

Abstract
We have used the substituted-cysteine accessibility method (SCAM) to map the residues in the sixth membrane-spanning segment of the CB2 cannabinoid receptor that contribute to the surface of the water-accessible binding-site crevice. Using a background of the mutant C2.59S which is relatively insensitive to the methanethiosulfonate (MTS) reagents, we mutated to cysteine, one at a time, 34 consecutive residues in TMH6 of the CB2 receptor. These mutant receptors were then expressed in HEK293 cells. By incubating HEK293 cells stably transfected with CB2 receptors with the small, charged, hydrophilic, thiol-specific reagent methanethiosulfonate ethylammonium (MTSEA), [3H]CP55940 binding was significantly inhibited for six mutant receptors. All six of the mutants that reacted with MTSEA were protected from the reaction when pretreated with the cannabinoid agonist WIN55212-2, suggesting that MTSEA modification occurred within the binding crevice. Therefore the side chains of the residues at these reactive loci (V6.51, L6.52, L6.54, M6.55, L6.59 and T6.62) are on the water-accessible surface of the binding-site crevice. These residues are extracellular to the TMH6 CWXP hinge motif. The pattern of accessibility is consistent with a α-helical conformation for this segment of TMH6. Molecular modeling studies performed in the context of the CB2 model show that V6.51, L6.52, L6.54, M6.55, L6.59 and T6.62 face into the CB2 binding pocket, further confirming our SCAM results. These results are similar to the accessibility patterns determined by SCAM studies of TMH6 in the opioid and dopamine D2 receptors.
Graphical Abstract

Cannabinoid receptors are members of the large family of G protein-coupled receptors (GPCRs). To date, two cannabinoid receptors, CB1 and CB2, have been cloned. In 1990, a complementary DNA from a rat cerebral cortex cDNA library that encodes the first cannabinoid receptor subtype (CB1) was cloned (1). Subsequently, the sequences of an amino terminus variant CB1 receptor (2), as well as the human and mouse CB1 sequences were reported (3, 4). The second cannabinoid receptor subtype, CB2, was first cloned in 1993 from a human promyelocytic leukemia cell HL60 cDNA library (5). The CB2 receptor in both rat (6, 7) and mouse (8) has been cloned as well. The CB1 receptor is located in the central nervous system as well as in the peripheral nervous system whereas the CB2 receptor has been found almost uniquely in immune cells (9, 10, 11). This distribution suggests a possible role for the CB2 receptor in mediating immunomodulatory, but not psychoactive effects of cannabinoids, for which CB1 is the prime target. The human CB2 receptor exhibits 68% identity to the human CB1 receptor within the transmembrane regions, and 44% identity throughout the whole protein (5). CB1 and CB2 receptors couple to multiple signal transduction pathways, including adenylate cyclase (12, 13) and mitogen-activated protein kinase (14, 15).
The structure-function relationships and dynamics of protein activity can be studied using a combination of cysteine substitution and covalent modification (16). An extension of this combination, the Substituted Cysteine Accessibility Method (SCAM), was first developed by Karlin and Akabas to map the channel-lining residues in the nicotinic acetylcholine receptor (17). Thereafter, this method was adapted to map the ligand binding crevice of dopamine D2 receptor, beta 2-adrenergic receptor, opioid receptors, and other GPCRs (18, 19, 20, 21, 22, 23, 24). The development of SCAM (17) has proved to be a powerful tool to gain information about the structure and dynamics of protein domains and has been utilized not only to study G-protein coupled receptors (18, 19, 20, 21, 22, 23, 24), but also ligand-gated ion channels (25, 26), voltage-gated ion channels (27), and transporters (28). Javitch and coworkers have extensively studied the dopamine D2 receptor using the SCAM (18, 19, 21, 22, 23).
In the SCAM method, after establishing an appropriate background in which all reactive native cysteines are mutated to serine, each residue in the transmembrane helix (TMH) of interest is mutated to cysteine, one at a time, and the mutant receptors are expressed in heterologous cells. Thereafter, the mutant receptors are treated with methanethiosulfonate (MTS) reagents, and the influence of MTS reagents on ligand binding is tested. The MTS reagents react 109 times faster with ionized thiolates than with un-ionized thiols (29) and only water-accessible cysteines are likely to be ionized to a significant extent. Therefore, MTS reagents react much faster with water-accessible cysteine residues than with cysteines facing the protein interior or lipid. If ligand binding to a cysteine substitution mutant is near normal, it is assumed that the structure of the mutant receptor, especially around the binding site, is similar to that of the wildtype receptor, and thus, that the substituted cysteine lies in a similar orientation to that of the wildtype receptor. Two criteria are used for identifying an engineered cysteine as forming the surface of the binding site crevice: (i) The reaction with an MTS reagent alters binding irreversibly and (ii) This reaction is retarded by the presence of agonists or antagonists.
In this study, we have used the SCAM to systematically map the residues on the water-accessible surface of TMH6 of the CB2 cannabinoid receptor. We also performed molecular modeling studies on the CB2 receptor, to complement our SCAM studies.
EXPERIMENTAL PROCEDURES
Materials
Reagents and enzymes used for recombinant DNA experiments were bought from Promega (Madison, WI). Lipofectamine, Dulbecco’s modified Eagles’s medium (DMEM), OPTIMEM medium, fetal bovine serum (FBS), geneticin, L-glutamine, penicillin/streptomycin, and trypsin were purchased from GIBCO-BRL (Gaithersburg, MD). [3H]CP55940 was purchased from Perkin Elmer (Shelton, CT), CP55940 was provided by the National Institute of Drug Abuse (Bethesda, MD), while WIN55212-2 was obtained from RBI/Sigma (Natick, MA). Forskolin was purchased from Sigma-Aldrich Corp (St. Louis, MO) and Ro 20-1724 from BIOMOL (Philadelphia, PA). HEK293 cells were obtained from the American Type Culture Collection (Rockville, MD). The MTS derivative MTSEA was purchased from Toronto Research Chemicals (North York, ON, Canada). GF/B filters were purchased from Whatman International (Maidstone, UK). Disposable glass tubes used for cannabinoid drug dilution and ligand binding assays were purchased from BVA Scientific (San Antonio, TX). These tubes were silanized by exposing them to dichlorodimethylsilane (Sigma Chemical Co., St. Louis, MO) vapor for 4 hours under vacuum.
Numbering of amino acid residues
The amino acid numbering system in which residues are indexed relative to the most conserved residue in that transmembrane helix (TMH) was used (30). In this system, the most conserved residue in the helix is assigned a position index of ‘0.50’. Each identifier starts with the TMH number, followed by the amino acid position relative to the reference amino acid in the helix, e.g. P6.50 is the most conserved residue in helix 6 of the CB2 receptor (Figure 1) and therefore the residues adjacent to it are F6.49 and V6.51.
Figure 1. Schematic representation of the human CB2 receptor.

The amino acid residues in TMH6 mutated to cysteine in this study are enclosed in a box.
Site-directed mutagenesis
A background of the mutant C2.59S (C89S) which is relatively insensitive to MTSEA (31) was used as plasmid DNA template. This 1.8-Kb full-length human CB2 mutant gene had been subcloned into pRC/CMV (Invitrogen, San Diego, CA) to construct the expression plasmid (32, 33). The QuikChange II in vitro Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used to mutate the CB2 background mutant receptor and the sequences of the mutant plasmids were confirmed by sequencing.
