

Abstract
Limited comparative studies suggest that the human genome is particularly enriched for recent segmental duplications. The extent of segmental duplications in other mammalian genomes is unknown and confounded by methodological differences in genome assembly. Here, we present a detailed analysis of recent duplication content within the mouse genome using a whole-genome assembly comparison method and a novel assembly independent method, designed to take advantage of the reduced allelic variation of the C57BL/6J strain. We conservatively estimate that ∼57% of all highly identical segmental duplications (≥90%) were misassembled or collapsed within the working draft WGS assembly. The WGS approach often leaves duplications fragmented and unassigned to a chromosome when compared with the clone-ordered-based approach. Our preliminary analysis suggests that 1.7%–2.0% of the mouse genome is part of recent large segmental duplications (about half of what is observed for the human genome). We have constructed a mouse segmental duplication database to aid in the characterization of these regions and their integration into the final mouse genome assembly. This work suggests significant biological differences in the architecture of recent segmental duplications between human and mouse. In addition, our unique method provides the means for improving whole-genome shotgun sequence assembly of mouse and future mammalian genomes.
The era of whole-genome sequencing has created an opportunity to assess fundamental biological processes of genome evolution in a global fashion (Eichler and Sankoff 2003). The comparative sequence of over 50 eukaryotic genomes is becoming available at various levels of completion. Concomitantly, the field of genome evolution is experiencing a renaissance of activity. Whereas many aspects of genome architecture and genome evolution are readily tractable by conventional computational analyses of working draft sequences, other aspects are more difficult to assay. The identification and characterization of highly homologous segmental duplications has been problematic in both clone-ordered and whole-genome shotgun sequencing-based strategies. Reliable estimates of recent segmental duplication content (defined as blocks of sequence ≥1 kb in length and showing ≥90% sequence identity) have been elusive and vary widely depending on the method of assembly and their method of detection (Bailey et al. 2001, 2002a; Eichler 2001; Cheung et al. 2003b).
Understanding the nature and pattern of recent segmental duplications is important for both practical and biological reasons. First, duplication of genomic sequence followed by subsequent mutation is one of the primary forces of functional and structural evolution (Muller 1936; Ohno et al. 1968). Delineation of the most recent duplication events at the genomic-sequence level, and particularly sequences located at their junctions (Bailey et al. 2003), may provide insight into their mechanism of origin. Second, genes embedded within duplicated sequence often identify regions of adaptive evolution within species. A catalog of such lineage-specific genes provides a roadmap for the identification of genes important for recent innovations in immunity, drug detoxification, and reproduction (Copley et al. 2003). Third, at a structural level, regions of highly homologous duplications are preferential sites of inversion, deletion, and translocation between species (Dehal et al. 2001; Stankiewicz et al. 2001; Armengol et al. 2003; Locke et al. 2003). This genomic instability extends to the level of human disease in which nearly two-dozen syndromes have been shown to be associated with duplication-mediated rearrangements (Lupski 1998; Ji et al. 2000; Samonte and Eichler 2002). Other large-scale duplication-mediated structural variants are associated with susceptibility to disease and may rise in frequency within the population to approach polymorphic levels of variation (Samonte et al. 1996; Sprenger et al. 2000; Osborne et al. 2001; Giglio et al. 2002; Gimelli et al. 2003). Interestingly, many of the duplications that predispose to rearrangement have been shown to represent a single loci in the mouse genome (Pentao et al. 1992; DiDonato et al. 1997; Ji et al. 1999; Probst et al. 1999). Finally, from the practical perspective, regions of large-scale duplication are particularly problematic for genotyping and mapping, as SNPs may be inadvertently assigned to a highly paralogous region (Eichler 2001; Bailey et al. 2002a; Estivill et al. 2002). Gene and SNP annotation significantly improve when more duplicated sequence is correctly integrated into the assembly (Eichler 1999; Collins et al. 2003). The annotation of segmental duplications is therefore an important aspect of genome sequence and assembly.
The detection of recent segmental duplications is sensitive to the quality of the underlying sequence assembly. Large blocks of highly homologous duplications pose challenges to both clone-ordered and whole-genome shotgun sequence-based assembly methods (Green 1997; Eichler 1999). The underlying problem is the same—the correct placement of sequence that is nearly identical at multiple positions within the assembly. Improper assembly may lead to either under- or over-representation of duplicated sequence. For example, failure to correctly merge allelic overlaps during clone-ordered approaches may lead to false positives (termed artifactual duplications). In such cases, the duplications are large and virtually identical at the sequence level. Initial estimates of the published working-draft level of the human genome, identified 280 Mb (or 10% of the genome) as artifactual duplications (Bailey et al. 2001). Alternatively, genomic duplications with high degree of sequence identity may be incorrectly collapsed as overlaps. These collapses have been anecdotally observed in both clone-ordered-based and wholegenome shotgun sequence assembly methods and lead to an underestimation of duplication content (Bailey et al. 2001, 2002a; Cheung et al. 2003a). A systematic analysis of this effect, however, could not be performed due to either the lack of public access to the underlying data and/or due to the fact that assemblies have combined both methods to different degrees (International Human Genome Sequencing Consortium [IHGSC] 2001; Venter et al. 2001; Mouse Genome Sequencing Consortium [MGSC] 2002). At least, four factors directly impact an assessment of the segmental duplication content within any genome assembly as follows: (1) the depth of sequencing (fold coverage), (2) the methodology of assembly, (3) the quality of common repeat annotation, and (4) level of allelic variation. All of these factors must be taken into account during an assessment of recent segmental duplication content.
To overcome some of the limitations associated with the detection of highly homologous duplication, we developed two independent in silico detection strategies. The first method termed whole-genome assembly comparison (WGAC) is a BLAST-based approach that performs an all-by-all comparison of assembled genomic sequence (Bailey et al. 2001). Similar methods have been developed by others (Cheung et al. 2003a; Schwartz et al. 2003). Such methods tacitly assume that the genome assembly is correct. The second approach termed whole-genome shotgun detection (WSSD) develops a model for distinguishing unique and duplicated sequence on the basis of the depth of coverage and the average degree of sequence identity of whole-genome shotgun sequence reads aligned to a reference genomic segment (Bailey et al. 2002a). In essence, duplicated regions will show an increased depth-of-coverage and a significant reduction in the average degree of sequence identity, due to the recruitment of both allelic and paralogous sequence reads. This method is independent of the assembly and offers high sensitivity and specificity to detect large ≥15 kb, highly homologous (≥95% sequence identity) duplications.
We provide an assessment of the duplication content of the mouse genome on the basis of these two fundamentally different methods. To increase our power to detect duplicated regions, we implemented a sequence quality filter that allowed us to take advantage of the reduced allelic variation. For the purpose of this study, we chose to analyze the published working-draft sequence of the C57BL/6J mouse genome (MGSCv3, 2.5 Gb), as well a smaller BAC-based assembly of only finished sequence (NCBI build 29, 440 Mb). It should be pointed out that the latter represents a small proportion of the mouse genome, in which clone choice selection may be biased. These two assemblies provide a direct comparison of the strengths and weaknesses of BAC-based clone-ordered versus whole-genome shotgun approaches for estimating global segmental duplication content. On the basis of the results of our analysis, we have constructed an integrated mouse segmental duplication database that will provide a frame-work for future evolutionary analyses. In addition, the resource should provide valuable information in directing finishing and sequencing efforts within the mouse.
RESULTS
Human Versus Mouse Genome Assembly Comparisons for Segmental Duplication
We compared the duplication content (≥90% sequence identity) of mouse (MGSCv3) and human draft genomes on the basis of the published sequence assemblies (IHGSC 2001; MGSC 2002). Both genomes were analyzed using the identical whole-genome sequence comparison (WGAC) method (Bailey et al. 2001), which assumes that no under- or over-representation has occurred within duplicated regions, and that the genomes are correctly assembled. Due to the presence of uncharacterized low-copy repeat sequences within the mouse genome, we specifically focused on the analysis of duplications in which pairwise alignment lengths exceeded 10 kb in length (Methods). We observed significant differences in the duplication content between man and mouse (Fig. 1). Overall, 0.7% (19 Mb) of MGSCv3 assembly showed evidence of duplication when compared with human, in which 4.5% (180 Mb) of the human genome was found to be duplicated for these alignment parameters (≥90% and ≥10 kb in length; Table 1; Supplemental Table 1; Supplemental Figure 1). The median length of the alignments was significantly shorter for the mouse (13.7 kb) in contrast to human (26.5 kb). The most striking feature was that the number of pairwise alignments differed by more than an order of magnitude (6552 human pairwise alignments as compared with 732 mouse pairwise alignments; Table 2). Further, the majority of the mouse alignments (425) involved the unassigned mouse chromosome, suggesting that the treatment of mouse duplications were problematic during the assembly.
