons through estrogen receptor-mediated ERK1/2 signal pathway. Chemosphere. 2014;96:129–137. doi: 10.1016/j.chemosphere.2013.09.063. [DOI] [PubMed] [Google Scholar]
- 30.Bray K, et al. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013;15(5):R91–108. doi: 10.1186/bcr3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H-Y, Yang YM, Stevens BM, Noble M. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol Med. 2013;5(5):723–736. doi: 10.1002/emmm.201202140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnelzer A, et al. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19(26):3013–3020. doi: 10.1038/sj.onc.1203621. [DOI] [PubMed] [Google Scholar]
- 33.Rosenblatt AE, et al. Inhibition of the Rho GTPase, Rac1, decreases estrogen receptor levels and is a novel therapeutic strategy in breast cancer. Endocr Relat Cancer. 2011;18(2):207–219. doi: 10.1677/ERC-10-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adam O, Hagel M, Theobald K, Böhm M, Laufs U. Inhibitory effect of estrogen on Rac1-expression in monocytes. Biochem Biophys Res Commun. 2009;386(1):45–49. doi: 10.1016/j.bbrc.2009.05.126. [DOI] [PubMed] [Google Scholar]
- 35.Laufs U, et al. Down-regulation of Rac-1 GTPase by Estrogen. J Biol Chem. 2003;278(8):5956–5962. doi: 10.1074/jbc.M209813200. [DOI] [PubMed] [Google Scholar]
- 36.Choi HJ, et al. Bcl3-dependent stabilization of CtBP1 is crucial for the inhibition of apoptosis and tumor progression in breast cancer. Biochem Biophys Res Commun. 2010;400(3):396–402. doi: 10.1016/j.bbrc.2010.08.084. [DOI] [PubMed] [Google Scholar]
- 37.Bergman LM, Birts CN, Darley M, Gabrielli B, Blaydes JP. CtBPs promote cell survival through the maintenance of mitotic fidelity. Mol Cell Biol. 2009;29(16):4539–4551. doi: 10.1128/MCB.00439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng Y, et al. Transcriptional down-regulation of Brca1 and E-cadherin by CtBP1 in breast cancer. Mol Carcinog. 2012;51(6):500–507. doi: 10.1002/mc.20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Agthoven T, et al. CITED2 and NCOR2 in anti-oestrogen resistance and progression of breast cancer. Br J Cancer. 2009;101(11):1824–1832. doi: 10.1038/sj.bjc.6605423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghoshal P, et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009;69(10):4380–4387. doi: 10.1158/0008-5472.CAN-08-3467. [DOI] [PubMed] [Google Scholar]
- 41.van Hoof A, Wagner EJ. A brief survey of mRNA surveillance. Trends Biochem Sci. 2011;36(11):585–592. doi: 10.1016/j.tibs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akimitsu N. Messenger RNA surveillance systems monitoring proper translation termination. J Biochem. 2008;143(1):1–8. doi: 10.1093/jb/mvm204. [DOI] [PubMed] [Google Scholar]
- 43.Karam R, et al. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27(30):4255–4260. doi: 10.1038/onc.2008.62. [DOI] [PubMed] [Google Scholar]
- 44.Yoon K, Kwack SJ, Kim HS, Lee B-M. Estrogenic endocrine-disrupting chemicals: Molecular mechanisms of actions on putative human diseases. J Toxicol Environ Health B Crit Rev. 2014;17(3):127–174. doi: 10.1080/10937404.2014.882194. [DOI] [PubMed] [Google Scholar]
- 45.Keen JC, et al. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. J Biol Chem. 2005;280(33):29519–29524. doi: 10.1074/jbc.M505317200. [DOI] [PubMed] [Google Scholar]
- 46.Ma Y, Hu C, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol Endocrinol. 2007;21(8):1905–1923. doi: 10.1210/me.2006-0397. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, Peterson RT, Schreiber SL. Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem Biophys Res Commun. 1998;247(3):827–832. doi: 10.1006/bbrc.1998.8792. [DOI] [PubMed] [Google Scholar]
- 48.Calin GA, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19(9):1191–1195. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Zhao LL, Funder JW, Liu JP. Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J Biol Chem. 1997;272(27):16729–16732. doi: 10.1074/jbc.272.27.16729. [DOI] [PubMed] [Google Scholar]
- 50.Sugaya K, Ishihara Y, Inoue S, Tsuji H. Characterization of ubiquitin-activating enzyme Uba1 in the nucleus by its mammalian temperature-sensitive mutant. PLoS One. 2014;9(5):e96666. doi: 10.1371/journal.pone.0096666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moudry P, et al. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle. 2012;11(8):1573–1582. doi: 10.4161/cc.19978. [DOI] [PubMed] [Google Scholar]
- 52.Ashley C, Pastushok L, McKenna S, Ellison MJ, Xiao W. Roles of mouse UBC13 in DNA postreplication repair and Lys63-linked ubiquitination. Gene. 2002;285(1-2):183–191. doi: 10.1016/s0378-1119(02)00409-2. [DOI] [PubMed] [Google Scholar]
- 53.Moynihan TP, et al. Characterization of a human ubiquitin-conjugating enzyme gene UBE2L3. Mamm Genome. 1996;7(7):520–525. doi: 10.1007/s003359900155. [DOI] [PubMed] [Google Scholar]
- 54.Cory S, Adams JM. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 55.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 56.Hu YJ, Diamond AM. Role of glutathione peroxidase 1 in breast cancer: Loss of heterozygosity and allelic differences in the response to selenium. Cancer Res. 2003;63(12):3347–3351. [PubMed] [Google Scholar]
