

Abstract
Somatic mitochondrial mutations are common in human cancers, and can be used as a tool for early detection of cancer. We have developed a mitochondrial Custom Reseq™ microarray as an array-based sequencing platform for rapid and high-throughput analysis of mitochondrial DNA. The MitoChip contains oligonucleotide probes synthesized using standard photolithography and solid-phase synthesis, and is able to sequence >29 kb of double-stranded DNA in a single assay. Both strands of the entire human mitochondrial coding sequence (15,451 bp) are arrayed on the MitoChip; both strands of an additional 12,935 bp (84% of coding DNA) are arrayed in duplicate. We used 300 ng of genomic DNA to amplify the mitochondrial coding sequence in three overlapping long PCR fragments. We then sequenced >2 million base pairs of mitochondrial DNA, and successfully assigned base calls at 96.0% of nucleotide positions. Replicate experiments demonstrated >99.99% reproducibility. In matched fluid samples (urine and pancreatic juice, respectively) obtained from five patients with bladder cancer and four with pancreatic cancer, the MitoChip detected at least one cancer-associated mitochondrial mutation in six (66%) of nine samples. The MitoChip is a high-throughput sequencing tool for the reliable identification of mitochondrial DNA mutations from primary tumors in clinical samples.
Detecting cancer in its earliest stages presents the opportunity to treat the disease before it spreads. Despite the tremendous advances made in understanding the pathogenesis and molecular aberrations in human cancer, early detection still remains a contentious issue. As an example, lung cancer is the most common cause of cancer deaths in the countries of North America and other developed countries, accounting for 29% of all cancer deaths (Greenlee et al. 2000). The poor prognosis of lung cancer is largely attributable to the lack of effective early detection methods, since over two-thirds of the patients have regional lymph-node involvement or distant disease at the time of presentation (Hirsch et al. 2001). Many of the currently available early detection strategies are invasive, laborious, or lack adequate sensitivity and specificity for discriminating cancer from confounding clinical scenarios. It is recognized that neoplastic cells carry unique genetic signatures that distinguish them from normal somatic cells, thereby permitting their detection in a heterogeneous background. Although aberrations within the neoplastic transcriptome and proteome have both been utilized for cancer diagnosis in clinical samples (Liotta and Petricoin 2000; Petricoin et al. 2002), the approach with the greatest success as well as ease of application has been the analysis of DNA alterations in cancer cells (Mao et al. 1996; Dong et al. 2001; Traverso et al. 2002).
A large number of mitochondrial DNA mutations have recently been reported in cancers at several anatomic sites (Polyak et al. 1998; Fliss et al. 2000; Bianchi et al. 2001; Jones et al. 2001; Parrella et al. 2001; Sanchez-Cespedes et al. 2001; Chen et al. 2002; Copeland et al. 2002). The frequency of mitochondrial mutations in these studies is high, with half to two-thirds of cancers harboring at least one somatic mutation. The mitochondrial genome is an ideal target for mutation detection in cancers for several reasons. First, mitochondrial mutations in cancer are not only common, but unlike nuclear genes, do not appear to be restricted by cancer type (Polyak et al. 1998; Fliss et al. 2000; Jones et al. 2001; Sanchez-Cespedes et al. 2001). Second, detection of mitochondrial DNA mutations in clinical samples (such as exfoliated cells in urine, or lavage fluids) offers a distinct advantage over nuclear DNA because of the high copy number of mitochondrial genomes in cancer cells. Fliss et al. (2000) determined that mitochondrial DNA was 19 to 220 times as abundant as mutated p53 nuclear DNA in matched body fluids from cancer patients. Similarly, Jones et al. (2001) confirmed the facile detection of mitochondrial DNA mutations in primary tumors with a 30% or less neoplastic cellularity, whereas known nuclear DNA mutations could not be detected in the nonenriched samples. Finally, the presence of mitochondrial DNA mutations in a proportion of preneoplastic lesions suggests that mutations occur early in multistep tumor progression (Jeronimo et al. 2001; Parrella et al. 2001; Ha et al. 2002), and hence, may be used as a tool for early detection of cancer in clinical samples, including body fluids and serum (Hibi et al. 2001; Jeronimo et al. 2001; Nomoto et al. 2002; Okochi et al. 2002).
Current strategies for using the mitochondrial genome as a screening tool in cancer are limited by the availability of a high-throughput platform for mutation detection. Even with the availability of sensitive and rapid mutation detection platforms such as automated capillary sequencers and denaturing high-performance liquid chromatography (HPLC; Medintz et al. 2001; Liu et al. 2002), the routine sequencing of 16.5 kb of mitochondrial DNA is an onerous task. Microarrays are inherently parallel devices that offer the promise of determining the genotypes at every site of interest with a limited level of effort (Hacia 1999). Chee et al. developed the first mitochondrial sequencing microarray in 1996, comprised of “tiled” oligonucleotide sequencing probes synthesized using standard photolithography and solid-phase DNA synthesis (Chee et al. 1996). This microarray platform, however, had several limitations, including the requirement for generating RNA by in vitro transcription of genomic DNA for chip hybridization, tiling of only a single strand of the target mitochondrial sequence on the chip, and absence of robust genotype assignment software. We have developed a “second-generation” sequencing microarray for high-throughput analysis of mitochondrial coding region mutations that vastly improves on the previous design, and present here the validation and potential application of our MitoChip towards cancer detection.
The MitoChip can sequence 29,366 bp of double-stranded DNA, which includes 980 bp of plasmid DNA sequence as control for chip hybridization. Both strands of the entire mitochondrial coding region (nucleotides 573 through 16024, i.e., 15,451 bp) are tiled once on the array. The forward and reverse strands of an additional 12,935 bp of the mitochondrial DNA (the coding region minus 12S and 16S RNA sequences) are tiled on the remaining features, and thus provide an inbuilt duplication of sequence data for ∼84% of the mitochondrial coding region (Table 1). The requirements for starting template are minimal (only 300 ng of genomic DNA), and an automated adaptive background genotype-calling scheme (Cutler et al. 2001) enables the detection of coding sequence variations that meet a threshold “quality score.” We have sequenced >2 million base pairs of mitochondrial DNA with the MitoChip, and demonstrate an overall frequency of 96.0% for successfully assigning base calls, with >99.99% reproducibility of base calls in replicate experiments. The ability of the MitoChip to detect mitochondrial mutations in samples of body fluids obtained from cancer patients attests to its promise as a tool for the early detection of cancer in clinical samples.