Figure 1.
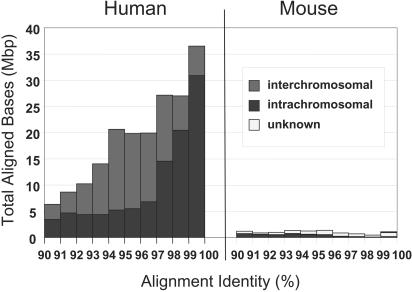
Whole-genome assembly comparison for mouse and human. We compared the sum of aligned bases (excluding gaps) for segmental duplications represented by alignments ≥10 kb in both the human genome (build 31) and the draft mouse genome (MGSCv3). Both the human and mouse genomes have alignments at all levels of identity; however, the human genome has a dramatically greater amount of aligned bases relative to the mouse (227,812 kbp vs. 10,042 kbp). The number of alignments increases geometrically relative to the number of copies. Mouse appears relatively rich in intrachromosomal duplications (black) and lacking in interchromosomal duplications (dark gray). However, many alignments are poorly characterized as indicated by the enrichment within the unplaced chromosome (chrUn—light gray).
Table 1.
Genome Fraction of Duplicated Sequence
|
WGAC (≥90%)
|
WSSD (≥95%)
|
||||||
|---|---|---|---|---|---|---|---|
|
Mouse MGSCv3
|
Mouse MGSCv3
|
||||||
| Human (build 31) | w/unplaced | w/o unplaced | Mouse build29 | BACs | w/unplaced | w/o unplaced | |
| >1 kb | 5.25% | N.D. | N.D. | 2.56% | N.D. | N.D. | N.D. |
| >5 kb | 4.78% | 1.95% | 1.01% | 2.00% | N.D. | N.D. | N.D. |
| >10 kb | 4.52% | 0.70% | 0.48% | 1.74% | 1.51% | 2.09% | 0.27% |
| >20 kb | 4.06% | 0.11% | 0.10% | 1.14% | 1.46% | 2.01% | 0.23% |
Whole-genome assembly comparison (WGAC) identified duplications ≥90%; whereas whole genome shotgun sequence detection (WSSD) only reliably identified duplications ≥95% and ≥10 kb. Genome assemblies analyzed included human build31 (2,860,784,610 bp); mouse MGSCv3 including unplaced contigs (2,475,067,632 bp) and excluding unplaced contigs (2,374,117,067 bp); mouse NCBI build 29 (439,076,820). Mouse build29 was hand curated removing high copy repeats (poorly characterized LINEs and LTRs). Similarly, missed allelic overlaps were also removed (Methods). WSSD detection was two tiered. All finished clones (4298 BACs totaling 706,309,797 bp) and MGSCv3 assembly segments (400 kb) were scanned for regions encompassing ≥10 kb with divergence ratios of ≥0.80 based on Megablast alignments. We reanalyzed positive sequences by realigning and rescoring with quality all reads between 98% and 100% identity using Needleman-Wunsch global alignment. Regions of high divergence were then reanalyzed. Regions encompassing ≥10 kb were then further defined in 1-kb windows to determine more precisely the boundaries of the duplication. The amount of duplication within the MGSCv3 WSSD was corrected for intervening gaps and 7,882,708 bases of major and minor centromeric satellite.
Table 2.
Human Versus Mouse Alignment Properties
| Alignments | Aligned bp | Mean size | Mean similarity | |
|---|---|---|---|---|
| Human | 6552 | 171,310,564 | 26,146 | 95.6% |
| inter | 3144 | 76,197,373 | 24,236 | 95.0% |
| intra | 3408 | 95,113,191 | 27,909 | 96.1% |
| Mouse build29 | 241 | 5,636,773 | 23,389 | 94.1% |
| inter | 6 | 115,152 | 19,192 | 95.9% |
| intra | 235 | 5,521,621 | 23,496 | 94.1% |
| Mouse MGSCv3 | 732 | 10,041,877 | 13,718 | 94.7% |
| inter | 40 | 520,693 | 13,017 | 95.4% |
| intra | 267 | 4,137,586 | 15,497 | 93.4% |
| unknown | 425 | 5,383,598 | 12,667 | 95.5% |
Only alignments ≥10 kb were analyzed. MGSCv3 is the published version of the draft mouse genome. Build29 is a partial genome assembly based on finished C57BL/6J clones only. Aligned bp is the sum of aligned bases for all pairwise alignments, and thus, is a redundant measure of duplicated sequence relative to the genome.
Whole-Genome Shotgun Versus Clone-Ordered Assembly of the Mouse Genome
Because two different methodologies, whole-genome shotgun versus clone-ordered-based sequencing, were used to assemble the human and mouse genomes, respectively, the apparent dearth of highly identical duplications within the mouse assembly may have resulted from collapse of whole-genome shotgun sequence reads during the assembly process (MGSCv3). To test this hypothesis, we compared the duplication content of the NCBI clone-ordered assembly (build 29) of mouse C57BL/6J BACS with that of the published mouse genome assembly (Table 1; Supplemental Table 2). An examination of 439 Mb of build 29, approximately one-fifth of the genome, predicted a significant increase in the length (mean 23.4 kb), frequency (1.74% of the genome), and the number of pairwise alignments (241 alignments) (Tables 1 and 2). If build 29 is representative of the entire mouse genome, these data predict that ∼60% (1 - 0.0070/0.0174) of segmental duplications may have been collapsed inadvertently during the assembly. Both WGAC analyses suggest that the intrachromosomal duplications predominate in mouse in contrast to the human, in which interchromosomal and intrachromosomal pairwise alignments are equally prevalent (Figs. 1 and 2). If we limit our analysis to more divergent duplications (<94% identity, which can be easily resolved by WGS assembly methods), there is virtually a complete absence of interchromosomal duplications with MGSCv3 (Fig. 2).
Figure 2.
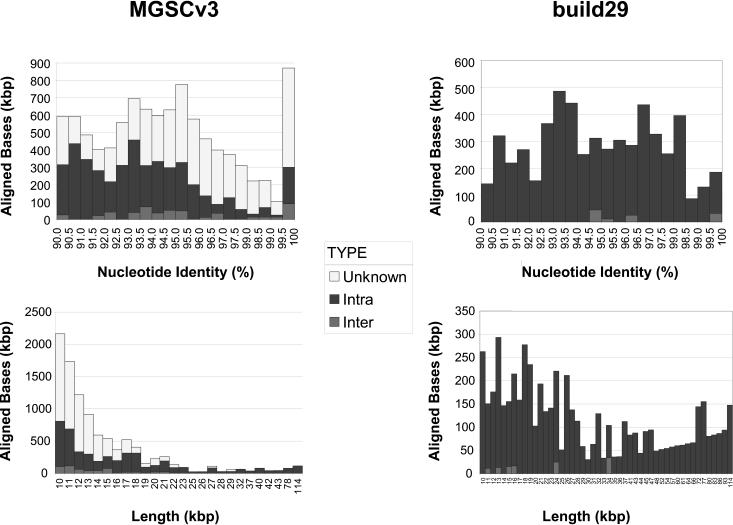
Whole-genome alignment (WGAC) statistics of the mouse draft and the build29 finished genome. Alignment statistics are binned in terms of percent identity or length (≥10 kb). We performed BLAST-based segmental duplication detection on MGSCv3 and the finished portion of build 29. The finished build 29 subset represents 439 Mb (17.7% of the draft assembly size). The abundance of aligned bases between 99.5%–100% that map to the unknown chromosome in MGSCv3 may represent highly similar duplication requiring further characterization. The build 29 pairwise were hand curated to remove uncharacterized interspersed transposable elements (Methods).
Whole-Genome Shotgun Sequence Detection of Mouse Duplications
As an independent approach to detect highly homologous mouse segmental duplications, we applied a previously described whole-genome shotgun sequence detection (WSSD) method (Table 3; Bailey et al. 2002a). This method assumes a random distribution of genome shotgun sequence reads and measures the depth-of-coverage and sequence identity of shotgun-sequence reads aligned to a given genomic sequence (Methods). Increases in the depth of coverage and decreases in the average percent sequence identity demarcate putative duplicated sequence regions, due to the recruitment of identical allelic reads as well as divergent paralogous sequence reads. Previous studies show that this method can reliably detect duplications in the human genome that exceed 10 kb in length and show ≥95% sequence identity (Bailey et al. 2002a). The power, however, is dependent on the depth and randomness of the whole-genome shotgun sequence library.
Table 3.