Cell transfection and culture
Human Embryonic Kidney 293 (HEK293) cells were grown as monolayers in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere consisting of 5% CO2 and 95% air, at 37°C. Expression plasmids containing the wildtype and mutant cannabinoid receptors were stably transfected into HEK293 cells using lipofectamine, according to manufacturer’s instructions. Briefly, 35 mm dishes of HEK293 cells at 70–80% confluence were treated with 1.5 μg of wildtype or mutant receptor cDNA in 10 μl of lipofectamine and 1 ml of OPTIMEM. Stably transfected cells were selected in culture medium containing 800 μg/ml geneticin. Having established cell lines stably expressing wildtype and mutant CB2 receptors, the cells were maintained in growth medium containing 400 ug/ml of geneticin until needed for experiments.
Cell Harvesting
Cells were washed twice with phosphate-buffered saline (PBS) containing 8.1 mM NaH2PO4, 1.5 mM KH2PO4, 138 mM NaCl, and 2.7 mM KCl, pH 7.2, and then dissociated in PBS containing 1 mM EDTA. Dissociated cells were collected by centrifugation at 1000 g for 5 min at 4°C. The cell pellets were resuspended in binding buffer (50 mM Tris-HCl, 200 mM sucrose, 5 mM MgCl2, 2.5 mM EDTA, and 0.5 mg/ml bovine serum albumin, pH 7.4) for treatment with MTSEA and ligand binding assays.
Treatment with MTSEA
MTSEA was always freshly prepared by dissolving in distilled water at 4°C. Aliquots (180 μl) of the cell suspensions were incubated with 20 μl MTSEA at the stated concentrations for 10 min at room temperature. Cell suspensions were then diluted 50-fold with binding buffer and centrifuged at 1000 g for 5 min. After discarding the supernatant, the cell pellets were resuspended in binding buffer for ligand binding assays.
Protection by WIN55212-2 against MTSEA Reaction
For the ligand binding crevice protection experiments, the cells expressing C2.59S background and cysteine-substitution mutant CB2 receptors were pre-incubated with cannabinoid ligand WIN55212-2 at two different concentrations (0.1 μM and 1 μM) for 40 min and then treated with MTSEA for 10 min at a concentration that causes >80% inhibition of ligand binding. After diluting with binding buffer and centrifuging at 1000 g for 5 min at 4°C, the cell pellets were resuspended in fresh binding buffer and incubated for 15 min at room temperature. Thereafter, the cell pellets were again centrifuged at 1000 g for 5 min at room temperature, resuspended in fresh binding buffer, and incubated for 15 min at room temperature. This process was repeated eight times to wash off the cannabinoid ligands. At the end of washing, the cells were resuspended in binding buffer for ligand binding assays. Protection was calculated as 1-[(inhibition of binding in the presence of WIN55212-2)/(inhibition in the absence of drug)].
Ligand Binding Assay
Cannabinoid ligand dilutions were made in binding buffer and then added to silanized assay tubes. [3H]CP55940 was used as radiolabeled ligand for competition binding assays. Nonspecific binding was determined in the presence of 1 μM unlabeled CP55940. Binding assays were performed in 0.5 ml of binding buffer containing 0.1 mg/ml BSA for 60 min at 30°C. Cells were incubated with unlabelled CP55940 in binding buffer in the presence of [3H]CP55940. Free and bound radioligands were separated by rapid filtration through GF/B filters. The filters were washed three times with 3 ml of cold wash buffer (50 mM Tris-HCl, pH 7.4, consisting of 1 mg/ml of BSA). The bound [3H]CP55940 was determined by liquid scintillation counting after overnight equilibration in 5 ml of scintillation fluid (CytoScint; ICN, Costa Mesa, CA). The assays were performed in duplicate, and the results represent the combined data from at least three independent experiments.
Determination of Second-Order Rate Constant
The second-order rate constant (kMTSEA) for the reaction of MTSEA with wildtype CB2 or each mutant was estimated according to the method used by Javitch and coworkers (19). In brief, each receptor was incubated with different concentrations of MTSEA for a fixed time. The results were fit to the equation: Y = (1 − plateau) e−kct + plateau. Y is the fraction of initial binding, plateau is the fraction of residual binding at saturating concentrations of MTSEA, k is the second-order rate constant (M−1s−1), c is the concentration of MTSEA (molar), and t is the fixed time (seconds). This equation was transformed into the following form: −ln[(Y − plateau)/(1 − plateau)]/t = kc. After plotting −ln[(Y − plateau)/(1 − plateau)]/t against c, k was obtained as the slope.
Data analysis
Data from ligand binding assays were analyzed and curves generated by using the GraphPad Prism program (GraphPad Software). The IC50 and EC50 values were determined through nonlinear regression analysis performed with Prism. Kd values were estimated from competition binding experiments using the following equation: Kd =IC50−L where L is the concentration of total radioligand (34).
Computer Model of CB2
Transmembrane Helix Bundle
A model of CB2 was created using the 2.8 Å crystal structure of bovine rhodopsin (Rho) (35) as a starting point. Model creation began with an alignment of the bovine Rho and human CB2 sequences. Fourier Transform methods were employed to guide the alignment of sequences in the TMH5 region, as CB2 lacks the highly conserved Pro residue normally used as the alignment guide in this sequence region (36). Changes to the general Rho structure that were necessitated by sequence divergences included: (1) the absence of helix kinking proline residues in TMH1 and TMH5; (2) a structural role for S2.54(84) due to the lack of a GG motif in TMH2 (31); as well as, (3) the possible difference in flexibility of TMH6 (37). Complete details of the creation of our transmembrane helix model of the CB2 are provided in our recent publication (31). Below we describe the addition of loop segments and N and C termini to this TMH bundle model to complete our model of the CB2.
Modeling of Loops and N- and C-Termini
Initial Construction and Refinement
Extracellular (EC-1 H(98)-S(102); EC-2 W(172)-N(188); EC-3 L(273)-K(278)) and intracellular (IC-1 H(62)-K(67); IC-2 P(138)-L(145); IC-3 A(216)-G(233)) loops, as well as the N (M(1)-S(29)) and C-termini (H(316)-C(360)) were added to the refined CB2 TMH bundle model using the Loopy program within the protein structure modeling suite, Jackal 1.5 (Xiang, J.Z. and Honig, B., Columbia University). The Modeller program was then used to refine loop structures (38, 39).
Special Considerations for EC-2 Loop
The EC-2 loop (W172- N188) is the largest extracellular loop in CB2. The conformation of this loop was calculated using the Biased Scaled Collective Variable in Monte Carlo (SCV-MC) method (40,41). The aqueous environment of the EC-2 loop was modeled with a recently developed implicit solvent model that is based on a screened Coulomb potential formulation (the SCP-ISM) (42,43). This loop has an internal C4.66(174)-C179 disulfide bridge, one which has been suggested to be present in CB2 via mutagenesis studies (44).
