

A robust and sensitive platform was developed for chemical-genomics in bacteria. Kinetic acquisitions of colony growth enable calculation of growth rates alongside conventional endpoint volume measurements, generating a wealth of chemical-genetic interactions. This kinetic platform is highly amenable to prokaryotic or eukaryotic strain collections.
Abstract
While genetic perturbation has been the conventional route to probing bacterial systems, small molecules are showing great promise as probes for cellular complexity. Indeed, systematic investigations of chemical-genetic interactions can provide new insights into cell networks and are often starting points for understanding the mechanism of action of novel chemical probes. We have developed a robust and sensitive platform for chemical-genomic investigations in bacteria. The approach monitors colony volume kinetically using transmissive scanning measurements, enabling acquisition of growth rates and conventional endpoint measurements. We found that chemical-genomic profiles were highly sensitive to concentration, necessitating careful selection of compound concentrations. Roughly 20,000,000 data points were collected for 15 different antibiotics. While 1052 chemical-genetic interactions were identified using the conventional endpoint biomass approach, adding interactions in growth rate resulted in 1564 interactions, a 50–200% increase depending on the drug, with many genes uncharacterized or poorly annotated. The chemical-genetic interaction maps generated from these data reveal common genes likely involved in multidrug resistance. Additionally, the maps identified deletion backgrounds exhibiting class-specific potentiation, revealing conceivable targets for combination approaches to drug discovery. This open platform is highly amenable to kinetic screening of any arrayable strain collection, be it prokaryotic or eukaryotic.
INTRODUCTION
Since the introduction of microwell plates, biomedical research laboratories have been growing bacteria in high throughput. Over the past decade, this technology has advanced tremendously, and arraying robots can now pin entire genomic libraries onto a single agar plate. In bacteria, high-throughput conjugation between genomic libraries, such as the celebrated Escherichia coli K-12 in-frame deletion (Keio) collection (Baba et al., 2006), and mutations in genes of interest yield elaborate genetic interaction profiles. These profiled “fingerprints” are a list of genomic interactions for the particular gene of interest, with synthetic sick or synthetic lethal interactions suggesting functional relationships between genes. Genetic interactions such as these lead to new gene clusters or can be compiled into interaction networks. This has led to significant advances in our understanding of biological systems (Typas et al., 2008; Brochado and Typas, 2013) and the functional relationships between genes and processes in E. coli. For nonessential processes, probing them in this manner works extremely well, but the method necessarily excludes essential genes that resist deletion. In contrast, antibiotics are elegant probes of these essential bacterial functions that can be exploited by exploring chemical-genomic interactions. This ultimately results in a mechanistic fingerprint for the chemical of interest (Liu et al., 2010; Nichols et al., 2011) and is of tremendous utility when hunting for the ever-elusive mechanism of action in chemical screening.
Complications arise in chemical genomics, however, due to the inoculum effect with small molecules. This dictates that increasing bacterial inoculum will generally increase the minimum inhibitory concentrations (MICs) of bioactive chemicals and is especially true in solid agar media. The latter are the growth media of choice for arraying genomic collections in high density and typically with tremendously high inocula. This makes generating mechanistic fingerprints challenging, as relatively precise concentrations of drug are required to elicit a response from a genomic library such as the Keio collection. Means to address this challenge include screening at several chemical concentrations and probing genomic libraries kinetically. The latter was pioneered on solid agar medium in the recent work by Takeuchi et al. (2014). Bacterial growth rate is a relatively inoculum-independent parameter, with true exponential growth occurring early in the bacterial growth cycle, making this approach more sensitive than conventional endpoint biomass measurement. Further, conventional endpoint reads sum all growth phases. Measuring colony biomass across growth curves, as described herein, provides the opportunity to study both growth rates and overall colony size.