Correlation Between WSSD and WGAC
|
Build29
|
MGSCv3 w/unplaced
|
||||||
|---|---|---|---|---|---|---|---|
| WSSD | WGAC | Bases | Fraction | Regions | Bases | Fraction | Regions |
| + | + | 4,882,018 | 49% | 46 | 11,437,113 | 16% | 159 |
| — | + | 4,058,785 | 41% | 146 | 6,883,852 | 10% | 531 |
| + | — | 967,960 | 10% | 24 | 53,380,584 | 74% | 786 |
| Total | 9,908,763 | 71,701,549 | |||||
A nonredundant set of duplications detected by WGAC (≤10 kb) was compared against the nonredundant set of duplications detected by WSSD (≤10 kb). The fraction represents the proportion of the duplicated sequence mapped to each of the three categories. A good correlation exists for build29 by these two methods. Most of the duplications in MGSCv3 that were detected only by WSSD mapped to the unknown chromosome and were highly fractured.
To test the utility of this method, we established a baseline for comparison by calibrating a collection of unique (2052 kb) and duplicated (952 kb) mouse BACs (Supplemental Table 3). For each reference sequence, both the depth of coverage and average percent sequence identity were measured for sequence reads within 5-kb windows. (Each window corresponded to 5 kb of genomic sequence in which known repetitive sequences were excluded). Because the C57BL/6J mouse represents a highly inbred strain with limited allelic variation, we examined more closely the degree of sequence variation. To improve our power, we considered only high-quality (phred quality score ≥30) bases during our calculation of sequence identity (Ewing et al. 1998). This high stringency effectively removed potential sequence errors from further consideration. This quality masking of alignments (Fig. 3; Methods) dramatically improved our ability to detect duplications on the basis of departures from 100% sequence identity to the aligned reference sequence. Departures from 100% sequence identity were observed infrequently in sequences lacking segmental duplications, and most often represented incompletely masked transposable elements within the mouse genome (Methods). Consequently, a small number of paralogous reads was occasionally recruited, especially in the vicinity of recent LTR sequence.
Figure 3.
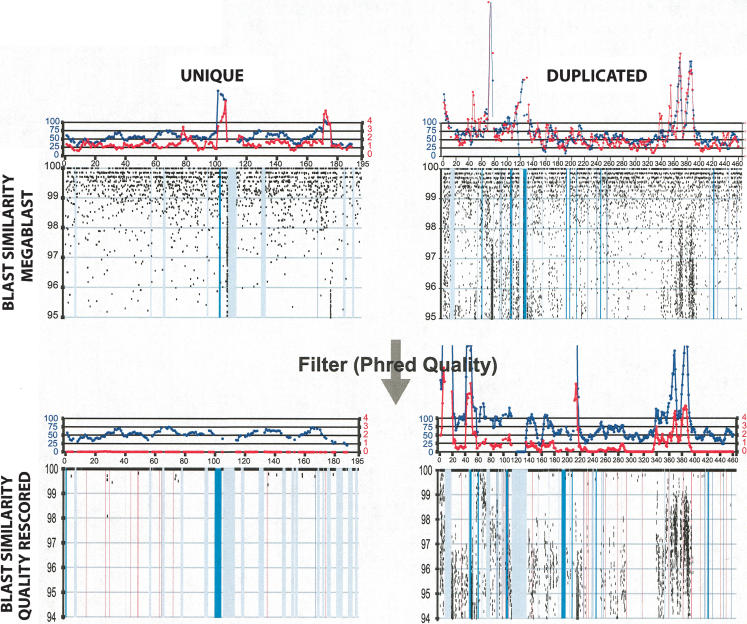
Examples of whole-genome shotgun sequence detection (WSSD). The calibration of our WSSD method was performed on a set of unique and duplicated sequences. Unique sequences were drawn from clones shown to be unique by both metaphase and interphase FISH (e.g., AL590991). Examples of duplicated sequence were drawn from recently described pericentromeric duplications (e.g., mmu5; Thomas et al. 2003). Detection parameters were optimized to differentiate unique from duplicated sequence. Black dots represent the similarity and position of individual sequence reads. Masked repetitive regions (LINE elements, purple; ERV elements, green; and simple sequence repeats, red) are shown as vertical bars. From previous studies of the human genome (Bailey et al. 2002a), read depth (blue line) provided the measure for duplication detection. Here, we also took advantage of the reduced level of allelic variation within the C57BL/6J strain to increase our power. Thus, single base-pair differences most likely signify either paralogous sequence or sequencing errors. By excluding errors (through the calculation of read identity using only high quality base positions), we could categorize each read as allelic (≥99.8% identity) or paralogous (<99.8% identity). Regions showing a divergent read ratio (red line) of >0.8 (paralogous: allelic) were deemed duplicated. A divergent read ratio of 1 would suggest one paralogous copy.
Figure 3 depicts a typical comparison of two mouse BACs containing known unique and duplicated sequence before and after quality masking. Due to the homogenous nature of the mouse genome, the ratio of the number of diverged sequence reads to identical sequence reads (termed the divergent read ratio) was used to provide a crude estimate for the copy number for a given reference segment. Among unique regions of the genome, this ratio should approximate zero. In contrast, a region of the mouse genome duplicated once would possess a divergent read ratio of one—as half of the reads map to a separate locus. This, of course, assumes that there will be at least 2-bp differences per read (∼700 bp on average) between duplicated loci for all paralogous reads. Identical sequence duplications could not be detected simply by the divergent read ratio. Significant differences in both the depth-of-coverage and the divergent read ratio were observed between unique and duplicated reference sequences (Fig. 3). Although both measures (depth-of-coverage and divergent read ratio) could effectively discriminate highly homologous (>95%) duplications, the divergent read ratio showed the greatest sensitivity (Supplemental Table 3) in our analysis.
We applied the whole-genome shotgun sequence detection (WSSD) strategy separately to MGSCv3 (2475 Mb) and to all available finished C57BL/6J BACs (706 Mb; 4298 BACs). This entailed a computational intensive analysis of 40.7 million reads against both reference genomes assessing the depth-of-coverage and divergent read ratio in 5-kb windows (overlapping 1 kb; see Methods). We identified all regions in which at least five consecutive windows were consistent with duplication (a divergent read ratio ≥0.8). The analysis predicts that 1.5% of the sequence of the mouse BACs and 2.0% of the MGSCv3 of the genome are duplicated (≥95% and ≥10 kb; Table 1). In MGSCv3, these correspond to 197 nonredundant regions assigned to chromosomes and 753 regions mapped to the unplaced mouse chromosome (Supplemental Table 4). Thus, the unplaced mouse chromosome showed the greatest abundance of putatively duplicated sequence (40% of the unplaced sequence appears duplicated by the WSSD method [Fig. 4; Supplemental Tables 1 and 4]). Much of the unplaced chromosome consists of known repetitive LTR elements and centromeric satellite sequences.
Figure 4.
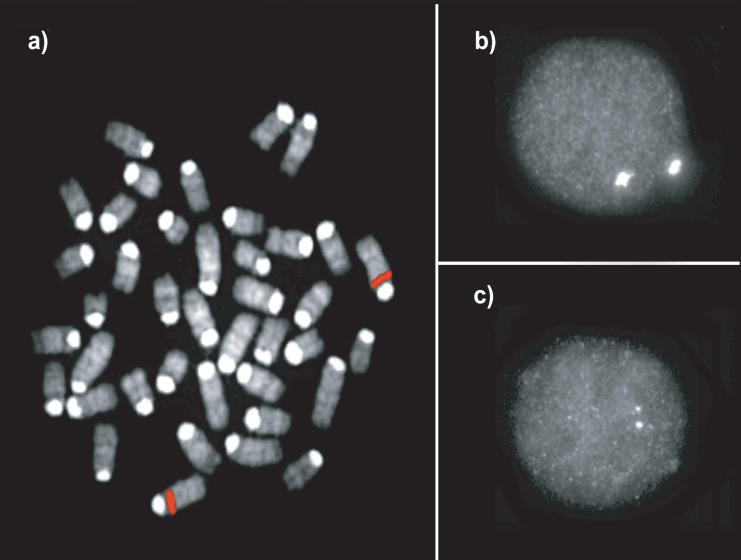
FISH confirmation. An example of (a) metaphase and (b) interphase FISH hybridization with a duplicated BAC clone (RP23–3D2; see Table 4) that was identified by the whole-genome shotgun detection strategy. Increased signal intensity was confirmed using (c) cohybridization with a unique probe (RP21-344N12) in the same nucleus as shown in b. Tandem segmental duplications were most frequently observed (Table 4). The results of all FISH experiments are available online (http://www.biologia.uniba.it/mouse/).