Special Considerations for IC-3 Loop
NMR experiments on a peptide fragment comprised of the CB1 sequence span from the intracellular end of TMH5 to the intracellular end of TMH6, in micelles suggested that sections of the CB1 IC-3 loop are alpha helical (45). These regions consist in a short alpha helical segment from A301 to R307 followed by an elbow region (R307-I309) and an alpha helical segment (Q310 to S316) up to a Ile-Ile-Ile (I317-I319) in IC-3. The initial portion of the CB2 IC-3 loop (AHQHVAS) bears high homology with the analogous sequence in the CB1 IC-3 loop (AHSHAVR) that Ulfers and co-workers found to be helical, while the rest of the IC-3 loops are quite divergent between CB2 and CB1. Based on these results, we replaced the initial Loopy built IC-3 loop with an intracellular alpha helix (A216-S222). The rest of IC-3 loop (L223-G233) was then re-built using Loopy and optimized using Modeller.
Final Energy Minimization
The energy of the CB2 transmembrane helix bundle model was minimized using the OPLS 2005 force field in Macromodel 9.1 (Schrödinger Inc., Portland, OR). An 8.0 Å extended non-bonded cutoff (updated every 10 steps), 20.0 Å electrostatic cutoff, 4.0 Å hydrogen bond cutoff were used in each stage of the calculation. The minimization was performed in two steps. The first step consisted of Conjugate Gradient minimization in a distance dependent dielectric until a gradient of 0.1 kcal/mol was reached. This stage of the calculation allowed the helix bundle to pack. To preserve the pitch of the transmembrane helices during this minimization, a 100 kcal/mol restraint was placed on all phi and psi angles in TMHs1-7 and Hx 8. N- and C-termini together with extra- and intracellular loops were then added to the minimized TMH bundle model as described above.
In the second stage of the calculation, the added loop and termini regions were allowed to relax during a 500 step Polak-Ribier conjugate gradient (PRCG) minimization. The transmembrane helix bundle atoms were defined as non-moving fixed atoms, but their non-bonded contributions to the system were retained as implemented in Macromodel. The loop and termini regions were unrestrained. An 8.0 Å extended non-bonded cutoff (updated every 10 steps), 20.0 Å electrostatic cutoff, 4.0 Å hydrogen bond cutoff were used in this calculation and the Generalized Born/surface area (GB/SA) continuum solvation model for water available in Macromodel was employed.
Measurements of TMH Geometry
The bend, wobble and face shifts of transmembrane helices were measured using Simulaid (46).
RESULTS
Ligand binding assay – Effects of cysteine substitution on agonist binding
MTSEA has been shown to be the most effective MTS reagent for inhibition of ligand binding to the CB2 cannabinoid receptor (31). We therefore used MTSEA in this study. Using the background of the C2.59(89)S mutant which is relatively insensitive to MTSEA (31), we mutated to cysteine, one at a time, 34 consecutive residues, R6.28 – T6.62 of TMH6. The cysteine-substituted mutants as well as the background mutant, were stably expressed in HEK293 cells and the Kd values characterizing the equilibrium binding of the radiolabelled agonist, [3H]CP55940 determined. The Kd values ranged from 0.58 – 19.79 nM (Table 1). All but eight of the cysteine-substitution mutants had Kd values significantly lower than that of the C2.59S background mutant. For the eight mutants R6.28C, L6.29C, L6.41C, A6.42C, V6.43C, L6.44C, F6.49C, and V6.51C, Kd values were not significantly different from that of the C2.59S mutant. Kd values could not be determined for D6.30C, W6.48C, P6.50C and S6.58C, which lacked detectable binding.
Table 1.
Parameters of [3H]CP55940 binding to wildtype and TMH6 cysteine-substituted CB2 receptors stably expressed in HEK293 cells.
| Receptor | Kd (95%CI), nM | Kd mut/Kd C2.59S | Bmax, fmol/104 cells | n |
|---|---|---|---|---|
| CB2 | 8.98 (5.60–14.39) | 0.4 | 3.13 ± 0.35 | 6 |
| C2.59S | 22.90 (17.23–30.44) | 1.0 | 7.84 ± 0.05 | 10 |
| R6.28C | 7.29 (2.95–18.61) | 0.32 | 2.97 ± 0.29 | 6 |
| L6.29C | 10.79 (6.35–18.34) | 0.47 | 6.40 ± 0.05 | 6 |
| D6.30C | ND | 6 | ||
| V6.31C | 2.25 (1.37–3.70)* | 0.10 | 4.34 ± 0.07 | 6 |
| R6.32C | 4.33 (2.40–7.82)* | 0.20 | 5.53 ± 0.10 | 6 |
| L6.33C | 1.29 (0.90–1.86)* | 0.06 | 2.41 ± 0.07 | 6 |
| A6.34C | 1.54 (1.00–2.36)* | 0.07 | 3.04 ± 0.08 | 6 |
| K6.35C | 1.31 (0.83–2.08)* | 0.06 | 1.99 ± 0.40 | 6 |
| T6.36C | 0.58 (0.40–0.82)* | 0.03 | 1.06 ± 0.04 | 6 |
| L6.37C | 5.10 (3.26–7.89)* | 0.22 | 2.42 ± 0.04 | 6 |
| G6.38C | 5.18 (3.97–6.74)* | 0.23 | 2.35 ± 0.08 | 6 |
| L6.39C | 5.51 (3.09–9.84)* | 0.24 | 2.08 ± 0.05 | 6 |
| V6.40C | 3.61 (2.45–5.33)* | 0.16 | 1.75 ± 0.30 | 6 |
| L6.41C | 19.79 (9.84–39.80) | 0.86 | 7.50 ± 0.80 | 6 |
| A6.42C | 13.55 (7.58–24.20) | 0.59 | 7.08 ± 0.29 | 6 |
| V6.43C | 12.83 (6.78–24.26) | 0.56 | 6.64 ± 0.17 | 6 |
| L6.44C | 9.93 (5.04–19.58) | 0.43 | 4.18 ± 0.08 | 6 |
| L6.45C | 9.56 (5.48–16.69)* | 0.42 | 4.47 ± 0.15 | 6 |
| I6.46C | 2.72 (1.86–3.99)* | 0.12 | 3.07 ± 0.04 | 6 |
| W6.48C | ND | 6 | ||
| F6.49C | 8.57 (4.25–17.29) | 0.37 | 4.26 ± 0.09 | 6 |
| P6.50C | ND | 6 | ||
| V6.51C | 10.97 (3.35–35.88) | 0.48 | 5.09 ± 0.21 | 6 |
| L6.52C | 1.31 (0.93–1.84)* | 0.06 | 2.13 ± 0.40 | 6 |
| A6.53C | 8.06 (4.43–14.66)* | 0.35 | 3.48 ± 0.06 | 6 |
| L6.54C | 0.99 (0.52–1.87)* | 0.04 | 1.73 ± 0.08 | 6 |
| M6.55C | 2.12 (1.05–4.29)* | 0.09 | 2.51 ± 0.11 | 6 |
| A6.56C | 8.02 (4.45–14.45)* | 0.35 | 3.04 ± 0.11 | 6 |
| H6.57C | 7.78 (4.69–12.90)* | 0.34 | 3.17 ± 0.08 | 6 |
| S6.58C | ND | 6 | ||
| L6.59C | 3.73 (2.23–6.23)* | 0.16 | 2.15 ± 0.08 | 6 |
| A6.60C | 8.55 (4.27–17.14)* | 0.37 | 3.51 ± 0.07 | 6 |
| T6.61C | 3.50 (2.12–5.76)* | 0.15 | 2.33 ± 0.20 | 6 |
| T6.62C | 1.58 (1.00–2.48)* | 0.07 | 2.49 ± 0.06 | 6 |
Kd and Bmax values were measured in competition binding experiments using [3H]CP55940 as radioligand (see Materials and Methods). Data are the mean±SEM of at least 6 independent experiments performed in duplicate. ND, no detectable binding.