High-throughput chemical-genetic crosses are done on solid agar plates, in which colony sizes are strongly influenced by spatial location in high-density arrays. This ultimately requires normalization of raw data such that results are comparable across treatments. Past studies have taken a multistep approach, beginning with completely removing larger colonies from the perimeter and then scaling remaining colony sizes to a common value above or below the cutoff threshold. Each colony is then normalized to its value across all treatments (Typas et al., 2008). Normalization techniques for standard high-throughput screening (Mangat et al., 2014) are useful in experiments run in liquid medium but do not work well for within-plate spatial variations in high-density solid media arrays. Other methods, such as the B-score (Brideau et al., 2003), can be used to normalize screening data, but the iterative nature of the method raises questions regarding overpolishing and consequences such as false negatives. Indeed, the biological effectiveness of certain chemical probes may not be correlated with the strongest phenotypic response (Zlitni et al., 2013; Stokes et al., 2014).
Here we present a high-throughput chemical-genomics platform for the E. coli gene deletion (Keio) collection that enables the identification of genetic enhancers (and suppressors) of the growth inhibitory phenotypes of bioactive chemicals. We utilize a simple, two-pass normalization that is not iterative and counters most spatial effects on high-density agar plates. High sensitivity is achieved by monitoring biomass accumulation throughout the growth cycle and by combining traditional endpoint measurements with calculated growth rates for every chemical-genetic cross. Elaborate chemical-genetic interaction maps can be generated, using interactions calculated from each of the acquired screening phenotypes. Further, highly populated nodes with edges spanning each antibiotic class suggest deep involvement in bacterial resistance strategies, whereas highly populated nodes within a single class suggest new screening targets for potent antimicrobial combinations. Of course, not all chemical-genetic interactions are lethal in nature. The simultaneous acquisition of multiple phenotypes enhances our ability to suggest relationships between chemicals and genetic mutations, even if the resulting crosses do not result in cell death or stasis.
RESULTS
Experimental validation and workflow
The scanning method used to analyze colonies on solid medium is transmissive, passing light through the colony and detecting it on the other side. This allows us to measure the absorbance of light by a bacterial colony. When colonies are cut out of the high-density array, vortexed in buffer, and plated across a traditional dilution series, the colony light absorbance (as measured via integrated density in ImageJ; fully described in Materials and Methods and Supplemental Figure S1) has a linear relationship with the number of colony-forming units (CFUs; Supplemental Figure S2). As such, this validates the solid medium biomass quantification method used in our workflow. This is especially important in downstream growth-rate calculations, as full-colony integrated densities track with cell number.
The general chemical-genomics workflow is shown in Figure 1a. Individual steps in the pipeline will be discussed at length below. Briefly, we first chose a diverse panel of antibiotic probes targeting a wide range of essential cell processes (Figure 1b). DNA replication, protein translation, folate biosynthesis, and cell wall biogenesis are all impacted by the drugs chosen. At sub-MICs, these effectively serve as knockdowns of those essential processes. We perform a potency assay on these drugs in liquid medium to identify the MIC. With the liquid MIC as a reference point, 12 concentrations are incorporated into solid media plugs (Supplemental Figure S3) to identify each solid medium MIC. The plug method of solid MIC determination uses the exact same inoculum and pressure settings as a high-density chemical-genomics experiment. Given how variable the solid medium MICs can be in relation to their liquid MIC counterparts, this is an immensely important step in the workflow. Next solid media slabs are prepared with 1/4 and 1/8 MICs of each drug and inoculated using copies of the E. coli Keio deletion collection (Baba et al., 2006) grown on 1536-density plates. Inoculated plates are placed on the aforementioned transmissive scanners and then imaged repeatedly over an 18-h period. An image analysis script is used to quantify colony biomass at each time point, and the resulting data are compiled into growth curves. Growth curves are normalized (Figure 2) and analyzed for endpoint biomass and maximum growth rate, and enhancement profiles are generated. Normalization is fully discussed in the Materials and Methods, and uses a two-pass function based on the interquartile medians of both rows and columns within a high-density colony array. This counters spatial edge and incubator effects. Downstream testing is, however, still necessary to assess nutrient competition effects potentially arising from empty positions in the plates (Baryshnikova et al., 2010). We examined the normalized colony sizes adjacent to empty positions in our plates and saw no significant size differences in comparison with randomly selected colonies after a one-way analysis of variance (F = 0.0754, p = 0.7838). Chemical enhancement in deletion backgrounds is defined using a multiplicative rule (Dixon et al., 2009), with enhancement occurring when the chemical-genetic interaction value is less than the product of each chemical and gene knockout individually (Figure 3). This multiplicative method of detecting sensitization is shown in Figure 3 and is further described in Materials and Methods. Strains enhancing chemical effects are combined into a network map for downstream analysis. This platform performs well for 1536- and 6144-density plates and also removes gradient spatial effects arising from plate positions within an incubator during normalization.