We experimentally validated our detection strategy by FISH. Previous analyses suggest good correlation between highs-equence identity duplications and the presence of multisite FISH signals (Bailey et al. 2001, 2002b; Cheung et al. 2001). We selected a total of 22 C57BL/6J BACs that contained putative duplications as determined by the WSSD strategy. Clone sequence identity was confirmed by BAC-end sequence analysis. The putative duplications within these clones ranged in size from 21 to 251 kb, whereas divergent read ratios ranged from 0.8 to 21.4 (Table 4). Both interphase and metaphase nuclei were prepared directly from C57BL/6J bone marrow and were hybridized using BAC clones as probes (Methods). The presence of strong multilocus signals and increased signal intensity were used to assess duplication status. A total of 77% (17/22) of the selected BACs were confirmed duplication positive by this assay, and 16/17 of these showed a clustered intrachromosomal configuration (see, Fig. 4 for example) within the mouse genome as opposed to a multichromosomal distribution pattern. These results are consistent with the in silico analysis of build 29, which indicates that duplicate copies are separated on average by only 57 kb. In general, BACs with lower divergent read ratios and a lower depth-of-coverage could not be confirmed as duplicated. Two additional BACs were assessed with divergent read ratios >0.8, but with slight increases in the depth-of-coverage. All of these scored negative by this assay. It should be emphasized that not all true duplications would be expected to be detected by FISH— particularly low-copy tandem duplications. This analysis, therefore, provides us with a conservative estimate of the false-positive rate using our WSSD criteria.
Table 4.
FISH Validation of a Subset of WSSD Duplication-Positive C57BL/6J BACs
|
WSSD duplicated regions
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Accession | Clone | Length | MGSCv3 best placement | Size (bp) | Read depth (#/5 kb) | Divergent read ratio | Copy # | Cytogenetic position | FISH results description |
| AL929496.8 | RP23-3D2 | 254072 | chrX:21967539 | 251,000 | 1,403.9 | 21.4 | 5 | chrX_A3 | Intra Cluster |
| AC124400.4 | RP24-357O21 | 191219 | chr14:35800505 | 48,512 | 469.4 | 10.0 | 3 | chr14_C | Intra Cluster |
| AC124511 | RP23-440I16 | 179914 | chr12:1838950 | 179,000 | 440.2 | 6.4 | 4 | chr12_A2 | Inter + Intra interspersed (pericentromeric) |
| AC122877.3 | RP23-115K1 | 186549 | chr14:42943941 | 88,459 | 204.0 | 4.9 | 4 | chr14_B | Intra Cluster |
| AC124452.4 | RP24-219N2 | 177600 | chr7:23230794 | 177,961 | 611.8 | 4.4 | 2 | chr7_A2 | Intra Cluster (pericentromeric) |
| AL671630.8 | RP23-327E13 | 173610 | chrX:12967423 | 149,000 | 384.7 | 3.1 | 3 | chrX_F3 | Intra Cluster |
| AC087780.19 | RP23-324b17 | 206924 | chr1:88368502 | 99,000 | 192.2 | 3.0 | 2 | chr1_C-D;A2 | Intra Interspersed |
| AL627326.16 | RP23-20A22 | 184504 | chr4:27153628 | 95,000 | 163.9 | 2.8 | 4 | chr13_A2 | Intra Interspersed (pericentromeric) |
| AC098741.2 | RP23-122J17 | 211681 | chr12:97659614 | 146,818 | 134.8 | 2.3 | 2 | chr12_F1 | Intra Cluster |
| AL691445.21 | RP23-67G3 | 189233 | chrX:58776354 | 99,000 | 128.8 | 1.9 | 1 | chrX_D | Unique |
| AL606987.11 | RP23-38314 | 212647 | chr4:144269068 | 206,000 | 447.1 | 1.8 | 5 | chr4_E2 | Intra Cluster (telomeric) |
| AL731676.5 | RP23-460B8 | 180106 | chrX:116982329 | 71,542 | 155.9 | 1.8 | 1 | chrX_F2,3 | Unique |
| AC122796.3 | RP24-347J6 | 166768 | chr7:23230794 | 21,117 | 85.0 | 1.5 | 3 | chr7_A3 | Intra Cluster |
| AL731663.12 | RP23-360J20 | 181033 | chr4:144011588 | 180,000 | 391.2 | 1.4 | 5 | chr4_E2 | Intra Cluster (telomeric) |
| AC087166.3 | RP23-354D10 | 210476 | chr12:109410281 | 49,000 | 87.7 | 1.3 | 1 | chr12_F2 | Unique |
| AL451076.14 | RP23-43O20 | 203581 | chrX:2344144 | 47,000 | 271.8 | 1.2 | 2 | chrX_A3 | Intra *weak 2nd signal |
| AC126273.3 | RP23-99P15 | 182698 | chr13:23632869 | 182,130 | 122.0 | 1.1 | 4 | chr13_A3 | Intra Cluster |
| AC026767.30 | RP23-328L8 | 193167 | chr7:48547102 | 76,116 | 83.9 | 1.1 | 2 | chr7_C | Intra Cluster |
| AC046145.16 | RP23-306D24 | 188606 | chr7:48296368 | 37,000 | 80.8 | 1.0 | 1 | chr7_B | Unique |
| AL732405.8 | RP23-149P4 | 193425 | chrX:87422908 | 45,000 | 109.0 | 1.0 | 2 | chrX_C,D | Intra Cluster |
| AL808115.7 | RP23-224M8 | 163408 | chr17:9981377 | 56,000 | 77.3 | 1.0 | 1 | chr17_A2 | Unique (pericentromeric) |
| AL713974.8 | RP23-135I10 | 191603 | chrX:40294317 | 77,001 | 72.0 | 0.8 | 2 | chrX_A5 | Intra Cluster |
A subset of mouse BAC clones with large (>20 kb) regions of duplication by WSSD detection were subsequently examined by FISH (Methods; 17/22 were confirmed as duplicated). The best placement within MGSCv3 was determined by similarity searches. Estimated copy number by FISH for tight tandem clusters was based on signal intensity compared with a unique hybridizing probe. The results of all FISH experiments are available online (http://www.biologia.uniba.it/mouse/).
A Comparison of Duplication Detection and Genome Assembly Methods
Table 3 compares different duplication detection methods and genome assembly strategies. In general, the whole-genome analysis comparison estimate of duplication content from build 29 is more consistent with the duplication estimate on the basis of whole-genome shotgun sequence detection (Table 1). Approximately 49% of the bases that were detected by WGAC (≥90% sequence identity and ≥10 kb) of build 29 were also positive by WSSD method. Because build 29 does not represent a complete genome sequence, regions that score positive by WSSD, but not WGAC, are expected (Supplemental Fig. 2). Regions that score positive by WGAC, but not WSSD, likely represent missing sequence overlaps in the assembly. In contrast, for MGSCv3, only 12% (159/954) of the potential duplicated regions were concordant between WSSD and WGAC (16% by duplication of the bases). These data confirm potential sequence collapse of segmental duplications during assembly of the mouse genome. If only WSSD regions are considered, then the duplication estimate for the draft genome begins to approximate the clone-ordered assembly. Finally, it should be noted that the average length of duplicated sequences within MGSCv3 is substantially shorter than that for build 29. Only eight alignments in the MGSCv3 were >30 kb in length (maximum 114 kb). The four largest alignments were highly similar (>95%) tandem duplications completely contained within the small number of finished BAC sequences that were incorporated into the assembly. The four other alignments outside of finished sequence were 30–40 kb in length and <95% identical.
Gene Content Analysis
To assess the gene content of mouse segmental duplications, we analyzed putative duplicated regions from finished BACs that showed evidence of duplication by WSSD. We limited our analysis to those genes that had an annotated NCBI coding sequence (RefSeq mRNAs) with at least two exons based on the build30 composite assembly (Methods). Only 0.50% of the RefSeq mRNA exons fall into duplicated regions, even though 1.7%–2.0% of the genome is predicted to be duplicated by our analyses (Table 5). We assessed protein domain assignments associated with each of these RefSeq genes (Methods). As in human, genes involved in immunity/defense (defensins, serpins, immunoglobulin containing proteins) and growth/development (B56 and hormone receptor containing proteins [Supplemental Table 5]) are highly enriched within recent segmental duplications. Many of these appear to be clusters of gene families at least within the limits of the current assembly and annotation methods. In contrast to humans, genes involved in DNA binding and transcriptional regulation are also enriched (KRAB and HMG box domain containing proteins).
Table 5.