Table 1.
Summary of MitoChip Design and Experiments
| Total double-stranded DNA sequenced per chip | 29,366 bp |
| Control (plasmid) DNA | 980 bp |
| Mitochondrial coding sequence (includes RCRS 573 through 16024) | 15,451 bp |
| Mitochondrial coding sequence tiled in dupicate (includes RCRS 573—648; RCRS 1603—1670; RCRS 3231—16024) | 12,935 bp |
| Total mitochondrial DNA sequenced per MitoChip | 28,386 bp |
| Total samples analyzed | 75 |
| Total mitochondrial DNA sequenced | 2,128,950 bp |
| Total mitochondrial base pairs assigned by GDAS | 2,044,000 bp |
| Total percentage bases called (%) | 96.0 |
| (range 89.12—97.83) |
RESULTS
Overall Genotype Calls
Seventy-five MitoChip assays, using DNA extracted from a variety of cancer cell lines, primary tumors, lymphocytes, and body fluid samples were performed (Supplemental Table 1). We amplified 300 ng of genomic DNA from these 75 samples in three overlapping fragments as described in the Methods section. The products were fragmented and hybridized to MitoChips to yield the equivalent to 2,128,950 base pairs of double-stranded mitochondrial DNA sequence (Table 1). Using adaptive background subtraction and a total threshold (Ttotal) quality score of 30 (Cutler et al. 2001), the Affymetrix GeneChip DNA Analysis Software (GDAS) assigned 2,044,000 base calls (mean base calls = 96.0%, range 89.12%–97.83%). There were no significant differences in the percentage of genotype calls between DNA extracted from lymphocytes, cell lines, primary tumors, or body fluids.
Reproducibility of Array-Based Sequencing
In order to assess within-chip and between-chip reproducibility (in effect, consistency and accuracy of the microarray data, respectively), we performed duplicate analyses on a subset of 13 cancer cell lines, beginning with the PCR amplification step from genomic DNA (Table 2). For the within-chip reproducibility, we evaluated the 12,935 bp of DNA represented in duplicate on each chip, and each of the 26 samples was considered an independent comparison. Overall, 311,814 / 336,310 (92.71%) bases were called at the duplicate positions across the 26 samples. We found only eight discordant base calls out of 311,814 or a within-chip error rate of 0.0025% (Supplemental Table 2). All eight bases were heteroplasmic, and associated with low quality scores (median quality score for discordant bases = 42.5; median quality score for all bases = 71.3, using threshold score of 30). For the between-chip reproducibility, the number of bases called in the first set of experiments was 350,010 (94.84%), and in the second set 355,086 (96.22%). Overall, GDAS assigned a common set of 345,094 base calls (93.51%) that were called in both sets. In this replicate set, we found discordant base calls at only 10 nucleotide positions, or a between-chip error rate of 0.0027%. Of the 10 discordant base pairs, eight were the same miscalls as in the within-chip replicate data (Supplemental Table 2), and as stated previously, most had low quality scores. The two additional “miscalls” in the between-chip replicate data corresponded to the same Revised Cambridge Reference sequence (RCRS) nucleotide position (i.e., there was no within-chip discordance), and it is possible that these “miscalls” represent true low-level heteroplasmy in the DNA sample detected by the MitoChip. In either case, the extremely low-level genotyping error in our series of replicate experiments confirms >99.99% reproducibility of base calls both within and between chips using array-based sequencing. In fact, if repeatability were to equal accuracy, this degree of repeatability would equate with a Phred score of at least 48 [assuming a binomial error probability of P = 10 / (2*345,094) = 0. 000013039 and Phred = –10 log (P)] (Nickerson et al. 1997; Ewing and Green 1998; Ewing et al. 1998). Using this data set of 26 chips, we also wanted to ascertain what proportion of base positions on the MitoChip had consistently poor hybridization characteristics; thus, 477 / 28,386 (1.7%) bases generated failed signals (“N”) across all 26 samples. Of these, the majority, 243 / 477 (51%) were C residues, often in regions containing two or more consecutive C bases, reflecting an inherent pitfall of microarray chemistry for suboptimal hybridization of GC-rich oligonucleotide residues.
Table 2.
Summary of Replicate Experiments
| Total samples analyzed in replicate | 13 |
| Total chips analyzed for replicate experiments | 26 |
| Within-chip reproducibility | |
| Mitochondrial base pairs tiled in duplicate per chip | 12,935 bp |
| Total within-chip duplicate base pairs analyzed | 26*12,935 = 336,310 bp |
| Total within-chip duplicate base pairs called | 311,814 bp (92.71%) |
| Discordant calls within chips | 8 bp (0.0025%) |
| Between-chips reproducibility | |
| Total mitochondrial base pairs tiled per chip | 28,386 bp |
| Total base pairs analyzed in one set of 13 chips | 13*28,386 = 369,018 bp |
| Total base pairs assigned in first set of chips | 350,010 bp (94.84%) |
| Total base pairs assigned in second set of chips | 355,086 bp (96.22%) |
| Total base pairs assigned in both sets | 345,094 bp (93.51%) |
| Discordant base calls between chips | 10 bp (0.0028%) |
Matched Normal Tumor Samples
We then sequenced matched normal samples (lymphocyte DNA) for four primary lung cancers in our series (JHU_MITO #9, 11, 12, and 13). A somatic mutation was defined as either a “homozygote” (i.e., homoplasmic) or a “heterozygote” (i.e., heteroplasmic) variation in the tumor that was also confirmed in the duplicate location on the same array, with wild-type (i.e., reference) genotype in the normal sample. Using these criteria, two of four (50%) tumor samples (JHU_MITO #9 and 12) harbored at least one mitochondrial coding sequence mutation (Supplemental Table 3), whereas the remaining two tumors did not demonstrate any somatic mutations, although comparison with lymphocyte DNA sequences revealed multiple germline variations compared to the RCRS (data not shown). A notable feature of this analysis was the considerable variability in the frequency of somatic mitochondrial mutations in the tumors analyzed (all four are primary lung cancers), with one case harboring as many as 24 mitochondrial mutations, and two cases harboring none.