Significantly different from the background (C2.59S)(p<0.05)
Reactions of MTSEA with the mutants
10 mM MTSEA significantly inhibited [3H]CP55940 binding to 6 of 31 cysteine-substitution mutants (Figure 2A). 3 mM MTSEA also significantly blocked binding to these 6 mutants, to different extents (Figure 2B).
Figure 2. Effects of MTSEA on specific [3H]CP55940 binding to mutant CB2 receptors.
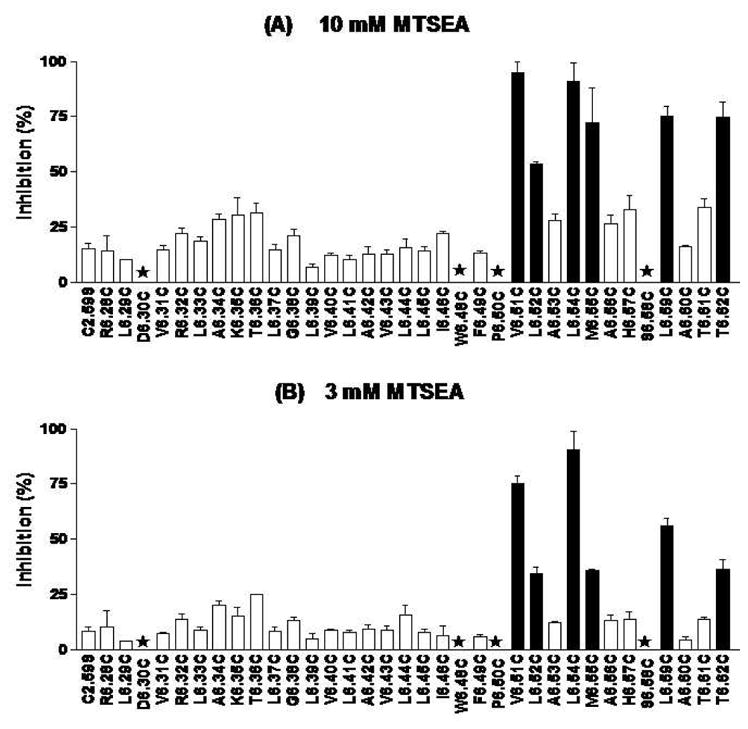
HEK293 cells stably transfected with C2.59S background and CB2 cysteine-substitution mutant receptors were treated with 3 mM and 10 mM MTSEA for 10 mins at room temperature. CP55940 was then used for competition binding with [3H]CP55940 on the MTSEA-treated cells. Data shown represent the mean±SEM of at least six independent experiments performed in duplicate. Solid black bars are mutants for which inhibition was significantly different (p < 0.05) from the background by one-way ANOVA plus Newman-keul’s post test. The stars signify mutants for which [3H]CP55940 binding was undetected.
Determination of Second-Order Rate Constant
To quantitate the susceptibility of MTSEA, we determined the second-order rate constants for the sensitive mutants (Table 2 and Figure 3). The rate constants of the mutant receptors (V6.51C, L6.52C, L6.54C, M6.55C, L6.59C and T6.62C) were 4–6 times higher than that of the background (C2.59S) mutant receptor, but comparable to that of the wildtype CB2 receptor. To confirm the difference between sensitive and insensitive mutants, we also calculated the second-order rate constants for several insensitive mutants for which [3H]CP55940 binding was not significantly inhibited by MTSEA. The second-order rate constants for these mutants (T6.36C, H6.57C and T6.61C) were no different from that of the C2.59S background mutant.
Table 2.
Rates of reaction of MTSEA (M−1 s−1) with cysteine-substitution mutants of the CB2 receptor stably expressed in HEK293 cells.
| Receptor | kMTSEA (M−1 s−1) | kMUT/kWT |
|---|---|---|
| CB2* | 1.22 ± 0.03 | 1.00 |
| C2.59S | 0.23 ± 0.03 | 0.19 |
| (T6.36C) | 0.34 ± 0.09 | 0.28 |
| V6.51C* | 1.20 ± 0.02 | 0.98 |
| L6.52C* | 1.21 ± 0.02 | 0.99 |
| L6.54C* | 1.29 ± 0.02 | 1.06 |
| M6.55C* | 1.24 ± 0.03 | 1.02 |
| (H6.57C) | 0.28 ± 0.01 | 0.23 |
| L6.59C* | 1.24 ± 0.03 | 1.02 |
| (T6.61C) | 0.24 ± 0.01 | 0.20 |
| T6.62C* | 1.23 ± 0.01 | 1.01 |
Second-order rate constants were determined as described in materials and methods. Cells were treated with four concentrations of MTSEA (1, 3, 10 and 30 mM) and [3H]CP55940 binding performed. Rate constants of all accessible mutants were determined. Data represent the means±SEM of at least four independent experiments performed in duplicate. kMUT/kWT was obtained by dividing the k value obtained for each cysteine mutant by the k value of the CB2 wildtype receptor (not the C2.59S background which does not react). The asterisk (*) denotes a statistically significant difference (p < 0.05) compared to the C2.59S background mutant, by one-way ANOVA plus Newman-keuls post test. The brackets ( ) show selected “insensitive” mutants that were not significantly more inhibited than the background (C2.59S) mutant.
Figure 3. Comparison of the second order rate constants for wildtype CB2, C2.59S background mutant and CB2 TMH6 cysteine-substitution mutant receptors.
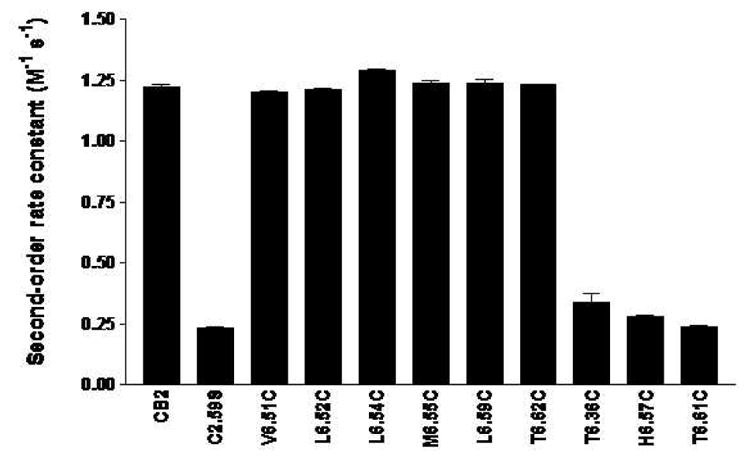
The experiments were performed as described in Methods. Data shown represent the mean±SEM of at least six independent experiments performed in duplicate.