Duplicated NCBI RefSeq Genes Within Finished BACs
| Exons
|
||||||
|---|---|---|---|---|---|---|
| mRNA | Protein | Name | D | U | Best domain | Chr |
| NM_007820 | NP_031846 | Cyp3a11 | 4 | 4 | p450 | 5 |
| NM_007844 | NP_031870 | Defcr-rs1 | 2 | 0 | defensin propep | Un |
| NM_007850 | NP_031876 | Defcr3 | 2 | 0 | defensins | Un |
| NM_007851 | NP_031877 | Defcr5 | 2 | 0 | defensins | 8 |
| NM_007852 | NP_031878 | Defcr6 | 2 | 0 | defensins | Un |
| NM_008252 | NP_032278 | LOC330026 | 1 | 0 | HMG box | 6 |
| NM_008459 | NP_032485 | Klra10 | 6 | 0 | lectin c | 6 |
| NM_009051 | NP_033077 | Rex2 | 6 | 0 | KRAB | 4 |
| NM_009243 | NP_033269 | Spi1-1 | 4 | 0 | serpin | 12 |
| NM_009244 | NP_033270 | Spi1-2 | 4 | 0 | serpin | 12 |
| NM_009246 | NP_033272 | Spi1-4 | 5 | 0 | serpin | 12 |
| NM_009486 | NP_033512 | V2r11 | 2 | 2 | ANF receptor | 14 |
| NM_009487 | NP_033513 | V2r11 | 3 | 2 | ANF receptor | Un |
| NM_009489 | NP_033515 | V2r14 | 6 | 0 | ANF receptor | 7 |
| NM_009490 | NP_033516 | V2r15 | 6 | 0 | Un | |
| NM_009493 | NP_033519 | V2r4 | 6 | 0 | ANF receptor | Un |
| NM_009529 | NP_033555 | Xmr | 9 | 0 | X | |
| NM_010648 | NP_034778 | Klra3 | 7 | 0 | lectin c | 6 |
| NM_010650 | NP_034780 | Klra8 | 6 | 0 | lectin c | 6 |
| NM_011120 | NP_035250 | Plfr | 1 | 4 | hormone | 13 |
| NM_011455 | NP_035585 | Spi13 | 6 | 0 | serpin | 13 |
| NM_011456 | NP_035586 | Spi14 | 7 | 0 | serpin | 13 |
| NM_013794 | NP_038822 | Klra1 | 1 | 6 | lectin c | 6 |
| NM_014194 | NP_055009 | Klra7 | 3 | 0 | lectin c | 6 |
| NM_017396 | NP_059092 | Cyp3a11 | 13 | 0 | p450 | 5 |
| NM_021365 | NP_067340 | Xlr4 | 9 | 0 | X | |
| NM_023743 | NP_076232 | Eif4enif1 | 2 | 17 | 11 | |
| NM_026206 | NP_080482 | 1600017N11Rik | 6 | 0 | hormone | 13 |
| NM_026492 | NP_080768 | 4930414C09Rik | 8 | 0 | KRAB | X |
| NM_027017 | NP_081293 | 3300002I08Rik | 3 | 1 | KRAB | 2 |
| NM_028561 | NP_082837 | 1700081O22Rik | 5 | 0 | Un | |
| NM_029203 | NP_083479 | 4930539I12Rik | 4 | 0 | homeobox | X |
| NM_031188 | NP_112465 | Mup1 | 7 | 0 | lipocalin | Un |
| NM_031390 | NP_113567 | Pramel3 | 5 | 0 | X | |
| NM_031493 | NP_113681 | Xlr5 | 6 | 0 | X | |
| NM_053124 | NP_444354 | Smarca5 | 2 | 0 | helicase C | 4 |
| NM_134252 | NP_599013 | Trpm8 | 4 | 21 | ion transport | 1 |
| NM_145078 | NP_659544 | 2610305D13Rik | 2 | 0 | 4 | |
Only the 38 RefSeq mRNAs containing duplicated (D) exons are listed. Overall, 0.50% (177/35,056) of exons from a total of 3834 RefSeq mRNAs within finished BACs were duplicated. Each gene was assigned the best-aligned domain by BLAST from the NCBI Conserved Domain Database (Methods). Chromosome assignment is based on BAC position within NCBI build30. The complete list of all 3834 genes is available (http://mouseparalogy.cwru.edu/).
DISCUSSION
The published mouse genome sequence (MGSCv3) represented one of the first attempts to publicly sequence and assemble a mammalian genome based largely on whole-genome shotgun sequence read data. A particular concern of such an approach has been the treatment of large high-copy repeats and segmental duplications that share a high degree of sequence identity (Green 1997; Weber and Myers 1997; Eichler 1998). The correct assembly of segmental duplications is not usually considered a high priority, especially during the draft phase of sequencing projects due to the perceived gene-poor content of such regions. In organisms such as humans, highly homologous segmental duplications are enriched for transcript and gene content (∼6%–7%). The resolution of such regions is, therefore, important to the genetics community and remains one of the most difficult tasks in the completion of the human genome. It is currently unknown whether the duplication-rich and gene-rich content of the human genome is characteristic of mammalian genome organization. An assessment of the duplication content and its relationship to the proteome are therefore critical issues in not only directing finishing efforts, but also in understanding the biology of the organism. Whereas it is typically expected that WGS sequence assemblies will underestimate the true duplication content (Bailey et al. 2001; Eichler 2001; Cheung et al. 2003a), analysis of private sequence assemblies of the mouse and human have been limited by accessibility to both the sequence and the underlying data (Venter et al. 2001; Mural et al. 2002). The availability of the public mouse genome sequence assembly (MGSCv3) and all of the underlying whole-genome shotgun sequence read data provide us with a unique opportunity to assess both duplication content as well as the quality of the assembly within these regions of the genome.
In this study, we examined the duplication content using two different approaches, a sequence assembly-based approach (termed WGAC) and a whole-genome shotgun sequence detection measure (termed WSSD). The latter, which is not dependent upon the assembly, was used previously as a robust method to detect large, highly identical duplications within the human (Bailey et al. 2002a). We implemented a modification of this approach, which was designed to take advantage of the reduced allelic variation of C57BL/6J. This entailed a quality assessment of the underlying read data to accurately calculate percent identity to determine the proportion of variant and identical reads within a region of the genome. As no allelic variation is expected, this provides a further estimate of copy number. We conservatively estimate (based on FISH verification of duplication content) an approximate sensitivity of 70% by this approach. The WSSD strategy therefore provides a very powerful tool for the identification of potentially collapsed duplicated regions within the mouse genome. We applied this detection method to two previous mouse genome assemblies that were constructed using independent strategies. MGSCv3 was constructed almost exclusively by paired-end sequence data derived from 40.7 million sequence reads from the same B6 strain female. MGSCv3 largely ignored mapping data or clone-based sequences during the assembly process (Table 3). In contrast, build 29 was largely based on the hand-curated assembly of sequence overlaps from a BAC-based tiling path of clones (∼400 Mb of sequence) and represented a relatively purist form of hierarchical clone-based sequencing.
There are a few important conclusions from this study with respect to genome assembly. A strict whole-shotgun sequence approach such as Arachne (Batzoglou et al. 2002) will collapse highly identical duplications. On the basis of FISH sensitivity of the WSSD detection method (Table 4,) we project that at least 50%–60% of the duplications are not resolved as duplicated copies within the assembly (Tables 1 and 3). It should be emphasized that the WSSD method is designed to reliably detect large (≥10 kb) and highly identical (>95%) duplications. This underrepresentation of duplications is exacerbated for large duplications ≥20 kb (Table 1). Such collapsed duplications are enriched on the unplaced chromosome (Fig. 5; Supplemental Table 4), confirming that duplications are difficult to map. Global estimates of segmental duplication content that do not take this collapse into account will underestimate the duplicated portion of the genome (Cheung et al. 2003a). For example, Cheung and colleagues estimated that 1.2% of the mouse genome as duplicated (≥90% and ≥5 kb) on the basis of an analysis of the Feb. 2003 assembly. Their estimate was solely dependent on the genome assembly, although sequence collapse was suspected. The working draft nature of the mouse genome, we believe, precludes such a precise estimate. Our combined analyses, however, suggest a range from 1.7% to 2.0% (for duplications ≥90% and ≥10 kb). This estimate is lower than what has recently been predicted for the rat genome (2.9%; Tuzun et al. 2004). The percentage of duplications in the mouse increases to 2.5% if the size thresholds are relaxed to 1 kb. Our analyses suggest that most of these represent uncharacterized lineage-specific retroposons rather than segmental duplications.
Figure 5.

Mouse segmental duplications. Segmental duplications detected by whole-genome shotgun sequence detection (WSSD, black bars) and whole-genome analysis comparison (WGAC, red/blue bars) are drawn to scale within the published mouse genome assembly (MGSC 2002). Chromosome lengths and the centromere positions are shown in purple. These data are available as part of an interactive mouse segmental duplication database (http://mouseparalogy.cwru.edu).