FIGURE 1:

Chemical-genomics platform. (a) The workflow for the chemical-genomics platform. Bioactive chemicals (in this case antibiotics) are first tested for liquid and solid potency. Next they are supplemented into the agar medium of choice, onto which the genomic library is arrayed. Plates are imaged kinetically, and quantified in ImageJ. Finally, data are normalized, analyzed, and used to build a chemical-genomic network map. The spectrum of antibiotics chosen is outlined in b, illustrating the various cell processes targeted by the drugs chosen. Drugs targeting cell wall biogenesis, folate biosynthesis, protein translation, and DNA replication are all represented in our chosen panel of antibiotics. The chosen drugs probe a range of essential cell processes to validate our chemical-genomics platform.
FIGURE 2:

Data normalization for edge effects. (a) Index plot depicting raw integrated density (colony volume) data for a 1536-well plate inoculated with 1536 Keio collection clones. The plate is organized according to column across the index plot, resulting in a familiar horseshoe appearance, due to spatial effects within the plate. These are especially clear in b, a three-dimensional plot of the raw data in a 1536-well plate. When plotted as a histogram (c), the data are heavily skewed, which makes statistical interpretation challenging. Our data normalization function turns the data shown in a into the index plot shown in d. The function divides individual colonies by the interquartile medians across rows and columns, removing the edge effects (e). Data are also then symmetrical about 1 (f), which is ideal for plate-to-plate comparisons, and downstream statistical analyses. The full R code is provided in the Supplementary Data, and the function is further explained in the Materials and Methods.
FIGURE 3:
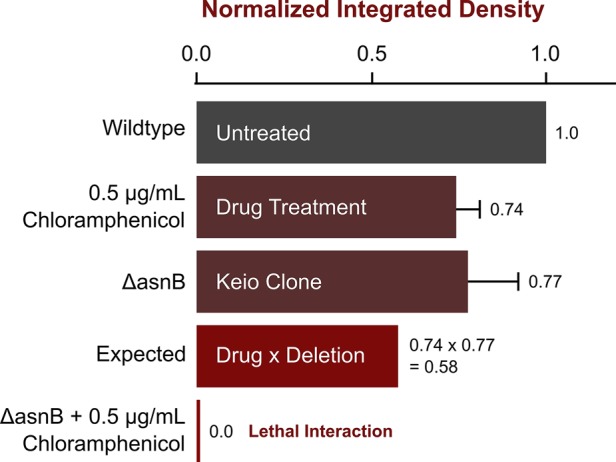
Multiplicative approach to identifying chemical-genetic interactions. We show here an example of how the multiplicative approach (Dixon et al., 2009) can be used to identify lethal or sick interactions in a chemical genomics pipeline. The integrated density values shown are in relation to the untreated wild-type E. coli K-12 BW25113 (Keio parent) strain. Drug-treated wild-type colonies (in this example, treated with 0.5 μg/ml chloramphenicol) show the effect of the drug on its own. Next the individual deletion background (ΔasnB in this case) is measured from a chemically untreated Keio collection plate. The multiplicative rule states that the expected size of this colony, assuming no interaction takes place, is the product of the single deletion and the chemical treatment of the wild type. The chemically treated deletion background is then compared with the expected value to determine whether there is a chemical-genetic interaction.