Protective Effect of WIN55212-2 against the Inhibitory Action of MTSEA
To determine whether the substituted cysteine residues that reacted with MTSEA are actually within or in the vicinity of the binding pockets, we examined if the CB2 receptor agonist WIN55212-2 could protect the substituted cysteines from the inhibitory action of MTSEA on [3H]CP55940 binding. The residues that constitute the surface of the binding-site crevice are a subset of the water-accessible residues. Pretreatment of MTSEA-sensitive cysteine-substituted mutants with WIN55212-2 significantly reduced the inhibitory effects of MTSEA on [3H]CP55940 binding for all the sensitive mutants, including V6.51C, L6.52C, L6.54C, M6.55C, L6.59C and T6.62C (Figure 4). The extent of protection varied for different mutants, with the higher WIN55212-2 concentration of 1 μM generally affording more protection than 0.1 μM WIN55212-2.
Figure 4. WIN55212-2 protection of the MTSEA effects on specific [3H]CP55940 binding to mutant CB2 receptors.

HEK293 cells stably transfected with C2.59S background and cysteine-substitution mutant receptors were incubated without or with 0.1 μM and 1 μM WIN55212-2 for 40min at 30OC and then reacted with 10 mM (V6.51C and L6.54C) or 30 mM (L6.52C, M6.55C, L6.59C and T6.62C) MTSEA (to inhibit >80% of binding) for 10 min at room temperature. The cells were then washed eight times, resuspended in binding buffer, and assayed for specific [3H]CP55940 binding as described under “materials and methods”. The data are expressed as a fraction of the binding measured in the absence of MTSEA treatment. Data shown represent the mean±SEM of at least four independent experiments performed in quadruplet. WIN55212-2 provided significant protection (p < 0.05 (*) by one-way ANOVA plus Newman-keul’s post test for all of the mutants.
Molecular Modeling
SCAM results reported here suggest that residues V6.51, L6.52, L6.54, M6.55, L6.59, and T6.62 are accessible to MTSEA from within the binding site crevice. Figure 5A illustrates the positions of these residues in the context of the CB2 model reported here. It is clear here that V6.51, L6.54, M6.55, L6.59 and T6.62 face into the CB2 binding pocket. While the C-α-C-β bond of residue L6.52 is in the TMH6-TMH5 interface of the CB2 model, the Leu side chain points into the binding site crevice when its chi-1 torsion angle is in g+ (as illustrated in Figure 5A). In addition, the flexibility of TMH6 may contribute to residue L6.52 being accessible to the binding site crevice.
Figure 5. Extracellular views of receptor models.

(A) An extracellular view of the CB2 model. This model includes both N- and C-termini, as well as extracellular and intracellular loops. TMH6 is highlighted in magenta and the residues labeled by MTSEA (V6.51, L6.52, L6.54, M6.55, L6.59 and T6.62) are highlighted in green. (B–D) Illustrated here are the positions of these same residues in the three reported crystal structures of the Class A GPCRs: bovine rhodopsin (5B) (35), the human beta-2-adrenergic (β-2-AR; 5C) (47.48), and the turkey beta-1-adrenergic (β-1-AR; 5D) (49) receptors. It is clear that the residues labeled by MTSEA in CB2 would be accessible in these other receptors as well.
Thus the modeling and SCAM studies reported here are in direct agreement with each other. Figure 5 also illustrates the positions of the same residues in the three reported crystal structures of Class A GPCRs (bovine rhodopsin (Figure 5B) (35), human beta-2-adrenergic (β-2-AR; Figure 5C) (47,48), and turkey beta-1-adrenergic (β-1-AR; Figure 5D) (49) receptors. It is clear here that the residues labeled in CB2 would be accessible in these other receptors as well. Figure 5 also illustrates a fundamental difference between the position of the extracellular end of TMH6 in the CB2 model vs. rhodopsin, β-2-AR and β-1-AR. The origin of this difference is explored in the Discussion section below.
DISCUSSION
Effects of Cysteine Substitution on [3H]CP55940 Binding
In this study, 22 of the cysteine-substitution mutants had Kd values lower than that of the C2.59S background mutant. These mutations therefore increased [3H]CP55940 binding affinity for the CB2 cannabinoid receptor. The rest of the cysteine-substitution mutants (R6.28C, L6.29C, L6.41C, A6.42C, V6.43C, L6.44C, F6.49C and V6.51C) had Kd values for [3H]CP55940 not significantly different from that of the C2.59S background mutant. [3H]CP55940 was used in this study because a suitable radiolabeled CB2 antagonist is currently not available to us. One possible complication with all mutagenesis experiments is that the overall structure of the mutant receptors may be changed from wildtype receptor structure. However, in this study, because the majority of the cysteine mutations caused either no change in the Kd value for CP55940 or decreased the Kd value, we believe that the overall structures of the mutant receptors are not compromised by the cysteine substitutions. Since TMH6 is the key helix involved in GPCR activation (50), it is possible that those mutations led to higher binding affinities (lowered Kd values) might have biased receptor populations towards activated state, thus increasing agonist affinity.
W6.48, D6.30, P6.50 and S6.58
Even though W6.48C, D6.30C, P6.50C and S6.58C receptor proteins were detected on cell membranes by Western blot analysis (data not shown), these mutant receptors lost their ability to bind [3H]CP55940. W6.48, D6.30, P6.50 are highly conserved residues whereas S6.58 is not a conserved amino acid among GPCRs.
In opioid receptors, the W6.48C mutation did not alter [3H]diprenorphine binding affinity (24). In the dopamine D2 receptor, the W6.48C mutant showed greatly reduced binding affinity (21). W6.48 is a highly conserved residue in rhodopsin-like GPCRs and is part of the very important CWXP hinge motif in TMH6 of Class A GPCRs. Mutating W6.48 of the CB1 cannabinoid receptor to alanine resulted in a significant reduction in ligand binding affinity in the presence of WIN55212-2 and SR141716A, but not CP55940 and anandamide (51). In our recent mutagenesis studies on the CB2 receptor, W6.48A and W6.48F mutant receptors completely lost their ligand-binding and signaling ability (52). These results, together with our W6.48C mutant binding results, suggest that a tryptophan residue at position 6.48 is important for ligand binding and proper function of CB2 receptor. This tryptophan at 6.48 may be necessary to maintain the proper geometry of the ligand binding pocket.
There has been no report on a D6.30C mutation for either the opioid or the dopamine D2 receptors. Previously, we have shown that the D to N mutation at position D6.30 of the CB2 receptor did not result in a loss of ligand binding (53). Residue 6.30 is part of an intracellular ionic lock (with R3.50/D3.49) in rhodopsin (35). The CB2 D6.30N mutant would retain the ability to hydrogen bond with R3.50 and may be the reason why the D6.30N mutation does not show deleterious effects on ligand binding. The D6.30C mutation may cause this connection with R3.50 to be lost, given that Cys residues are not strong hydrogen bonding residues. This may explain the deleterious effect of D6.30C mutation on ligand binding seen in this study.
In the D2 dopamine receptor, P6.50C reduced the binding affinity of the antagonist 3[H]-N-methylspiperone to the receptor (21). Similar to our P6.50C results, [3H]Diprenorphine binding was undetectable for P6.50C mutants of the mu, delta, and kappa opioid receptors in SCAM studies by Xu and coworkers (24). Our results suggest that the 6.50 locus is crucial for maintaining ligand binding and function of the CB2 receptor. P6.50 is the most conserved residue in TMH6 and is responsible for the helix kink. It might be expected that a P6.50C mutant would exhibit no binding given that this mutation will cause a drastic change in TMH6 geometry.