Two important conclusions can be derived from this analysis of matched normal and tumor samples. First, as stated, the MitoChip does not contain the D-loop, a region frequently mutated in human cancers (Fliss et al. 2000; Sanchez-Cespedes et al. 2001). This raises a justifiable concern that the absence of mitochondrial mutations in a MitoChip assay could simply reflect lack of sampling of the D-loop. However, in both cases without mitochondrial coding sequence mutations (JHU_MITO #11 and 13), comparison with previous sequence data of the entire mitochondrial genome confirmed the absence of mutations in the D-loop as well (D. Sidransky, unpubl.). Conversely, both cases where we detected coding sequence mutations also demonstrated D-loop mutations. Although our sample size is small, we predict that the proportion of cancers mutated only in the mitochondrial D-loop is unlikely to be a significant number, and by sampling the entire mitochondrial coding sequence, we greatly enhance the ability to detect at least one somatic mutation. The second observation relates to the presence of as many as 24 somatic mutations in a single tumor, providing additional evidence for a “mitochondrial DNA mutator” phenotype (Habano et al. 1999, 2000; Richard et al. 2000; Bianchi et al. 2001), akin to MSI+ cancers with mismatch repair deficiency (Boland et al. 1998). Previous studies with limited mitochondrial DNA sequencing provided conflicting results about the correlation between nuclear and mitochondrial genomic instability (Habano et al. 1999, 2000; Richard et al. 2000; Bianchi et al. 2001; Jeronimo et al. 2001); the existence of such an association, and the overall functional significance of a “mitochondrial DNA mutator” phenotype, needs to be borne out in a larger series of cases using known MSI+ and MSI–cancers. Moreover, other repair deficits could be manifest predominantly in the mitochondrial genome while sparing the nuclear genome.
Validation of Array-Based Sequencing Data
Several cancers included in this study have been previously analyzed by direct sequencing for mitochondrial DNA mutations, either within the entire 16.5-kb mitochondrial DNA sequence, or in selected mitochondrial genes (e.g., NAD4 and 5). A subset of these results have been published (Polyak et al. 1998; Fliss et al. 2000; Jones et al. 2001), and the remainder (D. Sidransky, pers. comm.) are part of ongoing studies in mitochondrial DNA mutations in cancer. Direct sequencing data, either by manual autoradiographic or by automated fluorescent techniques, were available for 18/35 cancers included in the present study, and the mitochondrial DNA mutations detected in these previous analyses were compared to the MitoChip results. For those nucleotide positions where discordance was noted between previous sequence and microarray data, manual autoradiographic sequencing was performed as an additional validation step, using the same PCR-amplified product as that used for the MitoChip hybridization (Supplemental Table 4). Overall, 63 mitochondrial DNA mutations were previously identified by conventional direct sequencing. Of these mutations, array-based sequencing confirmed 54 (86%), whereas nine nucleotide positions were discordant. Repeat manual sequencing confirmed the microarray data in six of nine instances, whereas at three nucleotide positions, previous sequence data were found to be correct. Overall, using the concordance of any two genotyping attempts as a “gold standard,” array-based sequencing correctly identified 60/63 (95%) previously reported mutations, but miscalled 3/63 (5%). Curiously, two of the three mutations miscalled by the MitoChip were at the same base pair position (11299 in the ND4 gene).
As an additional validation of microarray data, we performed manual sequencing in order to confirm a subset of previously unreported “new” mutations identified by array-based sequencing in the two primary lung cancers (JHU_MITO #9 and 12; Supplemental Table 5). Thus, Fliss et al. (2000) reported two mitochondrial DNA mutations (at nucleotides 5521 and 12345) in JHU_MITO #9, and as stated above (Supplemental Table 4), both mutations were confirmed by array-based sequencing. However, the MitoChip identified two additional, previously unreported somatic mutations at nucleotide positions 1463 (G→A) and 12308 (A→A+G), respectively. We tested and confirmed the heteroplasmic 12308 mutation by manual sequencing (the homoplasmic 1463 mutation was not examined). Similarly, the MitoChip identified 24 somatic mutations in JHU_MITO #12, although only the D-loop mutation data were previously published on this case. We performed manual sequencing for a subset of the unreported mitochondrial coding sequence mutations in this tumor. As shown in Supplemental Table 5, manual sequencing confirmed all seven of seven mutations identified by the MitoChip.
Serial Dilution Experiments
In order to the test the utility of the MitoChip in detecting mutations in heterogeneous clinical samples, we performed serial dilution experiments using one primary lung cancer (JHU_MITO #12) and its matched normal DNA sample. This particular case was chosen because there were a large number of somatic mutations in the tumor (Table 3). Five “mixed” samples, with a tumor:normal (T:N) DNA ratio of 1:1, 1:2, 1:4, 1:9, and 1:49 were prepared, PCR-amplified and hybridized to the MitoChip for comparison of sequence data with the pure tumor population. Strikingly, a heterozygous signal, corresponding to an aberrant clonal population, was detectable at as many as 19/24 (79%) mutated nucleotide positions in the 50-fold diluted tumor DNA sample (Table 3, Fig. 1). At five mutated nucleotide positions, no signal (“N”) was observed, reflecting the inability of the adaptive background scheme to assign a reliable genotype score for a heterozygous call at a Ttotal quality score of 30. However, of these five positions, the aberrant population was still detectable at the 10-fold diluted tumor DNA sample in three positions at a Ttotal quality score of 30; only two nucleotide positions “failed” dilution experiments (no signal in any mixed sample), and both were heteroplasmic even in the pure tumor population. Thus, the MitoChip demonstrated a powerful ability to detect an aberrant clonal population (i.e., bearing a somatic mitochondrial mutation) in manifold mixed and diluted samples, supporting its potential application as a cancer detection tool in clinical samples.
Table 3.
Serial Dilution Experiments for Mutation Detection With MitoChip
| RCRS base # | Sequence in lymphocyte DNA | Sequence in pure tumor DNA | Sequence in 1:50 diluted tumor DNA |
|---|---|---|---|
| 750 | G | A | A + G |
| 1007 | G | A | A + G |
| 2000 | C | T | C + T |
| 2352 | T | C | C + T |
| 4655 | G | A | A + G |
| 5262 | G | A | A + G |
| 5421 | G | A + G | No calla |
| 6524 | T | C | No calla |
| 8701 | A | G | A + G |
| 9540 | T | C | C + T |
| 9547 | G | G + T | No call |
| 10398 | A | G | A + G |
| 10873 | T | C | No calla |
| 11180 | G | A + G | A + G |
| 11350 | A | G | A + G |
| 12248 | A | G | A + G |
| 12705 | C | T | C + T |
| 13101 | A | C | A + C |
| 13197 | C | T | C + T |
| 13650 | C | C + G | No call |
| 14212 | T | C | C + T |
| 14766 | C | T | C + T |
| 15812 | G | A | A + G |
| 15904 | T | C | C + T |
Heterozygous call is present at these three nucleotide positions (5421 = A + G; 6524 = C + T; 10873 = C + T) in the 10-fold diluted tumor DNA sample
Figure 1.