Chemical concentration considerations
In solid medium, MIC values are typically higher than in liquid medium. The extent of this can be quite variable, however, with some water-soluble cationic compounds displaying solid MICs orders of magnitude higher than respective MICs in liquid medium (unpublished data). Our method of solid MIC determination allows us to find precise MICs on solid medium, using a set of universal inoculum conditions. As in chemical-genomics experiments, the colony volumes can be measured and full MIC curves generated from the images (Supplemental Figure S3). This is a crucial step in chemical genomics, as varying chemical concentrations elicit different genetic responses when crossed with the E. coli Keio collection. Many strain sensitivities are highly dose dependent, with some enhancement phenotypes only present at lower concentrations (Figure 4). For example, when stressed with 0.5 μg/ml of chloramphenicol, 36 strains become sensitized in E. coli that are not sick at 2 or 4 μg/ml concentrations. This includes genes encoding ascorbic acid catabolic enzymes from the ula operon, involved in l-ribulose 5-phosphate and d-xylulose 5-phosphate biosynthesis. At higher concentrations, ribosome-related knockouts are the prevailing strains sensitized to chloramphenicol. Notably, this includes knockouts encoding a ribosome rescue factor ArfA, 23S pseudouridine synthase RluD, and an integration host factor subunit IhfB. For drugs acting on the bacterial ribosome, however, it can be challenging to find sensitized strains due to the slow-growing nature of ribosome-related knockouts, even without drug in the medium. This is the case for strains such as ΔrimM. We observed that going higher than 1/4 MIC in concentration resulted in colonies that were very small, with fewer pixels to count in the downstream image analysis software. Further, this often resulted in inconsistent results. As such, 1/4 MICs seem to be high enough to illustrate enhancement and suppression profiles, without going so high as to complicate downstream imaging.
FIGURE 4:

Chemical-genomic interactions are concentration dependent. This heat map illustrates the Keio collection probed by a concentration gradient of chloramphenicol. The data are symmetrical, ∼1, and show the enhancement (red) or suppression (green) of chloramphenicol by each deletion background. They demonstrate that, across multiple concentrations, suppression and enhancement profiles of drugs can be dose dependent. As the drug concentration is increased, more strains are sensitive to its effects. Interestingly, however, some strains are only sensitive to lower concentrations. Using a range of chloramphenicol concentrations as an example, we see, curiously, several reproducible metabolic genes sensitive to 0.5 μg/ml (1/32 MIC) concentrations of the drug. These disappear in higher concentrations of chloramphenicol, with some ribosome-related deletions becoming more apparent as the dose increases. The figure highlights the value of determining an accurate solid MIC in a chemical-genomics pipeline.
Chemical-genetic interactions
Endpoint biomass reads are the traditional manner of evaluating synthetic lethal interactions between genes (Tong et al., 2001; Typas et al., 2008; Nichols et al., 2011). Acquiring full growth curves (Supplemental Videos 1–3) enables us to collect these conventional measures of chemical-genetic interaction while also garnering growth rates. Maximum growth rate is a more sensitive measure of chemical-genetic interaction than endpoint biomass (Figure 5), with growth rate yielding 50–200% more interactions with antibiotics than endpoint biomass. This increased sensitivity does, however, broaden the data distribution slightly; the mean interquartile range of endpoint treatments calculated in this manner is 0.21 (±0.13), while for growth rate it is 0.24 (±0.08). Interestingly, the SD of the hit interactions was quite low (averaging 0.023 for endpoint and 0.031 for growth rate), while the SD of data centered about the median was higher (averaging 0.077 for endpoint and 0.090 for growth rate). Our interactions replicated well and serve as a basis for downstream network assembly and analysis.
FIGURE 5:

Analysis of growth rate finds more genetic enhancers of antibiotic action than endpoint biomass measurement. The figure shows a general example of conventional endpoint biomass values plotted against growth rate, with both measures compared with their expected values based on the multiplicative rule. Shown here are Keio collection deletion strains growing on 1/4 MIC inhibitory concentrations of the DNA replication drug ciprofloxacin. This plot separates strains with smaller colony sizes and slow growth rates, seen in the lower-left quadrant of the plot, from strains with average colony size but with slower growth. Screening for growth rate is much more sensitive than screening for endpoint biomass alone, yielding many more interactions. This gives a much more thorough glimpse into chemical-genetic interactions for a compound of interest that are not necessarily lethal or static in nature.