The S6.58C mutant lost ligand binding ability. S6.58 is an important residue at the extracellular end of TMH6 in CB2. Modeling studies suggest that this residue hydrogen bonds with EC-3 loop residues D(275) and S(274) and stabilizes the position of the extracelluar end of TMH6. Because Cys has reduced hydrogen bonding tendency, the S6.58C mutation may result in a de-stabilization of the extracellular position of TMH6, perturbing receptor structure and resulting in loss of ligand binding.
Residues in TMH6 that are Accessible in the Binding-Site Crevice
Several mechanisms can possibly explain the inhibition of ligand binding when an MTS reagent reacts with an accessible cysteine. These include steric block, electrostatic repulsion, and/or an indirect structural effect on the binding site. Our SCAM results show that [3H]CP55940 binding to 6 out of 30 mutants in TMH6 was sensitive to MTSEA. Cannabinoids are well known for their hydrophobic properties. WIN55212-2 was used for protection experiments, because among all the cannabinoids that we tested, this compound is the easiest to wash off in protection assays. WIN55212-2 protected the binding of [3H]CP55940 from the inhibitory action of MTSEA for all of these sensitive cysteine mutants. WIN55212-2 could protect directly substituted cysteine residues on the surface of the binding pocket from reaction with MTSEA, as well as protect substituted cysteine residues deeper in the binding-site crevice by blocking the passage of MTSEA from the extracellular medium. The residues identified by our results as being on the water-accessible surface of the binding-site crevice of the CB2 receptor include V6.51, L6.52, L6.54, M6.55, L6.59 and T6.62. As is clear in Figure 5, these residues form an arc that describes the degree to which the extracellular portion of TMH6 is pushed into the TMH bundle.
The loss of binding to the four cysteine-substitution mutants (W6.48C, D6.30C, P6.50C and S6.58C) suggests that the structures of these mutant receptors, especially around the binding site, have probably been distorted by the mutation such that the substituted cysteine no longer lies in a similar orientation to that of the wildtype receptor. Two of the mutant residues that lost their ligand-binding ability (W6.48C and S6.58C) are predicted by modeling studies reported here to face into the binding site crevice. W6.48 of the mu and kappa opioid receptors, and K6.58, W6.58 and E6.58 respectively of the mu, delta and kappa opioid receptors (24) were also identified as being in the water-accessible binding site crevice. P6.50 is predicted by modeling to face lipid and mutation of this residue to Cys can be expected to introduce structural perturbations in the TMH bundle. P6.50 was not identified as being in the water-accessible crevice of the mu, delta and kappa opioid receptors (24), however, this residue could be labeled in the dopamine D2 receptor (21). The other CB2 residue which lost its binding (D6.30C) is located at the intracellular end of TMH6 and therefore would not have been available for modification by MTSEA.
Most of the residues identified in our study as being on the water-accessible surface of the binding-site crevice of CB2 were also found to be accessible in the mu, delta and kappa opioid receptors, as well as in the dopamine D2 receptor. Just like our results for V6.51, L6.54, M6.55 and L6.59, Xu and coworkers (24) identified I6.51, Y6.54, V6.55 and A6.59 of the mu receptor, I6.51, F6.54, V6.55 and T6.59 of the delta receptor, and I6.51, F6.54, I6.55 and A6.59 of the kappa receptor as being on the water-accessible surface of the binding-site crevice. Javitch and coworkers (21) identified F6.51, F6.52, T6.54, H6.55 and I6.59 of the dopamine D2 receptor, residues which we also identified in CB2 as being on the water-accessible surface of the binding-site crevice. Three of their residues, V6.40, F6.44 and I6.56 were insensitive to MTSEA in our studies. Unlike ours, their W6.48C did not lose binding and was accessible in the binding-site crevice of the dopamine D2, and the mu and kappa opioid receptors, but not in the delta opioid receptor. H6.52C, which showed no detectable binding for all three opioid receptors, was identified as being on the water-accessible surface of the binding-site crevice of CB2, just like F6.52 of the dopamine D2 receptor. CB2 residue 6.62 was also labeled in the work reported here. Modeling studies predict that residue 6.62 is not part of TMH6, but is at the beginning of the EC-3 loop. However, this residue does face the ligand binding pocket (Figure 5) and this is likely the reason why this residue was reactive to MTSEA.
Rate constants of MTSEA Reaction
The second order rate constants of the sensitive cysteine mutants were similar in magnitude. There was, however, a 4–6-fold significant (p < 0.05) difference between the rate constants of the sensitive and insensitive mutants, signifying that the reaction of MTSEA with cysteines in the binding-site crevice is accelerated approximately 4–6-fold. This increase in second order rate constants between the most reactive CB2 TMH6 residue and the least reactive residue, though statistically significant, is much less than the 100-fold increase in second order rate constants reported for the mu, kappa and delta opioid receptors (24) or the dopamine D2 receptor (21).
There is one important structural difference in CB2 that may contribute to the lower MTSEA reactivity rates seen. Class A GPCRs such as rhodopsin, β-2-AR and β-1-AR have a Cys in the EC-2 loop and a Cys at 3.25 that form a disulfide bridge. As evidenced in the bovine rhodopsin (35), human β-2-AR (47,48) and turkey β-1-AR (49) crystal structures, this disulfide bridge causes the EC-2 loop region C-terminal to the conserved disulfide bond to be deeper in the binding-site crevice than is the N-terminal part of EC-2 loop. The position of the EC-2 loop in each of these structures is illustrated in Figure 6(B–D). Here the EC-2 loop is colored green and shown in molecular surface display (probe radius = 1.4 Å). It is clear here that in each case, the EC-2 loop protrudes into the binding site crevice between TMH3 and TMH6. This protrusion will limit how close the TMH bundle can pack together. At the same time, this protrusion will not block water penetration into the binding site crevice, but actually will facilitate such penetration since the TMH bundle is prevented from closer packing with itself. Shi and Javitch found that the pattern of accessibility of EC-2 loop residues in the dopamine D2 receptor was consistent with a structure similar to that of bovine rhodopsin. These investigators concluded that the EC-2 loop likely contributes to the binding site in the D2 receptor (54). While no EC-2 loop SCAM studies have been reported for the opioid receptors, each of these receptors also has a Cys in the EC-2 loop and a Cys at residue 3.25. Thus the structure of the opioid receptors in the EC-2 loop region is likely to be similar to rhodopsin, the beta-2-adrenergic, the beta-1-adrenergic, and the dopamine D2 receptors. In contrast to these other Class A GPCRs, the CB2 receptor lacks the Cys at 3.25 and therefore cannot mimic these other receptors. It is likely that an internal EC-2 C4.66(174)-C179 disulfide bridge (44) forms in CB2. Modeling studies suggest that because the CB2 EC-2 loop does not penetrate as deeply into the binding pocket (see Figure 6A), the CB2 TMH bundle can more closely pack with itself. One of the effects of this increased packing may be diminished water penetration and therefore reduced ability to label residues deeper in the binding pocket, as well as a slowing of MTSEA reaction kinetics even for residues higher in the binding site crevice.
Figure 6. EC-2 Loop Positions and Penetration into the Binding Pocket.