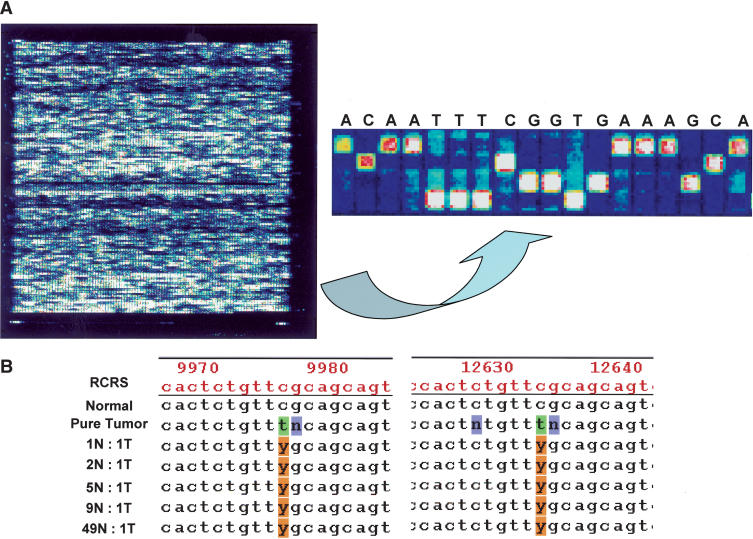
(A, left panel) Pictorial depiction of MitoChip hybridization data (.DAT file) after scanning in an Affymetrix Microarray suite; control tiles at the four corners of the chip permit automated grid alignment, which generates a .CEL file for subsequent batch analysis in GDAS. (right panel) Higher-magnification view of the tiling pattern on the MitoChip demonstrating the four alternative oligonucleotides (25mers with the 13th base being A, C, G, or T) for each RCRS base position, and the sequence-specific hybridization occurring at each position. All base calls are homozygous in the illustrated panel. (B) Serial dilution experiments performed with a primary lung cancer (JHU_MITO #12) and its corresponding normal DNA sample demonstrate the ability of the MitoChip to detect an aberrant clonal population in 50-fold-diluted tumor DNA. The sequence output is generated in GDAS, and the mutation detected corresponds to RCRS13197 C>T mutation in the tumor sample (Table 3). As illustrated, the mutation is detected at both positions for RCRS13197 on the MitoChip. Note that nucleotides immediately 3′ and 5′ of the mutated base position often result in a “no call” (N) due to poor hybridization quality scores caused by the mismatched base. The numbers depicted on the GDAS chromatogram correspond to the tiled base positions on the MitoChip and not the actual RCRS position. A convenient Excel-based conversion table linking the duplicate MitoChip positions to the RCRS nucleotide position is available from the authors on request. (N, normal; T, Tumor; Y, C+T in IUPAC ambiguity code)
Analysis of Mitochondrial Mutations in Clinical Samples
One of our eventual goals for the MitoChip is its use as a tool for early detection; therefore, we analyzed a series of matched primary tumors and body fluid specimens, in order to determine whether chip-based mutation detection is a feasible strategy in clinical samples. DNA from a total of 10 primary tumors (five bladder cancers and five pancreatic cancers) and matched body fluid specimens (urine and pancreatic juice, respectively) were examined. We were able to successfully amplify 5–6.5-kb-long PCR fragments from all but one pancreatic juice sample, and thus, nine tumor-fluid pairs (n=18 samples) were used in the final analyses. The average GDAS call rate for the DNA obtained from the fluid samples was essentially identical to that of the primary tumor DNA (94.1% vs. 94.4%, respectively), demonstrating that DNA obtained from these clinical samples is feasible for use in chip-based assays. Lymphocyte DNA was not available for comparison in the nine cases; however, because germline heteroplasmy is a rare event, we considered any heteroplasmic base call in the tumor DNA as prima facie evidence of a somatic mitochondrial mutation. Using this criterion, six of nine (66%) body fluid samples demonstrated one or more heteroplasmic mutations (range 1–3 mutations per case) that were identical to the sequence change seen in the matched primary tumor DNA (Fig. 2). By sample type, four of five (80%) urine samples and two of four (50%) pancreatic juice specimens demonstrated identical heteroplasmic mutations as seen in the primary tumor DNA. In two pancreatic cancer cases, no heteroplasmic sequences were seen in either the body fluid or the corresponding primary tumor DNA, and in one bladder cancer, we were unable to detect heteroplasmic sequences in the urine despite their presence in the primary tumor DNA.
Figure 2.
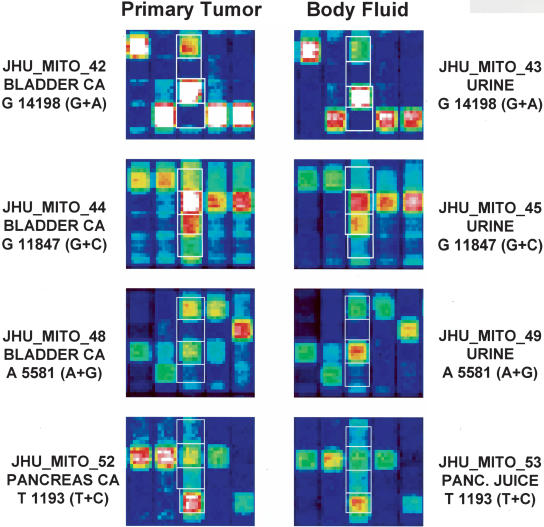
Analysis of primary tumors and matched body fluid samples (urine or pancreatic juice) obtained from patients with bladder cancer (n=5) and pancreatic cancer (n=4). In each case illustrated, a heteroplasmic mitochondrial mutation identical to one seen in the primary tumor DNA (left hemi-panel) is also found in the fluid DNA (right hemi-panel). The JHU_MITO ID number, type of specimen, the mitochondrial nucleotide position involved, and the specific base change are detailed for each tumor-fluid pair illustrated.