Consistent with past antibiotic stress profiling of the Keio collection (Nichols et al., 2011), genes involved in energy generation, drug efflux, and ribosome function were enriched in all treatments (Supplemental Data S1). The nuo operon (encoding NADH:ubiquinone oxoreductase I) is required for aerobic and anaerobic respiration, and deletions in any of these genes result in very small colonies, as does deletion of genes in the ubi operon. Deletion of the gene encoding the outer membrane porin TolC, involved in active efflux, also leads to sensitization to most antibiotics. Similarly, perturbation of the outer membrane with deletions in lipopolysaccharide (LPS) biosynthetic genes also leads to drug sensitivity, in particular to erythromycin, DNA replication inhibitors, and rifampicin. The marC knockout was a slow-growing strain under most drug stresses and strongly enhanced protein translation inhibitors, despite forming overall colony sizes that were often similar to the untreated controls. Other strains with the slow-growth phenotype under our drug stresses include those deletions from the cys and cyd operons and the AcrAB-TolC efflux system. Many relatively uncharacterized genes are also present in almost all drug treatments; more than 30% of the most frequently arising hits when screening for a slow-growth phenotype are genes with unknown or poorly characterized functions. Interestingly, treatment with d-cycloserine, fosfomycin, and, to a lesser extent, rifampicin resulted in a large number of nonreproducible suppressors that were observed late in the 18-h growth measurement. Over several days, this is a common occurrence in most treatments, but in the cases of these drugs it was observed to happen within 18 h.
When probed with antibiotics targeting the bacterial cell wall (Supplemental Figure S4), ∆pal and ∆lpoB mutants were sensitized in endpoint biomass (Supplemental Data S2). Pal is an outer membrane–anchored protein associated with peptidoglycan and is part of the TolC-Pal envelope complex. In addition, deletion of lpoB, encoding the outer membrane lipoprotein LpoB, also enhanced drugs targeting the bacterial cell wall. The LpoB protein forms a complex with PBP-1B and is required for peptidoglycan cross-linking (Typas et al., 2010). Interestingly, d-cycloserine and fosfomycin were enhanced by mutations in rffE, rffG, and rffM. These encode proteins in enterobacterial common antigen (ECA) synthesis, which forms a relatively minor constituent of the K-12 surface polysaccharide matrix containing between one and four trisaccharide repeats (Meier-Dieter et al., 1992). With respect to growth rate, ∆rodZ strains exhibit a defective growth-rate phenotype. RodZ is a cytoskeletal protein that is required to maintain a rod-shaped morphology and interacts with division protein MreB. In addition, deletions in rseA, encoding the anti-sigma factor for σE, also grow slowly in the presence of cell wall drugs. This is interesting, given that these drugs compromise cell wall structural integrity and σE has a key role in the osmotic stress response (Bianchi and Baneyx, 1999). When treated with d-cycloserine or fosfomycin, which target peptidoglycan precursor biosynthetic processes, ∆envC mutants grow exceptionally slowly. EnvC is a sensory histidine kinase, regulating outer membrane porin expression in response to osmotic stress. Deletion of the outer membrane lipid transporter (Mla) is also an enhancer of d-cycloserine or fosfomycin treatment, with ∆mlaA and ∆mlaC deletions negatively impacted by these drugs.
Consistent with the role of the outer membrane as a permeability barrier, protein translation inhibitors targeting the 50S subunit of the ribosome were largely enhanced in endpoint biomass evaluations by mutations in genes common to LPS biosynthetic processes such as rfaE, waaF, waaG, waaI, waaP, and lpcA (Supplemental Figure S5 and Supplemental Data S3). Mutations in components of the Mla outer membrane phospholipid transporter also sensitized the respective strains to erythromycin and chloramphenicol. There were few unique endpoint biomass interactions shared across drugs targeting the 30S ribosomal subunit. Shared between spectinomycin and streptomycin were interactions with gpp, rplI, and yafQ. Gpp is a pyrophosphatase that catalyzes the conversion of guanosine 5′-triphosphate 3′-diphosphate (pppGpp) to the guanosome 5′-diphosphate (ppGpp) nucleotide. RplI is the 50S ribosomal subunit protein L9, and YafQ is a toxin of the DinJ-YafQ toxin/antitoxin complex, which interacts with the 50S ribosomal subunit and cleaves mRNA. When assaying the growth rates under protein translation inhibitor stress, there were also relatively few hits common among the various drugs tested. Instead, various combinations of LPS core biosynthetic genes (lpcA, rfaE, waaBDGIPQZ), 30S or 50S ribosomal subunit proteins (rpsTU, rplIKY), and efflux machinery were observed across the various drugs.