As expected, clone-ordered-based approaches for sequence assembly appear to more effectively resolve duplication overlaps, although artifactual duplications are more frequently encountered. Build 29 shows the best correlation between duplications confirmed by WGAC and WSSD in our analysis (Table 3). In contrast, 74% of the duplications within the whole-genome shotgun sequence detection could only be detected by WSSD, and most of these mapped to the unplaced chromosome. If the experimental cytogenetic data is used to estimate false positives (22%), we conclude that 57% of the large duplications (≥95% and ≥10 kb) have not yet been resolved within the assembly. Our data suggest that a combined approach using whole-genome shotgun sequence detection to identify regions of duplication within a WGS assembly followed by targeted high-quality BAC clone sequencing could provide the most affordable and effective means for resolving these complex regions of the genome. In this study, we pinpoint a small fraction (∼1%) of the mouse genome that should be targeted for finished sequence within BAC clones. These regions are unlikely to be properly assembled and mapped, irrespective of increased depths of whole-genome shotgun sequencing. We have constructed an integrated mouse segmental duplication database (http://mouseparalogy.gene.cwru.edu), which will provide a framework for directing finishing and sequencing efforts within these areas.
Biologically, some interesting differences in the pattern and organization of segmental duplications can be deduced when compared with human. Our analysis shows that only 0.54% of the annotated RefSeqs fall into duplicated regions, even though 1.5%–2.0% of the genome is predicted to be duplicated by the WSSD method. This is in contrast to the human, where 6.1% of the RefSeqs fell into duplicated regions, with 5.2% of the genome predicted to be duplicated (Bailey et al. 2002a). Preliminary analysis of the rat genome also suggests that recent duplicated regions are gene poor (Tuzun et al. 2004). It is currently unclear whether rodent duplications occur preferentially in gene-poor regions, or whether gene annotation is the limiting factor. In the original publication of the working-draft sequence of the mouse genome, it was hypothesized that the mouse had less segmental duplication content due to the remarkably large N50 supercontig size that was observed during sequence assembly. It was known that highly homologous tandem gene families such as Sp100-4s were, in fact, dramatically collapsed (MGSC 2002). Our preliminary data suggest that the mouse is reduced for segmental duplication, but in particular, the effects are much more local, taking the form of tandem duplications. This architecture of the mouse genome may facilitate a more robust sequence assembly based strictly on whole-genome shotgun sequence. Within the human genome, segmental duplications are interspersed over large genomic distances, leading to a much higher rate of misassignment, missed overlaps, and mapping and sequencing inconsistencies. This study and other recent work (Cheung et al. 2003b; Tuzun et al. 2004) indicate that interspersed segmental duplications are rare among rodent species. Rodent segmental duplications appear largely tandem or tightly clustered in their organization. The molecular basis for this difference in hominoid and rodent genome architecture is unknown, although the burst of primate Alu retroposition activity ∼35 million years ago may correlate with the expansion and dispersion of hominoid segmental duplications (Bailey et al. 2003).
The identification of coding sequence within these duplicated regions of the mouse genome has some interesting practical and biological implications. The mouse has been an invaluable tool for dissecting gene function, due to the ability to directly manipulate the genome and assess the phenotypic consequences in vivo (van der Weyden et al. 2002). However, genes within these duplicated regions will require additional analysis. Initial targeting might be technically compromised by the presence of duplicated sequence. In addition, phenotypic analysis could be complicated if the duplicated genes still share expression domains and functions (Copley et al. 2003). These duplicated regions are obvious candidates for evolution of rodent-specific genes, as has been recently documented in human (Johnson et al. 2001; Paulding et al. 2003). The direct implications of the genes within duplicated regions are unclear, as many remain uncharacterized gene predictions. Even in the case of well-known genes, available phenotypic data are limiting. The identification of such regions, however, provides a platform to begin experimental dissection of the genes embedded within segmental duplications in the context of the evolutionary history of the organism.
METHODS
Whole-Genome Assembly Comparison of Mouse Draft Genome (MGSCv3)
To analyze mouse segmental duplications, we applied a BLAST-based whole-genome assembly comparison (Bailey et al. 2001). This method was designed to detect highly similar (≥90% identity) primate-specific segmental duplications (≥1 kb). We applied this method to the mouse, but detected an excess of smaller putative segmental duplications (870,969 seeding alignments ≥500 bp and ≥88% identity, 10-fold greater than standard human analysis). Upon inspection, the vast majority of these alignments corresponded to incompletely masked high-copy repeats (mainly LTR and LINE elements). In mouse, both LTR and L1 elements show increased activity as well as complicated evolutionary histories (DeBerardinis et al. 1998; Mears and Hutchison III 2001; Cheung et al. 2003b). For instance, L1 elements show mosaic sequence structures due to frequent evolutionary gene conversion events (Mears and Hutchison III 2001). Because our detection algorithm extends seeding alignments into adjacent high-copy repeats, partially masked repeats will be lengthened to include the entire element. Although we have not searched directly, part of the inability to completely mask L1s may be due to increased rates of transduction of the 3′ flanking sequence (Kazazian Jr. 2000; Mears and Hutchison III 2001). To circumvent the overabundance of high-copy repeats, we increased the length threshold for alignments (seeding length ≥2500 bp; Supplement 1). At this threshold, many uncharacterized transposable element alignments were still present. To avoid these larger transposable elements, we set a 10-kb threshold for most analyses— avoiding the inclusion of all, but possibly, the largest full-length endogenous retroviral elements.
Whole-Genome Alignment Comparison: NCBI Build 29 Finished Clone-Based Sequence
We applied our BLAST-based analysis to the finished sequence contigs that were incorporated as part of an NCBI BAC-based sequence assembly (build 29). The nonredundant finished portion of this sequence assembly constitutes ∼18% (439,076,820/2.5 Gb genome) of the mouse genome. As this represents only one-fifth of the genome, we were able to apply our standard human parameters with a reasonably small number of alignments (Bailey et al. 2001). We detected 26,083 seeding alignments, ≥500 bp and ≥88% identity. These seeding alignments were then trimmed to precisely define the junctions. Optimal global alignments (ALIGN, Myers-Miller algorithm) were used as the basis for computation of alignment statistics. Alignments were merged over gaps (<10 kb size). A total of 3930 alignments (≥1 kbp and ≥90%) were retained for further analysis. To better assess the true duplication content of build 29, we hand curated the alignment set to remove prevalent interspersed high-copy repeats. High-copy repeats were removed on the basis of three criteria, based on visual inspection in Parasight (J.A. Bailey, unpubl.) as follows: (1) a size (<10 kb) consistent with known LINE and LTR insertion sequences; (2) at least 10 copies within build 29 (at least 50 copies in the genome); (3) a distribution to multiple chromosomes (at least three) consistent with genome-wide interspersed transposition. In a few cases, similarity to known transposable elements or evidence for protein similarity to reverse transcriptase was used as a basis to exclude alignments. We removed 2235 alignments (corresponding to 12 related clusters), leaving 1155 alignments.
It has been shown previously that clone-ordered genome assemblies are more apt to overestimate segmental duplication content (as much as threefold) due to a failure to correctly merge sequence overlaps (Bailey et al. 2001). For example, if sequences from two BACs have not been recognized as allelic, our whole-genome analysis comparison will identify such pairwise alignments as segmental duplications with an extraordinary high degree of sequence identity. We removed 49 pairwise alignments in which the sequence identity was >99.99%, and which mapped to the boundaries of sequence contigs. In addition, we excluded three large, highly identical (>99.99%) alignments that mapped internally to finished sequence contigs, but in which duplication junctions corresponded precisely to the boundaries of the BAC clone within the tiling path. This left a total of 1103 alignments ≥1 kb and ≥90% identity. As a final precaution, we confirmed their allelic nature by the lack of increased WSSD coverage (see below).
Whole-Genome Shotgun Sequence Detection of Duplications
We assessed duplication content using a whole-genome shotgun sequence detection strategy previously developed during the analysis of the human genome (Bailey et al. 2002a). For a given genomic sequence, this method essentially assesses the depth-of-coverage and the average degree of sequence identity within a random set of whole-genome shotgun sequence. In regions of duplications, a statistically significant increase in the depth-of-coverage and significant decrease in the average degree of sequence identity due to the additional recruitment of paralogous reads will be observed. We considered three groups of sequences as follows: (1) A calibration set of characterized unique (2052 kb) and duplicated sequences (954 kb) from the mouse genome, (2) all finished C57BL/6J BACs within NCBI GenBank (Jan 7, 2003 from the NCBI build30 freeze), and (3) all MGSCv3 sequence (processed in 400-kb nonoverlapping segments).