The differences in EC-2 Loop penetration into the binding pockets of (A) CB2, (B) bovine rhodopsin (35), (C) the human β-2-AR (47,48) and (C) the turkey β-1-AR (49) are ilustrated here. Class A GPCRs such as rhodopsin, β-2-AR and β-1-AR (but not CB2) have a Cys in the EC-2 loop and a Cys at 3.25 that form a disulfide bridge. In (B) the bovine rhodopsin (35), (C) the human β-2-AR (47,48) and (D) the turkey β-1-AR (49) crystal structures, this disulfide bridge causes the EC-2 loop region C-terminal to the conserved disulfide bond to be deeper in the binding-site crevice than is the N-terminal part of EC-2 loop. Here the EC-2 loop is colored green and shown in molecular surface display (probe radius = 1.4 Å). It is clear that in 6B-6D, the EC-2 loop protrudes into the binding site crevice between TMH3 and TMH6. This protrusion will limit how close the TMH bundle can pack together. In contrast, the CB2 receptor (A) lacks the Cys at 3.25. It is likely that an internal EC-2 C4.66(174)-C179 disulfide bridge (44) forms in CB2. Because the CB2 EC-2 loop does not have as deep a penetration into the binding pocket, the CB2 TMH bundle can more closely pack with itself, likely resulting in decreased water penetration, as well as a slowing of MTSEA reaction kinetics even for residues higher in the binding site crevice.
Geometry of TMH6
The pattern of residues in TMH6 of the CB2 cannabinoid receptor accessible in the binding-site crevice is consistent with TMH6 having an alpha-helical conformation extracellular to P6.50 (37) (Figure 5). These results are consistent with TMH6 residues found to be accessible to the binding site by SCAM studies of the dopamine D2 receptor (21) and the mu, delta and kappa opioid receptors (24).
The difference in the EC-2 loop between CB2 and rhodopsin, β-2-AR and β-1-AR (Figure 6) may also impact the direction in which the extracellular portion of TMH6 points. Figure 5 shows that the TMH6 wobble angle in the CB2 model causes the extracellular end of TMH6 to point into the TMH bundle towards TMH4 (Figure 5A), while the extracellular end of TMH6 in rhodopsin, β-2-AR or β-1-AR actually points out of the bundle past TMH5 (Figure 5 B–D). This is reflected in the wobble angles for TMH6 in bovine rhodopsin (35), human β-2-AR (47,48), turkey β-1-AR (49) and the model of human CB2 which are −83.7°, −73.0°, −65.5° and −121.6° respectively. The much larger magnitude wobble angle for CB2 TMH6 is possible because the EC-2 loop in CB2 does not penetrate deeply into the binding pocket, enabling the top of TMH6 to occupy a region precluded by the EC-2 loop in rhodopsin, β-2-AR and β-1-AR.
Conclusions
In this study, we used the substituted cysteine accessibility method to identify novel CB2 receptor-binding site residues and to elucidate the secondary structure of TMH6 of CB2. The cannabinoid agonist WIN55212-2 protected V6.51, L6.52, L6.54, M6.55, L6.59 and T6.62 from modification by the MTS reagent, indicating that these six residues line the CB2 binding site. Our molecular modeling studies indicate that these same six residues face into the CB2 binding pocket (V6.51, L6.52, M6.55, L6.54, L6.59 and T6.62), thereby corroborating our SCAM studies.
Abbreviations
- GPCR
G protein-coupled receptor
- HEK293
Human embryonic kidney cells
- MTS reagents
Methanethiosulfonate reagents
- MTSEA
Methanethiosulfonate ethylammonium
- Rho
Rhodopsin
- SCAM
Substituted cysteine accessibility method
- TMH
Transmembrane helix
Footnotes
This work was supported in part by National Institutes of Health Grants DA11551 and EY13632 to ZHS and grants DA03934 and KO5 DA021358 to PHR.
References
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Shire D, Carillon C, Kaghad M, Calandra B, Rinaldi-Carmona M, Le Fur G. An amino-terminal variant of the central cannabinoid receptor resulting from alternative slicing. J Biol Chem. 1995;270(8):3726–3731. doi: 10.1074/jbc.270.8.3726. [DOI] [PubMed] [Google Scholar]
- 3.Gerard CM, Mollereau C, Vassart G, Parmentier M. Molecular cloning of a human brain cannabinoid receptor which is also expressed in testis. Biochem J. 1991;279:129–134. doi: 10.1042/bj2790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abood ME, Ditto KE, Noel MA, Showalter VM, Tao Q. Isolation and expression of a mouse CB1 cannabinoid receptor gene. Comparison of binding properties with those of native CB1 receptors in mouse brain and N18TG2 neuroblastoma cells. Biochem Pharmacol. 1997;53(2):207–214. doi: 10.1016/s0006-2952(96)00727-7. [DOI] [PubMed] [Google Scholar]
- 5.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 6.Griffin G, Tao Q, Abood M. Cloning and pharmacological characterization of the Rat CB2 cannabinoid receptor. J Pharmacol Exp Ther. 2000;292:886–894. [PubMed] [Google Scholar]
- 7.Brown SM, Wager-Miller J, Mackie K. Cloning and molecular characterization of the rat CB2 cannabinoid receptor. Biochem Biophys Acta. 2002;1576(3):255–264. doi: 10.1016/s0167-4781(02)00341-x. [DOI] [PubMed] [Google Scholar]
- 8.Shire D, Calandra B, Rinaldi-Carmona M, Oustric D, Pessegue B, Bonnin-Cabanne O, Le Fur G, Caput D, Ferrara P. Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Biochem Biophys Acta. 1996;1307(2):132–136. doi: 10.1016/0167-4781(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 9.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74(2):129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 10.Buckley NE, Hansson S, Harta G, Mezey E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience. 1998;82:1131–1149. doi: 10.1016/s0306-4522(97)00348-5. [DOI] [PubMed] [Google Scholar]
- 11.Friedman H, Klein TW, Newton C, Daaka Y. Marijuana, receptors and immunomodulation. Adv Exp Med Biol. 1995;373:103–113. doi: 10.1007/978-1-4615-1951-5_15. [DOI] [PubMed] [Google Scholar]
- 12.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Comparison of the pharmacology and signal transduction of the human CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- 13.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs-linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouaboula M, Poinot-Chazel C, Bourrié B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouaboula M, Poinot-Chazel C, Marchand J, Canat X, Bourrié B, Rinaldi-Carmona M, Calandra B, Le Fur G, Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor: involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem. 1996;237:704–711. doi: 10.1111/j.1432-1033.1996.0704p.x. [DOI] [PubMed] [Google Scholar]
- 16.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of Rigid-Body Motion of Transmembrane Helicies for Light Activation of Rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 17.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 18.Javitch JA, Li X, Kaback J, Karlin A. A cysteine residue in the third membrane-spanning segment of the human D2 dopamine receptor is exposed in the binding-site crevice. Proc Natl Acad Sci USA. 1994;91(22):10355–10359. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Javitch JA, Fu D, Chen J. Residues in the fifth membrane-spanning segment of the dopamine D2 receptor exposed in the binding-site crevice. Biochemistry. 1995;34(50):16433–16439. doi: 10.1021/bi00050a026. [DOI] [PubMed] [Google Scholar]