Stressing cells with the topoisomerase inhibitors nalidixic acid, norfloxacin, and ciprofloxacin results in unique enhancement profiles as well (Supplemental Figure S6 and Supplemental Data S4). Highly sensitive to this class of antibiotics in endpoint biomass are ∆xseA, ∆xseB, and ∆recC. XseA and XseB are large and small subunits of E. coli exonuclease VII, respectively (Vales et al., 1982, 1983), and are known to be required for nalidixic acid resistance (Chase and Richardson, 1977). Furthermore, RecC is a component of exonuclease V, showing a clear trend in deletion strains sensitized to (fluoro)quinolones. Interestingly, for each individual quinolone, several Keio strains involved in peptidoglycan recycling and cell division also potentiate the respective drugs. This is of particular interest, given the tendency of DNA replication inhibitors to cause filamentous cell growth in E. coli. Among these strains are pbpG, lpcA, mpaA, nlpD, emtA, oppD, and rodZ. DNA replication inhibitors caused slow growth in ∆nlpI and ∆paaA strains across drug treatments. Slow growth in ∆nlpI mutants lacking cell division lipoprotein NlpI (Ohara et al., 1999) was an interesting observation, given the tendency of DNA replication inhibitors to cause an elongated morphological phenotype. Slow-growing ∆paaA strains in the presence of quinolones were much more enigmatic, as PaaA is involved in phenylacetate catabolism in E. coli.
Chemical-genetic interaction mapping
Chemical-genetic interaction maps were first generated from endpoint biomass accumulation compiled for each antibiotic treatment (Figure 6). This network highlights the functional relationships between nonessential genes and antibiotics and, as previously noted, helps to identify genes unique to the various drug classes assayed. While this is useful in itself, combining these data with kinetically acquired rates is especially powerful. The result of this union is seen in Figure 7. Because these are the small or slow-growing colonies on drug-treated media, many genes are shared between both phenotypes. The growth rate–enriched map in Figure 7 was further clustered based on the Markov cluster (MCL) algorithm (Van Dongen, 2008) implemented in BioLayout3D, and the resulting computed clusters were analyzed for common cell processes using their GO terms (Supplemental Figure S7). Interestingly, drug classes did not completely separate using the MCL clustering method, and a wide variety of GO terms were listed in each cluster (summarized in Supplemental Figure S7). Nevertheless, for endpoint biomass analyses, there were 1052 interactions across all drugs tested, and this number of interactions nearly increased to 1564 interactions when combining endpoint biomass and growth-rate phenotypes. Given the vast number of orphan genes in E. coli, this aids in describing relationships between these poorly annotated genes and essential processes, particularly in the context of antimicrobial action and resistance.
FIGURE 6:

Escherichia coli chemical-genomic interaction map based on endpoint biomass. Interactions are shown, for the panel of antibiotics chosen at 1/4 MICs, with the E. coli Keio deletion collection. Interactions describe sick or lethal enhancement of each antibiotic probe and are based on the 2.5σ cutoff described in the Materials and Methods. The network was prepared using BioLayout3D, with major nodes for antibiotic classes annotated in the legend. Genes common across chemicals are easily identified in this manner (such as ubiG, rpoS, and nuoB), as are drug-sensitive strains within each particular drug class. The network is displayed as an edge-weighted force-directed Fruchterman-Reingold layout and can be further mined in BioLayout3D.
FIGURE 7:

Chemical-genetic interaction map that combines drug sensitivities in biomass accumulation with sensitivities in growth rate for the Keio collection against our panel of antibiotics. Nodes representing conventional endpoint biomass sensitivities are shown in gray, nodes representing slow-growing strains are shown in green, and interactions in both are shown in red. Interactions for both phenotypes are defined in the Materials and Methods. Interestingly, there are ∼50% more unique interactions in the combined map than in the endpoint map shown in Figure 6. The network is displayed in the same manner as in Figure 6, as an edge-weighted force-directed Fruchterman-Reingold layout.