DISCUSSION
Hybridization-based methodologies for high-throughput mutational analysis using oligonucleotide microarrays have been developed recently. Light-directed combinatorial chemical synthesis approaches enable the manufacture of high-density microarrays of >105 distinct species, typically 25 nucleotides in length, on 1.2 × 1.2 cm2 glass surfaces (Fodor et al. 1991). Hacia et al. (1998) exploited the power of chip-based mutation screen by analyzing 62 coding exons of the ATM gene for all possible sequence variations in the heterozygous state, using a highly parallel iterative strategy for hybridization. In a blinded mutational analysis scanning >200 kb of 22 genomic DNA samples, they accurately detected 17 of 18 distinct heterozygotes and eight of eight distinct homozygous sequence variants in the assayed region. These included missense mutations as well as frameshift insertion-deletions. In addition, five previously unreported sequence changes, not found by other mutational scanning technologies on the same samples, were detected by chip-based sequencing. Similarly, Wen et al. (2000) analyzed 108 ovarian tumors with the p53 GeneChip microarray (Affymetrix, Inc.) that interrogates ∼1262 base pairs of exons 2–11 of TP53, and compared the results of chip-based sequencing with gel-based DNA analysis. The p53 microarray detected 14 additional mutations not identified by gel-based sequencing. Comparable results were obtained by Ahrendt et al. (1999) in the analysis of TP53 mutations in lung cancers and by Wikman et al. (2000) in bladder cancers. For example, Ahrendt et al. (1999) reported that as many as 14 of the 58 (24%) tumors determined to be wild-type by direct sequencing had a mutation detected by the p53 microarray.
A major pitfall of using the mitochondrion as a reliable cancer detection tool has been the inability to develop an accurate yet high-throughput sequencing technique for mitochondrial mutation detection. Current direct sequencing platforms, including manual or automated fluorescent techniques, have several technological limitations and are time-consuming (Ahrendt et al. 1999). Although simple STS-based strategies have been developed for the hypervariable D310 region of the mitochondrial D-loop as a cancer detection tool, this assay is limited by its low frequency of occurrence in some of the major human cancer types (e.g., ovarian cancer, 0%; prostate, 0%; lung, 16%; thyroid, 6%; Sanchez-Cespedes et al. 2001; Tong et al. 2003). Chee et al. (1996) described a “first-generation” mitochondrial sequencing chip that had less than half the number of features on the current MitoChip (∼130,000 vs. ∼300,000), required a greater number of PCR assays (including an in vitro transcription step), and did not use an adaptive background scheme for assigning genotypes; in addition, no within- or between-chip reproducibility studies or dilution experiments were conducted to validate the performance of the chip, especially in the context of cancer diagnosis. We have developed a second-generation mitochondrial sequencing microarray that encompasses the entire mitochondrial coding sequence. The MitoChip has multiple inherent advantages that make it an ideal platform for mutation detection: (1) we minimized the DNA requirements—as little as 300 ng of genomic DNA is sufficient for the assay; (2) we considerably reduced the number of PCR reactions (currently 12–32 in most dye terminator or manual sequencing protocols)—only three long PCR reactions can amplify the mitochondrial coding sequence; (3) we utilized an adaptive background genotype-calling scheme for assigning genotypes—this scheme is entirely automated, yet allows a sliding threshold quality score that can be varied by the operator for filtering genotype calls, and significantly reduces the possibility of false positive or false negative heterozygous calls at a given nucleotide position; and (4) in addition to both strands of the entire coding sequence being arrayed once, the MitoChip also contains both strands from 84% of the coding sequence in duplicate; thus effectively, for these 12,395 bp, a genotype assigned at a given position represents the combined data from four independent hybridization events (two forward and two reverse strands)—this redundancy imparts considerable stringency in mutation detection. Finally, we have greatly reduced the time frame required to perform mitochondrial mutation analysis—75 tumors encompassing >2 million bases of mitochondrial DNA were sequenced and analyzed by a single investigator in 10 weeks; this effort would have consumed many months of an investigator's time using current direct sequencing platforms.
The ability of the MitoChip to detect identical heteroplasmic DNA mutations in body fluid specimens as are present in the matched primary tumor samples argues strongly for the use of chip-based technology as a tool for early detection of cancer in clinical samples (Fig. 2). Although the percentage of tumor cells present in such clinical samples is not known, the dilution experiments we performed demonstrate that the MitoChip can easily detect an aberrant clonal population of tumor cells with as little as 2% contribution to the admixture of normal and tumor DNA (Table 3); thus heterogeneous biological samples such as bodily fluids, lavage specimens, fine-needle aspirates, or biopsies can potentially be analyzed for cancer-associated mitochondrial DNA mutations. In addition, large series of a range of human cancers can now be conveniently analyzed to determine the existence of either tumor type-specific changes that could be used in diagnostics (such as determination of the source of a metastasis of unknown primary), or even cancer-specific changes that could be used as a “universal” indicator of altered cellular phenotype.
In summary, our findings show that mitochondrial sequencing can be a high-throughput tool for testing in clinical samples. Although cancer detection using this platform remains a major objective, we also envision that the MitoChip will greatly facilitate the conduct of large-scale epidemiologic studies of the human mitochondrial genome, the study of mitochondrial genotype-phenotype associations, and our understanding of the pathogenetic basis of nonneoplastic diseases linked to mitochondrial dysfunction.