- 20.Javitch JA, Fu D, Liapakis G, Chen J. Constitutive activation of the beta2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J Biol Chem. 1997;272(30):18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- 21.Javitch JA, Ballesteros JA, Weinstein H, Chen J. A cluster of aromatic residues in the sixth membrane-spanning segment of the dopamine D2 receptor is accessible in the binding-site crevice. Biochemistry. 1998;37(4):998–1006. doi: 10.1021/bi972241y. [DOI] [PubMed] [Google Scholar]
- 22.Javitch JA, Ballesteros JA, Chen J, Chiappa V, Simpson MM. Electrostatic and aromatic microdomains within the binding-site crevice of the D2 receptor: contributions of the second membrane-spanning segment. Biochemistry. 1999;38(25):7961–7968. doi: 10.1021/bi9905314. [DOI] [PubMed] [Google Scholar]
- 23.Javitch JA, Shi L, Simpson MM, Chen J, Chiappa V, Visiers I, Weinstein H, Ballesteros JA. The fourth transmembrane segment of the dopamine D2 receptor: accessibility in the binding-site crevice and position in the transmembrane bundle. Biochemistry. 2000;39(40):12190–12199. doi: 10.1021/bi001069m. [DOI] [PubMed] [Google Scholar]
- 24.Xu W, Li J, Chen C, Huang P, Weinstein H, Javitch JA, Shi L, de Riel JK, Liu-Chen LY. Comparison of the amino acid residues in the sixth transmembrane domains accessible in the binding-site crevices of mu, delta, and kappa opioid receptors. Biochemistry. 2001;40(27):8018–8029. doi: 10.1021/bi002490d. [DOI] [PubMed] [Google Scholar]
- 25.Akabas MH, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the alpha-subunit. Biochem. 1995;34:12496–12500. doi: 10.1021/bi00039a002. [DOI] [PubMed] [Google Scholar]
- 26.Beck C, Wollmuth LP, Seeburg PH, Sakmann B, Kuner T. NMDAR channel segments forming the extracellular vestibule inferred from the accessibility of substituted cysteines. Neuron. 1999;22:559–570. doi: 10.1016/s0896-6273(00)80710-2. [DOI] [PubMed] [Google Scholar]
- 27.Koch SE, Bodi I, Schwartz A, Varadi G. Architecture of Ca2+ Channel Pore-lining Segments Revealed by Covalent Modification of Substituted Cysteines. J Biol Chem. 2000;275:34493–34500. doi: 10.1074/jbc.M005569200. [DOI] [PubMed] [Google Scholar]
- 28.Lambert G, Forster IC, Stange G, Kohler K, Biber J, Murer H. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na(+)/P(i) cotransporter. J Gen Physiol. 2001;117:533–546. doi: 10.1085/jgp.117.6.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA. Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry. 1986;25:5595–5601. doi: 10.1021/bi00367a038. [DOI] [PubMed] [Google Scholar]
- 30.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three Dimensional Models and Computational Probing of Structure Function Relations in G Protein-Coupled Receptors. In: Conn PM, Sealfon SC, editors. Methods in Neuroscience. Vol. 25. 1995. pp. 366–428. Chapter 19. [Google Scholar]
- 31.Zhang R, Hurst DP, Barnett-Norris J, Reggio PH, Song ZH. Cysteine 2.59(89) in the second transmembrane domain of human CB2 receptor is accessible within the ligand binding crevice: evidence for possible CB2 deviation from a rhodopsin template. Mol Pharmacol. 2005;68(1):69–83. doi: 10.1124/mol.104.007823. [DOI] [PubMed] [Google Scholar]
- 32.Song ZH, Bonner TI. A Lysine Residue of the Cannabinoid Receptor Is Critical for Receptor Recognition by Several Agonists but Not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- 33.Song ZH, Slowey CA, Hurst DP, Reggio PH. The Difference Between the CB(1) and CB(2) Cannabinoid Receptors at Position 5.46 Is Crucial for the Selectivity of WIN55212-2 for CB(2) Mol Pharmacol. 1999;56:834–840. [PubMed] [Google Scholar]
- 34.DeBlasi A, O’Reilly K, Motulsky HJ. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol Sci. 1989;10:227–229. doi: 10.1016/0165-6147(89)90266-6. [DOI] [PubMed] [Google Scholar]
- 35.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 36.Bramblett RD, Panu AM, Ballesteros JA, Reggio PH. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995;56:1971–1982. doi: 10.1016/0024-3205(95)00178-9. [DOI] [PubMed] [Google Scholar]
- 37.Barnett-Norris J, Hurst DP, Buehner K, Ballesteros JA, Guarnieri F, Reggio PH. Agonist alkyl tail interaction with cannabinoid CB1 receptor V6.43/I6.46 groove induces a Helix 6 active conformation. Int J Quantum Chem. 2002;88:76–86. [Google Scholar]
- 38.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 39.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hassan SA, Mehler EL, Weinstein H. Structure calculation of protein segments connecting domains with defined secondary structure: A simulated annealing Monte Carlo combined with biased scaled collective variables technique. In: Schlick T, Gan HH, editors. Computational Methods for Macromolecules: Challenges and Applications. Springer Verlag; New York: 2002. pp. 197–231. [Google Scholar]
- 41.Barnett-Norris J, Hurst DP, Reggio PH. 2003 Symposium on the Cannabinoids. International Cannabinoid Research Society; Cornwall, Ontario: 2003. The influence of cannabinoid receptor second extracellular loop conformation on the binding of CP55940; p. 79. [Google Scholar]
- 42.Hassan SA, Guarnieri F, Mehler EL. A General Treatment of Solvent Effects Based on Screened Coulomb Potentials. J Phys Chem B. 2000b;104:6478–6489. [Google Scholar]
- 43.Hassan SA, Guarnieri F, Mehler EL. Characterization of Hydrogen Bonding in a Continuum Solvent Model. J Phys Chem B. 2000a;104:6490–6498. [Google Scholar]
- 44.Gouldson P, Calandra B, Legoux P, Kerneis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Mutational analysis and molecular modelling of the antagonist SR 144528 binding site on the human cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 45.Ulfers AL, McMurry JL, Kendall DA, Mierke DF. Structure of the third intracellular loop of the human cannabinoid 1 receptor. Biochemistry. 2002;41:11344–11350. doi: 10.1021/bi0259610. [DOI] [PubMed] [Google Scholar]
- 46.Mezei M. In: SIMULAID: a collection of utilities designed to help setting up molecular simulations. Mezei M, editor. New York: 2004. [Google Scholar]
- 47.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-Resolution Crystal Structure of an Engineered Human {beta}2-Adrenergic G Protein Coupled Receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383–7. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 49.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci U S A. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McAllister SD, Rizvi G, Anavi-Goffer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Abood ME. An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J Med Chem. 2003;46(24):5139–52. doi: 10.1021/jm0302647. [DOI] [PubMed] [Google Scholar]
- 52.Zhang R, Reggio PH, Song Z-H. 2004 Symposium on the Cannabinoids. International Cannabinoid Research Society; Paestum, Italy: 2004. Mutagenesis of Aromatic Microdomains at Human CB2 Cannabinoid Receptor. [Google Scholar]
- 53.Nebane NM, Kellie B, Song ZH. The effects of charge-neutralizing mutation D6.30N on the functions of CB1 and CB2 cannabinoid receptors. FEBS Lett. 2006;580(22):5392–5398. doi: 10.1016/j.febslet.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 54.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A. 2004;101(2):440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]