DISCUSSION
The presented chemical-genomics platform is sensitive, with growth-rate acquisitions increasing the information obtained experimentally. While endpoint biomass reads on their own can identify sick endpoint biomass interactions, not all chemical-genetic interactions result in a lethal or sick phenotype when grown to stationary phase. Screening for growth rate can capture these otherwise elusive interactions by identifying slow-growing strains. This is a more sensitive measure but is also more strongly influenced by any variability in chemical concentration. Indeed, this reality stresses the need for precise control of chemical concentrations, especially when the delicate relationship between chemical MIC and initial inoculum can alter the chemical-genetic fingerprint. This may account for some of the differences observed between our data and screens from other groups (Tamae et al., 2008; Liu et al., 2010; Nichols et al., 2011). For example, the inoculation methods used by Tamae et al. (2008) utilize a liquid-to-solid method, resulting in a very different inoculum than our study. Further, both groups use a different means of calculating sensitivity scores, resulting in data that are challenging to cross-evaluate. Variations in genetic interaction data across screening labs has been previously reported and are expected (Michaut and Bader, 2012). This holds especially true in chemical genomics, in which bioactive chemicals can influence a multitude of cell functions. Nonetheless, this suggests that a set of standard conditions is immensely important. Beginning with library maintenance, labs tend to have idiosyncratic approaches to strain upkeep, inoculation, and image and data analysis. To help reduce variability, we chose to maintain a 1/4 MIC dose of antibiotic based on a solid MIC determination that mimics the conditions of the chemical-genomics experimental method. Optimizing in this manner maintains consistency in the face of the high and variable inoculum on solid medium colony arrays.
The Keio collection contains roughly 3800 nonessential deletion strains, with a small subpopulation of strains that grow slowly or poorly even without drug treatment. This was reported in the recent work by Takeuchi et al. (2014), who pioneered the initial time-lapsed scanning procedure, and the large-scale chemical-genomics work by Nichols et al. (2011). This includes deletions in the nuo operon, which encode subunits or peptide chains in the NADH:ubiquinone oxidoreductase enzyme. These genes, along with members of the ubiquinone biosynthetic pathway (ubi operon) and ATP synthase complex (atp operon), frequently arise as hits in chemical-genetic and gene–gene interaction screening (unpublished data). Frequent enhancement in these genes across drug treatments suggests that target-based screens to inhibit their respective proteins may be a screening strategy for antimicrobial potentiators (Liu et al., 2010; Ejim et al., 2011). Their use in generating unique response profiles for bioactive chemicals, however, is limited, given their frequency of occurrence. Where an untapped wealth of information lies is in the large number of uncharacterized or poorly annotated genes that arise as hits when screening for enhancers of slow growth. Indeed, against a broad spectrum of antibiotics, we see systemic relationships between hundreds of uncharacterized genes and essential cell processes targeted by antibiotics.
Data generated by screening our panel of antibiotics represent responses to stressing a broad range of essential processes in E. coli (Figure 1b). Cell wall–targeting drugs are enhanced by mutations encoding proteins interacting with peptidoglycan, such as ∆lpoB and ∆pal. Cells devoid of LpoB are known to be sensitive to β-lactams (Paradis-Bleau et al., 2010), while pal deletions are known to cause periplasmic leakage and sensitivity to drugs (Cascales et al., 2000). These mutations cause structural integrity disruptions in the cell wall and, as such, are unable to withstand even sub-MICs of cell wall–targeting antibiotics. Similarly, ∆rodZ strains lose their typical rod shape, becoming spherical under microscopy. This has an effect on growth rate when exposed to cell wall drugs, but since the peptidoglycan cross-links are not compromised outside of curvature stresses, there is no significant phenotype in endpoint biomass. Additionally, ∆envC knockouts cause cells to become elongated, again with the end effect of slower growth when stressed with cell wall–targeting drugs but not significantly impacting endpoint biomass. Variations in the outer membrane sensitize E. coli to this antibiotic class as well, with several members of the rff operon involved in ECA biosynthesis appearing necessary for cell wall drug resistance. Even in the inner leaflet of the outer membrane, phospholipid composition also appears to play a role antimicrobial resistance.