METHODS
Design of Human Mitochondrial Custom Resquencing Array
The human mitochondrial Revised Cambridge Reference sequence as modified by Andrews et al. (1999; available online at MITOMAP: A Human Mitochondrial Genome Database, http://www.mitomap.org, 2003) was used as the reference DNA for selection of genomic regions. As stated, both strands of the entire mitochondrial coding region (nucleotides 573 through 16024, i.e., 15,446 bp) are tiled once on the Custom Reseq array, and the forward and reverse strands of an additional 12,935 bp of the mitochondrial DNA (i.e., the coding region minus 12S and 16S RNA sequences) are tiled in duplicate. The mitochondrial regulatory D-loop (nucleotides 16024 through 576) is not tiled on the MitoChip for two reasons: (1) this region is particularly GC-rich, which often leads to suboptimal hybridization on oligonucleotide microarrays (see Results section above), and (2) the most common mutation observed in this the D-loop are insertion/deletion mutations of a poly-C tract (known as D310); again, this class of mutations is usually poorly detected by current microarray hybridization chemistry. The MitoChip was fabricated using standard photolithography and solid-phase DNA synthesis by Affymetrix as described (Pease et al. 1994; Lipshutz et al. 1999). Briefly, each chip consists of ∼300,000 “features” with a feature size of 24×20 μm. A feature consists of ∼106 copies of a 25-bp-long oligonucleotide probe of defined sequence. To query any given site from the human mitochondrial reference sequence, four features are tiled on the MitoChip. The four features differ only by the central or 13th base, which consists of each of the four possible nucleotides. In the process of scanning the MitoChip, the scanner measures the fluorescence intensity for each feature by dividing each feature into 56 equalsized pixels (Cutler et al. 2001). The 26 pixels located at the border of the features are masked, and their fluorescence intensity values are not used in any subsequent calculations. The fluorescence intensity at the remaining 30 pixels constitutes the raw data measured by the detector. The raw data are then used for generating the genotype call at each site on the mitochondrial reference sequence for both the forward and reverse strands.
Cancer and Normal DNA Samples
The MitoChip was evaluated using DNA from 44 cancer samples (cell lines and primary tumors) obtained from a variety of anatomic sites. The list of tumors included pancreatic cancer (six cell lines, one xenograft, four primary cancers), lung cancer (six primary tumors), colon cancer (five cell lines), bile duct cancer (nine cell lines), bladder cancer (five primary cancers), breast, cervix, urothelial, thyroid and lung cancers (one cell line each), osteosarcoma (one cell line), melanoma (one cell line), and lymphoma (one cell line; see Supplemental Table 1). For all primary cancers, DNA was extracted from cryostat-embedded snap-frozen sections. Matched normal (lymphocyte DNA) samples were assayed for four primary lung cancers (JHU_MITO # 9, 11, 12, and 13). DNA was also extracted from 10 body fluid samples, including five urine and five pancreatic juice specimens, and analyzed in conjunction with the matched primary tumors; all urine and pancreatic juice specimens were obtained peri-operatively from patients undergoing surgical resection for a diagnosis of cancer. DNA from one pancreatic juice failed to amplify, and hence nine pairs of tumor-fluid samples were eventually analyzed with the MitoChip (see Supplemental Table 1). To assess reproducibility of MitoChip data, 13 cancer cell lines (Supplemental Table 1) were analyzed in independent replicate experiments, beginning with the PCR amplification step. Finally, serial dilution experiments were performed using one primary lung cancer (JHU_MITO #12) and its matched normal DNA sample in ratios of 1:1, 1:2, 1:4, 1:9, and 1:49 tumor:normal DNA, in order to determine the ability of the MitoChip to detect mutations in heterogeneous samples. Thus, a total of 75 individual MitoChip assays were performed.
Selection of Primers and PCR Amplification
Primers for PCR amplification were selected using the Amplify 1.2 program (http://engels.genetics.wisc.edu/amplify/) as described (Cutler et al. 2001). The entire mitochondrial DNA sequence was amplified in three overlapping long PCR fragments, each containing 100 ng of genomic DNA (no enrichment for mitochondrial DNA fraction was performed). Amplification was accomplished in 100-μL PCRs carried out in thin-walled polypropylene plates using the high-fidelity TaKaRa LA Taq (TaKaRa Biomedicals) as described (Cutler et al. 2001). Primer sequences and PCR conditions are freely available on request. As a control for PCR amplification and subsequent hybridization, a 7.5-kb plasmid DNA (Tag IQ-EX template) was amplified concomitantly with the test samples, using forward and reverse primers included in the CustomSeq Control kit (Affymetrix). Overall, long PCR amplifications for the three fragments were successful in ∼99% of samples on the first or second attempt (as stated above, one of five pancreatic juice samples did not amplify, although the matched tumor did; thus, counting this eventually discarded pair, 76/77 samples amplified successfully on the first or second attempt). Residual primers and nucleotides were removed using a QIAQuick PCR Clean up kit (QIAGEN), and the purified PCR products were resuspended in 30 μL volume of EB buffer (Affymetrix). The yield of each PCR reaction (ng/μL) was determined spectrophotometrically, and the specificity of the reaction was confirmed by agarose gel electrophoresis.
PCR Pooling, DNA Fragmentation, Labeling, and Chip Hybridization
To obtain optimal performance across the microarray, we pooled equimolar amounts from the three amplified fragments to ensure that an equal number of targets existed for each probe. DNA fragmentation was performed with a 15-μL master mix containing Affymetrix fragmentation reagent (calculated as 0.2 U DNAse I/μg DNA), 5 μL OnePhorAll buffer (Amersham Life Sciences), and EB Buffer. Fragmented DNA was labeled by adding 1.5 μL Biotin-N6-ddATP (Perkin Elmer Life Sciences) and 1 μL of 20 U/μL rTdT enzyme (New England Biolabs). Prehybridization, hybridization, washing, and scanning of the MitoChip were performed as described in the Affymetrix CustomSeq Resequencing protocol. Following hybridization, the chips were washed on the Affymetrix fluidics station using the preprogrammed CustomSeq Resequencing wash protocol [Affymetrix Microarray Suite (MAS) version 5.2 Beta]. The MAS 5.2 Beta performs automatic grid alignment (Cutler et al. 2001), an essential prerequisite for accurate interpretation of microarray hybridization data. The raw pixel data (.DAT file) generated is thus digitized into a .CEL file for subsequent batch analysis.
Automated Batch Analysis of Microarray Data
Batch analysis of .CEL files is performed on the Affymetrix GeneChip DNA Analysis Software (GDAS) version 1.0, using a modification of a previously described adaptive background genotype-calling scheme (ABACUS; Cutler et al. 2001). Briefly, the adaptive background scheme uses an objective statistical framework to assign each genotype call a “quality score.” The algorithm develops a series of statistical models under the assumption of the presence or absence of various genotypes in the target sample. The likelihood of each statistical model for a given genotype is calculated independently for both the forward and reverse strands, and is combined for the overall likelihood of the model. A “quality score,” which is the difference between the log (base 10) likelihood of the best fitting and the second-best fitting model is assigned to each genotype on the sequencing array. A site genotype is “called” when one model fits the data sufficiently better than all other models; genotypes deemed as unreliable are designated as N. The optimum total threshold quality score (Ttotal) was determined empirically to be 30 (Cutler et al. 2001), and this threshold score was used in the present study as well. As detailed in the original ABACUS description, the “adaptive” nature of this scheme uses hybridization data from multiple arrays across multiple runs to factor in the uneven background occurring due to differing levels of cross-hybridization at each site, and thus, significantly reduce the occurrence of miscalls. Once the batch analysis is completed, the GDAS software generates a report containing individual and total numbers and percentages of base calls within the batch, and a detailed case-by-case list of genotype variations vis-à-vis the reference sequence.