- 57.Jenssen TK, Kuo WP, Stokke T, Hovig E. Associations between gene expressions in breast cancer and patient survival. Hum Genet. 2002;111(4-5):411–420. doi: 10.1007/s00439-002-0804-5. [DOI] [PubMed] [Google Scholar]
- 58.Zhuo P, et al. Molecular consequences of genetic variations in the glutathione peroxidase 1 selenoenzyme. Cancer Res. 2009;69(20):8183–8190. doi: 10.1158/0008-5472.CAN-09-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, et al. Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia. 2011;13(5):461–471. doi: 10.1593/neo.101744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chinnadurai G. The transcriptional corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res. 2009;69(3):731–734. doi: 10.1158/0008-5472.CAN-08-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Birts CN, et al. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol Cell. 2010;103(1):1–19. doi: 10.1042/BC20100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Birts CN, et al. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol Cell. 2010;103(1):1–19. doi: 10.1042/BC20100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rennstam K, et al. Numb protein expression correlates with a basal-like phenotype and cancer stem cell markers in primary breast cancer. Breast Cancer Res Treat. 2010;122(2):315–324. doi: 10.1007/s10549-009-0568-x. [DOI] [PubMed] [Google Scholar]
- 65.Pece S, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167(2):215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding X, et al. Numb induces e-cadherin adhesion dissolution, cytoskeleton reorganization, and migration in tubular epithelial cells contributing to renal fibrosis. Curr Mol Med. 2015;15(4):368–379. doi: 10.2174/1566524015666150505162015. [DOI] [PubMed] [Google Scholar]
- 67.Brockschmidt A, et al. KIAA1797/FOCAD encodes a novel focal adhesion protein with tumour suppressor function in gliomas. Brain. 2012;135(Pt 4):1027–1041. doi: 10.1093/brain/aws045. [DOI] [PubMed] [Google Scholar]
- 68.Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell. 2001;104(4):605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 69.Xiao F, et al. Genome instability in blood cells of a BRCA1+ breast cancer family. BMC Cancer. 2014;14:342–352. doi: 10.1186/1471-2407-14-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miranda-Carboni GA, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008;22(22):3121–3134. doi: 10.1101/gad.1692808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allende-Vega N, et al. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene. 2013;32(1):1–14. doi: 10.1038/onc.2012.38. [DOI] [PubMed] [Google Scholar]
- 72.Bezzi M, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013;27(17):1903–1916. doi: 10.1101/gad.219899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu J, et al. From the Cover: Neutralization of terminal differentiation in gliomagenesis. Proc Natl Acad Sci USA. 2013;110(36):14520–14527. doi: 10.1073/pnas.1308610110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010;24(21):2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, et al. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26(3):374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Slorach EM, Chou J, Werb Z. Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev. 2011;25(5):471–484. doi: 10.1101/gad.1998111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radisky DC, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436(7047):123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pinney SM, et al. Serum biomarkers of polyfluoroalkyl compound exposure in young girls in Greater Cincinnati and the San Francisco Bay Area, USA. Environ Pollut. 2014;184:327–334. doi: 10.1016/j.envpol.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lawson DA, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Plaks V, et al. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Reports. 2013;3(1):70–78. doi: 10.1016/j.celrep.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2(1):90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fata JE, et al. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306(1):193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zamah AM, Hassis ME, Albertolle ME, Williams KE. Proteomic analysis of human follicular fluid from fertile women. Clin Proteomics. 2015;12(1):5–17. doi: 10.1186/s12014-015-9077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dwivedi RC, et al. Practical implementation of 2D HPLC scheme with accurate peptide retention prediction in both dimensions for high-throughput bottom-up proteomics. Anal Chem. 2008;80(18):7036–7042. doi: 10.1021/ac800984n. [DOI] [PubMed] [Google Scholar]
- 85.Tang WH, Shilov IV, Seymour SL. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res. 2008;7(9):3661–3667. doi: 10.1021/pr070492f. [DOI] [PubMed] [Google Scholar]
- 86.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 87.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kanehisa M. 2002. The KEGG database. Novartis Found Symp 247:91–101; discussion 101–103, 119–128, 244–152.
- 90.Ashburner M, et al. The Gene Ontology Consortium Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.