Each reference mouse genome sequence was compared by Megablast against the entire set of mouse WGS (whole-genome shotgun sequence reads [40,782,208 sequences; 31,117,512,375 bp]). Reference sequences were initially lowercase, masked for repeat elements showing <5% divergence from the consensus sequence, with the exception of LTR and LINE elements, which were masked at 15% and 10% divergence from consensus, respectively. This increased our sensitivity in removing lineage-specific elements. Megablast alignments were performed using lowercase masking parameters (-D 3 -J F -P 93 -U T -F m -s 220), which allows for greedy-algorithm extension into adjacent repetitive regions. The quality of the query sequence (genomic piece) was assumed to be high quality. Aligned bases from the read with a PHRED score of <30 (error rate >1/1000) were ignored in determining the percent identity. This process corrects for sequencing errors in an unbiased way (regardless of match or mismatch). The program paralogy_detector was then run on every segment. Alignments were only considered if they were >400 bp, represented 90% of the read, and had at least 300 bp within the unique regions, with a rescored similarity of >94% and ≥200 high-quality aligned bases. We chose 94% as the limit of read recruitment, because most duplications with lower percent identity are effectively resolved during WGS assembly (E.E. Eichler, unpubl.). Furthermore, detecting more divergent duplications, while possible, would increase the computational cost. To accurately determine sequence identity, we realigned alignments with optimal global alignment algorithm (Needleman-Wunsch).
A read-based detection method has been previously based on number of reads in 5-kb windows (1-kb overlap slide). In general, mouse unique sequence read depth showed slightly increased variability (40.3 +/- 13.5 reads per 5-kb reference) when compared with a similar analysis performed with human data (50.4 +/- 12.8 reads per 5-kb reference; Bailey et al. 2002a). Duplicated sequences showed significant departures from the calibrated unique sequence (100 +/- 116 reads per 5-kb reference). Because the mouse is inbred and has limited allelic variation, we exploited this fact to develop a more sensitive metric on the basis of the number of reads that diverged from the expected allelic levels (100%; termed the divergent read ratio). Divergent reads were defined as those which showed <99.8% identity to the reference sequence (>1 high-quality mismatch per average read); 99.8% allows for alignment errors, such as stretches of polypyrimidine and purine stretches, to be processed without requiring manual curation of each alignment. Windows were combined to delineate duplicated regions. Whereas this method will not detect identical duplications, it will reliably detect duplications with <99% sequence identity. On the basis of our calibration set of known unique and duplicated regions of the mouse genome, significant differences were detected between these two sets of data (Supplemental Table 3). This approach increased sensitivity for detecting large single-duplication events — including recent, but low-frequency tandem duplications. We initially examined all regions (≥10 kb) in which the divergent read ratio exceeded 0.5. Once again, 10 kb was selected as a length criterion due to the upper-limit insertion size of most retroelements. Because of a high number of false positives from this initial pass and the high thresholds observed among known duplicated sequences in the mouse (Supplemental data), we later raised the threshold to 0.8. A theoretical divergent read ratio of 1.0 does not account for the loss of reads due to insertions and large-scale differences between duplicated copies. In the case of the human analysis, a statistical model was developed because there were at least two-dozen duplications whose sequence properties had been experimentally validated. Establishing such a statistical model is not possible given the limited data available on well-characterized mouse duplications. All computational analyses (Tables 1 and 3) and later experimental analyses were based upon this final set of data.
FISH Analysis
FISH experiments were performed as previously described (Lichter et al. 1990). Both interphase and metaphase nuclei were prepared directly from bone marrow of C57BL/6J mice. Briefly, 3 d prior to tissue harvest, mice were injected peritoneally with 10 μL/g of body weight of yeast solution (2 g yeast, 2 g dextrose, 5 g milk powder, 25 mL water at 40°C). The procedure was repeated after 24 h. One day prior to sacrifice, animals were injected with 5 μL of colchicine 0.025%/g of body weight. Immediately after sacrifice, femur epiphyses were extracted, washed with physiological saline solution, and the bone marrow removed. After 10 min of centrifugation, the supernatant was discarded and cell material was resuspended in a hypotonic solution (75 mM KCl solution) at 37°C. After 15 min, fixative was added and metaphase and interphase nuclei were prepared.
A subset of mouse BAC clones with large (>20 kb) regions of duplication as determined by WSSD detection were subsequently examined by FISH. Metaphase nuclei were examined to identify interchromosomal or intrachromosomal duplications that were interspersed by 5 Mb or more. More intense FISH signals, which localized to a single site, were subsequently examined by interphase nuclei. Interphase analyses were controlled for replication by comparing cells at both G1 and G2 stages of arrest. At least 10 interphase nuclei were examined for each preparation. The number of interphase nuclei signals and signal intensity was compared with unique hybridizing clones to provide a relative estimate of copy number. Because probe signal intensity may vary due to sequence property differences, copy-number estimates provided in Supplemental Table 3 should be considered approximate.
Gene Content Analysis
We characterized the coding content of the duplicated regions from finished BACs that were detected by WSSD. We utilized information from NCBI's genome annotation pipeline (http://www.ncbi.nlm.nih.gov/genome/guide/build.html) on NCBI Mouse Build 30 (http://www.ncbi.nlm.nih.gov/genome/guide/mouse/MmStats.html). Build 30 represented the first attempt at a composite assembly. Only finished sequence was integrated into the MGSCv3, using a very conservative algorithm (http://www.ncbi.nlm.nih.gov/genome/seq/NCBIContigInfo.html). Duplicated regions on finished BAC clones were converted into contig coordinates. One BAC in the duplicated list was actually derived from 129/SvEvTac. Reference sequences (RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/) that lie entirely within these coordinates were then identified and tagged as `duplicated.' A distinction was made between curated genes (accession type NM_/NP_) and those detected by automated methods (accession type XM_/XP_). Whereas all genes had an annotated coding sequence, automated accessions (XM_) were excluded from analysis for consistency with previous human analysis (Bailey et al. 2002a).
As part of the annotation pipeline, the proteins from translated RefSeq mRNAs are compared by BLAST (Altschul et al. 1990) to the Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml). The best alignment (by bitscore) of a protein to a domain was retained. We examined the domain assignment for each protein within the duplicated portion of the genome and compared the unique portion of the genome (Supplemental Table 5).
Acknowledgments
We thank the large-scale sequencing centers (Baylor College of Medicine, Cold Spring Harbor Laboratory, Genome Therapeutics Corporation, Harvard Partners Genome Center, Joint Genome Institute, The NIH Intramural Sequencing Center, The UK-MRC Sequencing Consortium, The University of Oklahoma Advanced Center for Genome Technology, The University of Texas Southwest, The Whitehead Institute for Biomedical Research, The Washington University Genome Sequencing Center, and the Wellcome Trust Sanger Institute) for access to all large-scale finished sequence, genome assembly, and trace sequence data from the mouse genome prior to publication. We thank Ilya Dondoshansky for modifying megaBLAST output into a form that significantly increased the speed of analysis. We thank Royden Clark and Ulrich Neuss for technical assistance. This work was supported, in part, by NIH grants GM58815 and HG002385 to E.E.E., a NIH Career Development Program in Genomic Epidemiology of Cancer (CA094816) to J.A.B., Telethon, CEGBA (Centro di Eccellenza Geni in campo Biosanitario e Agroalimentare), MIUR (Ministero Italiano della Universita' e della Ricerca; Cluster C03, Prog. L.488/92) to M.R., the W.M. Keck Foundation, and the Charles B. Wang Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2238404.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Armengol, L., Pujana, M.A., Cheung, J., Scherer, S.W., and Estivill, X. 2003. Enrichment of segmental duplications in regions of breaks of synteny between the human and mouse genomes suggest their involvement in evolutionary rearrangements. Hum. Mol. Genet. 12: 2201-2208. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Yavor, A.M., Massa, H.F., Trask, B.J., and Eichler, E.E. 2001. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 11: 1005-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002a. Recent segmental duplications in the human genome. Science 297: 1003-1007. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Yavor, A.M., Viggiano, L., Misceo, D., Horvath, J.E., Archidiacono, N., Schwartz, S., Rocchi, M., and Eichler, E.E. 2002b. Human-specific duplication and mosaic transcripts: The recent paralogous structure of chromosome 22. Am. J. Hum. Genet. 70: 83-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, J.A., Liu, G., and Eichler, E.E. 2003. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 73: 823-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzoglou, S., Jaffe, D.B., Stanley, K., Butler, J., Gnerre, S., Mauceli, E., Berger, B., Mesirov, J.P., and Lander, E.S. 2002. ARACHNE: A whole-genome shotgun assembler. Genome Res. 12: 177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, V.E., Nowak, N., Jang, W., Kirsch, I.R., Zhao, S., Chen, X.-N., Furey, T.S., Kim, U.-J., Kuo, W.-L., Olivier, M., et al. 2001. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 409: 953-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, J., Estivill, X., Khaja, R., MacDonald, J.R., Lau, K., Tsui, L.C., and Scherer, S.W. 2003a. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 4: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, J., Wilson, M.D., Zhang, J., Khaja, R., MacDonald, J.R., Heng, H.H., Koop, B.F., and Scherer, S.W. 2003b. Recent segmental and gene duplications in the mouse genome. Genome Biol. 4: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, F.S., Green, E.D., Guttmacher, A.E., and Guyer, M.S. 2003. A vision for the future of genomics research. Nature 422: 835-847. [DOI] [PubMed] [Google Scholar]