Manual Sequencing of Mitochondrial DNA
In order to validate a subset of mutations identified by the MitoChip, manual autoradiographic sequencing was performed using previously described primers and conditions on a slab gel platform (Polyak et al. 1998; Fliss et al. 2000).
Acknowledgments
We thank Drs. Bert Vogelstein, Victor Velculescu, Jessa Jones, and Scott Kern for providing DNA samples used in this study, and Drs. Michael Zwick and David Cutler for their advice on microarrays. Supported by the NIH Specialized Programs of Research Excellence in Gastrointestinal Cancer (CA62924) Pilot Projects Grant (to A.M.), by a Maryland State Cigarette Restitution Fund grant (A.M.), and by a generous grant from the Michael Rolfe Foundation for Pancreatic Cancer Research. A.C. is a paid member of the Affymetrix, Inc., Scientific Advisory Board. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2228504.
Footnotes
[Supplemental material is available online at www.genome.org. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: B. Vogelstein, V. Velculescu, J. Jones, and S. Kern.]
References
- Ahrendt, S.A., Halachmi, S., Chow, J.T., Wu, L., Halachmi, N., Yang, S.C., Wehage, S., Jen, J., and Sidransky, D. 1999. Rapid p53 sequence analysis in primary lung cancer using an oligonucleotide probe array. Proc. Natl. Acad. Sci. 96: 7382-7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, R.M., Kubacka, I., Chinnery, P.F., Lightowlers, R.N., Turnbull, D.M., and Howell, N. 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23: 147. [DOI] [PubMed] [Google Scholar]
- Bianchi, N.O., Bianchi, M.S., and Richard, S.M. 2001. Mitochondrial genome instability in human cancers. Mutat. Res. 488: 9-23. [DOI] [PubMed] [Google Scholar]
- Boland, C.R., Thibodeau, S.N., Hamilton, S.R., Sidransky, D., Eshleman, J.R., Burt, R.W., Meltzer, S.J., Rodriguez-Bigas, M.A., Fodde, R., Ranzani, G.N., et al. 1998. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 58: 5248-5257. [PubMed] [Google Scholar]
- Chee, M., Yang, R., Hubbell, E., Berno, A., Huang, X.C., Stern, D., Winkler, J., Lockhart, D.J., Morris, M.S., and Fodor, S.P. 1996. Accessing genetic information with high-density DNA arrays. Science 274: 610-614. [DOI] [PubMed] [Google Scholar]
- Chen, J.Z., Gokden, N., Greene, G.F., Mukunyadzi, P., and Kadlubar, F.F. 2002. Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res. 62: 6470-6474. [PubMed] [Google Scholar]
- Copeland, W.C., Wachsman, J.T., Johnson, F.M., and Penta, J.S. 2002. Mitochondrial DNA alterations in cancer. Cancer Invest. 20: 557-569. [DOI] [PubMed] [Google Scholar]
- Cutler, D.J., Zwick, M.E., Carrasquillo, M.M., Yohn, C.T., Tobin, K.P., Kashuk, C., Mathews, D.J., Shah, N.A., Eichler, E.E., Warrington, J.A., et al. 2001. High-throughput variation detection and genotyping using microarrays. Genome Res. 11: 1913-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S.M., Traverso, G., Johnson, C., Geng, L., Favis, R., Boynton, K., Hibi, K., Goodman, S.N., D'Allessio, M., Paty, P., et al. 2001. Detecting colorectal cancer in stool with the use of multiple genetic targets. J. Natl. Cancer Inst. 93: 858-865. [DOI] [PubMed] [Google Scholar]
- Ewing, B. and Green, P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8: 186-194. [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175-185. [DOI] [PubMed] [Google Scholar]
- Fliss, M.S., Usadel, H., Caballero, O.L., Wu, L., Buta, M.R., Eleff, S.M., Jen, J., and Sidransky, D. 2000. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 287: 2017-2019. [DOI] [PubMed] [Google Scholar]
- Fodor, S.P., Read, J.L., Pirrung, M.C., Stryer, L., Lu, A.T., and Solas, D. 1991. Light-directed, spatially addressable parallel chemical synthesis. Science 251: 767-773. [DOI] [PubMed] [Google Scholar]
- Greenlee, R.T., Murray, T., Bolden, S., and Wingo, P.A. 2000. Cancer statistics, 2000. CA Cancer J. Clin. 50: 7-33. [DOI] [PubMed] [Google Scholar]
- Ha, P.K., Tong, B.C., Westra, W.H., Sanchez-Cespedes, M., Parrella, P., Zahurak, M., Sidransky, D., and Califano, J.A. 2002. Mitochondrial C-tract alteration in premalignant lesions of the head and neck: A marker for progression and clonal proliferation. Clin. Cancer Res. 8: 2260-2265. [PubMed] [Google Scholar]
- Habano, W., Sugai, T., Yoshida, T., and Nakamura, S. 1999. Mitochondrial gene mutation, but not large-scale deletion, is a feature of colorectal carcinomas with mitochondrial microsatellite instability. Int. J. Cancer 83: 625-629. [DOI] [PubMed] [Google Scholar]
- Habano, W., Sugai, T., Nakamura, S.I., Uesugi, N., Yoshida, T., and Sasou, S. 2000. Microsatellite instability and mutation of mitochondrial and nuclear DNA in gastric carcinoma. Gastroenterol. 118: 835-841. [DOI] [PubMed] [Google Scholar]
- Hacia, J.G. 1999. Resequencing and mutational analysis using oligonucleotide microarrays. Nat. Genet. 21: 42-47. [DOI] [PubMed] [Google Scholar]
- Hacia, J.G., Sun, B., Hunt, N., Edgemon, K., Mosbrook, D., Robbins, C., Fodor, S.P., Tagle, D.A., and Collins, F.S. 1998. Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res. 8: 1245-1258. [DOI] [PubMed] [Google Scholar]
- Hibi, K., Nakayama, H., Yamazaki, T., Takase, T., Taguchi, M., Kasai, Y., Ito, K., Akiyama, S., and Nakao, A. 2001. Detection of mitochondrial DNA alterations in primary tumors and corresponding serum of colorectal cancer patients. Int. J. Cancer 94: 429-431. [DOI] [PubMed] [Google Scholar]
- Hirsch, F.R., Franklin, W.A., Gazdar, A.F., and Bunn Jr., P.A. 2001. Early detection of lung cancer: Clinical perspectives of recent advances in biology and radiology. Clin. Cancer Res. 7: 5-22. [PubMed] [Google Scholar]
- Jeronimo, C., Nomoto, S., Caballero, O.L., Usadel, H., Henrique, R., Varzim, G., Oliveira, J., Lopes, C., Fliss, M.S., and Sidransky, D. 2001. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 20: 5195-5198. [DOI] [PubMed] [Google Scholar]
- Jones, J.B., Song, J.J., Hempen, P.M., Parmigiani, G., Hruban, R.H., and Kern, S.E. 2001. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 61: 1299-1304. [PubMed] [Google Scholar]
- Liotta, L. and Petricoin, E. 2000. Molecular profiling of human cancer. Nat. Rev. Genet. 1: 48-56. [DOI] [PubMed] [Google Scholar]
- Lipshutz, R.J., Fodor, S.P., Gingeras, T.R., and Lockhart, D.J. 1999. High density synthetic oligonucleotide arrays. Nat. Genet. 21: 20-24. [DOI] [PubMed] [Google Scholar]
- Liu, M.R., Pan, K.F., Li, Z.F., Wang, Y., Deng, D.J., Zhang, L., and Lu, Y.Y. 2002. Rapid screening mitochondrial DNA mutation by using denaturing high-performance liquid chromatography. World J. Gastroenterol. 8: 426-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, L., Schoenberg, M.P., Scicchitano, M., Erozan, Y.S., Merlo, A., Schwab, D., and Sidransky, D. 1996. Molecular detection of primary bladder cancer by microsatellite analysis. Science 271: 659-662. [DOI] [PubMed] [Google Scholar]
- Medintz, I.L., Paegel, B.M., Blazej, R.G., Emrich, C.A., Berti, L., Scherer, J.R., and Mathies, R.A. 2001. High-performance genetic analysis using microfabricated capillary array electrophoresis microplates. Electrophoresis 22: 3845-3856. [DOI] [PubMed] [Google Scholar]
- Nickerson, D.A., Tobe, V.O., and Taylor, S.L. 1997. PolyPhred: Automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 25: 2745-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto, S., Yamashita, K., Koshikawa, K., Nakao, A., and Sidransky, D. 2002. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin. Cancer Res. 8: 481-487. [PubMed] [Google Scholar]
- Okochi, O., Hibi, K., Uemura, T., Inoue, S., Takeda, S., Kaneko, T., and Nakao, A. 2002. Detection of mitochondrial DNA alterations in the serum of hepatocellular carcinoma patients. Clin. Cancer Res. 8: 2875-2878. [PubMed] [Google Scholar]
- Parrella, P., Xiao, Y., Fliss, M., Sanchez-Cespedes, M., Mazzarelli, P., Rinaldi, M., Nicol, T., Gabrielson, E., Cuomo, C., Cohen, D., et al. 2001. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 61: 7623-7626. [PubMed] [Google Scholar]
- Pease, A.C., Solas, D., Sullivan, E.J., Cronin, M.T., Holmes, C.P., and Fodor, S.P. 1994. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl. Acad. Sci. 91: 5022-5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petricoin, E.F., Ardekani, A.M., Hitt, B.A., Levine, P.J., Fusaro, V.A., Steinberg, S.M., Mills, G.B., Simone, C., Fishman, D.A., Kohn, E.C., et al. 2002. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359: 572-577. [DOI] [PubMed] [Google Scholar]
- Polyak, K., Li, Y., Zhu, H., Lengauer, C., Willson, J.K., Markowitz, S.D., Trush, M.A., Kinzler, K.W., and Vogelstein, B. 1998. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 20: 291-293. [DOI] [PubMed] [Google Scholar]
- Richard, S.M., Bailliet, G., Paez, G.L., Bianchi, M.S., Peltomaki, P., and Bianchi, N.O. 2000. Nuclear and mitochondrial genome instability in human breast cancer. Cancer Res. 60: 4231-4237. [PubMed] [Google Scholar]
- Sanchez-Cespedes, M., Parrella, P., Nomoto, S., Cohen, D., Xiao, Y., Esteller, M., Jeronimo, C., Jordan, R.C., Nicol, T., Koch, W.M., et al. 2001. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 61: 7015-7019. [PubMed] [Google Scholar]
- Tong, B.C., Ha, P.K., Dhir, K., Xing, M., Westra, W.H., Sidransky, D., and Califano, J.A. 2003. Mitochondrial DNA alterations in thyroid cancer. J. Surg. Oncol. 82: 170-173. [DOI] [PubMed] [Google Scholar]
- Traverso, G., Shuber, A., Levin, B., Johnson, C., Olsson, L., Schoetz Jr., D.J., Hamilton, S.R., Boynton, K., Kinzler, K.W., and Vogelstein, B. 2002. Detection of APC mutations in fecal DNA from patients with colorectal tumors. N. Engl. J. Med. 346: 311-320. [DOI] [PubMed] [Google Scholar]
- Wen, W.H., Bernstein, L., Lescallett, J., Beazer-Barclay, Y., Sullivan-Halley, J., White, M., and Press, M.F. 2000. Comparison of TP53 mutations identified by oligonucleotide microarray and conventional DNA sequence analysis. Cancer Res. 60: 2716-2722. [PubMed] [Google Scholar]
- Wikman, F.P., Lu, M.L., Thykjaer, T., Olesen, S.H., Andersen, L.D., Cordon-Cardo, C., and Orntoft, T.F. 2000. Evaluation of the performance of a p53 sequencing microarray chip using 140 previously sequenced bladder tumor samples. Clin. Chem. 46: 1555-1561. [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.mitomap.org; 2003, MITOMAP: A Human Mitochondrial Genome Database. [DOI] [PMC free article] [PubMed]
- http://engels.genetics.wisc.edu/amplify/; the Amplify 1.2 program.