- Copley, R.R., Goodstadt, L., and Ponting, C. 2003. Eukaryotic domain evolution inferred from genome comparisons. Curr. Opin. Genet. Dev. 13: 623-628. [DOI] [PubMed] [Google Scholar]
- DeBerardinis, R.J., Goodier, J.L., Ostertag, E.M., and Kazazian Jr., H.H. 1998. Rapid amplification of a retrotransposon subfamily is evolving the mouse genome. Nat. Genet. 20: 288-290. [DOI] [PubMed] [Google Scholar]
- Dehal, P., Predki, P., Olsen, A.S., Kobayashi, A., Folta, P., Lucas, S., Land, M., Terry, A., Zhou, C.L.E., Rash, S., et al. 2001. Human chromosome 19 and related regions in mouse: Conservative and lineage specific evolution. Science 293: 104-111. [DOI] [PubMed] [Google Scholar]
- DiDonato, C.J., Chen, X.N., Noya, D., Korenberg, J.R., Nadeau, J.H., and Simard, L.R. 1997. Cloning, characterization, and copy number of the murine survival motor neuron gene: Homolog of the spinal muscular atrophy-determining gene. Genome Res. 7: 339-352. [DOI] [PubMed] [Google Scholar]
- Eichler, E.E. 1998. Masquerading repeats: Paralogous pitfalls of the Human Genome. Genome Res. 8: 758-762. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Repetitive conundrums of centromere structure and function. Hum. Mol. Genet. 8: 151-155. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Segmental duplications: What's missing, misassigned, and misassembled—And should we care? Genome Res. 11: 653-656. [DOI] [PubMed] [Google Scholar]
- Eichler, E.E. and Sankoff, D. 2003. Structural dynamics of eukaryotic chromosome evolution. Science 301: 793-797. [DOI] [PubMed] [Google Scholar]
- Estivill, X., Cheung, J., Pujana, M.A., Nakabayashi, K., Scherer, S.W., and Tsui, L.C. 2002. Chromosomal regions containing high-density and ambiguously mapped putative single nucleotide polymorphisms (SNPs) correlate with segmental duplications in the human genome. Hum. Mol. Genet. 11: 1987-1995. [DOI] [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175-185. [DOI] [PubMed] [Google Scholar]
- Giglio, S., Calvari, V., Gregato, G., Gimelli, G., Camanini, S., Giorda, R., Ragusa, A., Guerneri, S., Selicorni, A., Stumm, M., et al. 2002. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am. J. Hum. Genet. 71: 276-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli, G., Pujana, M.A., Patricelli, M.G., Russo, S., Giardino, D., Larizza, L., Cheung, J., Armengol, L., Schinzel, A., Estivill, X., et al. 2003. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum. Mol. Genet. 12: 849-858. [DOI] [PubMed] [Google Scholar]
- Green, P. 1997. Against a whole-genome shotgun. Genome Res. 7: 410-417. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium (IHGSC). 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Ji, Y., Walkowicz, M.J., Buiting, K., Johnson, D.K., Tarvin, R.E., Rinchik, E.M., Horsthemke, B., Stubbs, L., and Nicholls, R.D. 1999. The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is deficient in mice with neuromuscular and spermiogenic abnormalities. Hum. Mol. Genet. 8: 533-542. [DOI] [PubMed] [Google Scholar]
- Ji, Y., Eichler, E.E., Schwartz, S., and Nicholls, R.D. 2000. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res. 10: 597-610. [DOI] [PubMed] [Google Scholar]
- Johnson, M.E., Viggiano, L., Bailey, J.A., Abdul-Rauf, M., Goodwin, G., Rocchi, M., and Eichler, E.E. 2001. Positive selection of a gene family during the emergence of humans and African apes. Nature 413: 514-519. [DOI] [PubMed] [Google Scholar]
- Kazazian Jr., H.H. 2000. Genetics. L1 retrotransposons shape the mammalian genome. Science 289: 1152-1153. [DOI] [PubMed] [Google Scholar]
- Lichter, P., Tang, C.J., Call, K., Hermanson, G., Evans, G.A., Housman, D., and Ward, D.C. 1990. High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247: 64-69. [DOI] [PubMed] [Google Scholar]
- Locke, D.P., Archidiacono, N., Misceo, D., Cardone, M.F., Dechamps, S., Roe, B.A., Rocchi, M., and Eichler, E.E. 2003. Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biol. 4: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski, J.R. 1998. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14: 417-422. [DOI] [PubMed] [Google Scholar]
- Mears, M.L. and Hutchison III, C.A. 2001. The evolution of modern lineages of mouse L1 elements. J. Mol. Evol. 52: 51-62. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium (MGSC). 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Muller, H.J. 1936. Bar duplication. Science 83: 528-530. [DOI] [PubMed] [Google Scholar]
- Mural, R.J., Adams, M.D., Myers, E.W., Smith, H.O., Miklos, G.L., Wides, R., Halpern, A., Li, P.W., Sutton, G.G., Nadeau, J., et al. 2002. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296: 1661-1671. [DOI] [PubMed] [Google Scholar]
- Ohno, S., Wolf, U., and Atkin, N. 1968. Evolution from fish to mammals by gene duplication. Hereditas 59: 169-187. [DOI] [PubMed] [Google Scholar]
- Osborne, L.R., Li, M., Pober, B., Chitayat, D., Bodurtha, J., Mandel, A., Costa, T., Grebe, T., Cox, S., Tsui, L.C., et al. 2001. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat. Genet. 29: 321-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulding, C.A., Ruvolo, M., and Haber, D.A. 2003. The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc. Natl. Acad. Sci. 100: 2507-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentao, L., Wise, C., Chinault, A., Patel, P., and Lupski, J. 1992. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 2: 292-300. [DOI] [PubMed] [Google Scholar]
- Probst, F.J., Chen, K.S., Zhao, Q., Wang, A., Friedman, T.B., Lupski, J.R., and Camper, S.A. 1999. A physical map of the mouse shaker-2 region contains many of the genes commonly deleted in Smith-Magenis syndrome (del17p11.2p11.2). Genomics 55: 348-352. [DOI] [PubMed] [Google Scholar]
- Samonte, R., Conte, R., Ramesh, K., and Verma, R. 1996. Molecular cytogenetic characterization of breakpoints involving pericentric inversions of human chromosome 9. Hum. Genet. 98: 576-580. [DOI] [PubMed] [Google Scholar]
- Samonte, R.V. and Eichler, E.E. 2002. Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 3: 65-72. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13: 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger, R., Schlagenhaufer, R., Kerb, R., Bruhn, C., Brockmoller, J., Roots, I., and Brinkmann, U. 2000. Characterization of the glutathione S-transferase GSTT1 deletion: Discrimination of all genotypes by polymerase chain reaction indicates a trimodular genotype–phenotype correlation. Pharmacogenetics 10: 557-565. [DOI] [PubMed] [Google Scholar]
- Stankiewicz, P., Park, S.S., Inoue, K., and Lupski, J.R. 2001. The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res. 11: 1205-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J.W., Schueler, M.G., Summers, T.J., Blakesley, R.W., McDowell, J.C., Thomas, P.J., Idol, J.R., Maduro, V.V., Lee-Lin, S.Q., Touchman, J.W., et al. 2003. Pericentromeric duplications in the laboratory mouse. Genome Res. 13: 55-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun, E., Bailey, J., and Eichler, E.E. 2004. Recent segmental duplications in the working draft assembly of the brown Norway rat. Genome Res. 14: 493-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden, L., Adams, D.J., and Bradley, A. 2002. Tools for targeted manipulation of the mouse genome. Physiol. Genomics 11: 133-164. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, R.J., Mural, P.W., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Weber, J.L. and Myers, E.W. 1997. Human whole-genome shotgun sequencing. Genome Res. 7: 401-409. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://mouseparalogy.gene.cwru.edu; Eichler Lab Mouse Segmental Duplication Database.
- http://www.biologia.uniba.it/mouse/; FISH experiments of WSSD duplication-positive clones.
- http://www.ncbi.nlm.nih.gov/genome/guide/build.html; NCBI's Genome Annotation Pipeline.
- http://www.ncbi.nlm.nih.gov/genome/guide/mouse/MmStats.html; Mouse Build 30 Statistics.
- http://www.ncbi.nlm.nih.gov/RefSeq/; NCBI Reference Sequence Database.
- http://www.ncbi.nlm.nih.gov/genome/seq/NCBIContigInfo.html; NCBI Assembly Process.
- http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml; NCBI Conserved Domain Database.