Delving deeper into protein translation drugs, both endpoint biomass and growth-rate hits were predominantly related to cell envelope permeability. For endpoint, LPS core biosynthetic genes rfaE, waaFGIP, and lpcA all significantly potentiated most translation inhibitors. Each of these deletions was slow growing as well, with waaBDQZ also arising as a hit for growth rate. This follows the paradigm of LPS involvement in the outer membrane as a main permeability barrier in Gram-negatives (Nikaido, 2003; Delcour, 2009). Streptomycin and spectinomycin activity was enhanced by ∆rplI mutants, which are strains deficient in 50S ribosomal protein L9 that accumulate immature small subunits (Naganathan et al., 2015). These drugs were also potentiated by ∆yafQ mutants, which encode the toxin of the DinJ-YafQ toxin–antitoxin system. Deletions in yafQ result in biofilm production that impacts antibiotic resistances, with ∆yafQ becoming susceptible to the aminoglycoside tobramycin and resisting the tetracycline doxycycline (Harrison et al., 2009). This is a pattern similar to that seen with the aminoglycoside streptomycin (and aminocyclitol spectinomycin) and tetracycline. Interestingly, deletions related to ribosome structure were predominately observed as growth-rate phenotypes. Impaired translation tends to correlate with slow growth with some exceptions (Ruusala et al., 1984), and under translation stress, slow growth rates were anticipated. With respect to efflux, the AcrAB-TolC pump was especially well represented in protein translation and DNA replication–inhibitor antibiotics. Of the drugs tested, chloramphenicol, erythromycin, tetracycline, rifampicin, nalidixic acid, and norfloxacin are known to be actively pumped out in E. coli (Nikaido, 1996; Nishino and Yamaguchi, 2001). Conversely, Nikaido et al. (1998) observed in Salmonella typhimurium that cell wall drugs are typically poor substrates for the AcrAB-TolC system, unless they possess hydrophobic side chains.
DNA replication inhibitors exhibited strong endpoint biomass phenotypes upon deletion of endonculease endA or exonuclease VII subunits xseA and xseB. The EndA protein resides in the periplasm, and it is unclear why deletions in its gene would sensitize E. coli to subinhibitory concentrations of quinolones. Deletions in xseA and xseB are known to be sensitive to nalidixic acid (Chase and Richardson, 1977) and ciprofloxacin (Tamae et al., 2008). With respect to permeability, ciprofloxacin and norfloxacin were minimally affected by deletions in genes responsible for LPS core sugars, while deleting rfaE resulted in a strong enhancement of nalidixic acid. This is attributed to the hydrophobic nature of nalidixic acid (logP = 1.01), whereas ciprofloxacin and norfloxacin (logP = −0.81 and −0.92, respectively) are more soluble (Hirai et al., 1986). Across the various DNA replication drugs tested, deletions in genes related to cell division or envelope integrity were common. This was previously reported (Piddock and Walters, 1992) and attributed to an overactive SfiA division inhibitor during SOS response while in the presence of quinolones. It is therefore understandable why a growth-rate defect exists in ΔnlpI strains under DNA damage from replication drugs. Conversely, why ∆paaA mutants are enhancers of slow growth in the presence of DNA replication inhibitors is more puzzling. PaaA is the monooxygenase subunit of the ring 1,2-phenylacetyl-CoA epoxidase, involved in phenylacetate catabolism (Grishin et al., 2011) and used to metabolize environmental aromatic compounds.
In general, growth rate offers a means of identifying more subtle chemical-genetic interactions, but it is important to note that along with this increased sensitivity comes a broader data distribution. Interactions with deletion mutants affecting growth rate are especially interesting in the context of known slow-growth phenotypes, such as interference with ribosome biogenesis, or with aspects of metabolism; an interaction, but not necessarily resulting in a lethal phenotype under screening conditions. Also, the kinetic approach to colony screening does reduce throughput, but the platform can be adjusted to acquire the desired data type. Contrasting our platform to pooling and deep-sequencing approaches to chemical genomics, such as in the case of Lee et al. (2014), our approach is less prone to complex and inevitable competition effects (Hibbing et al., 2010). Further, suppression mutants in our screen are very evident and relatively nondestructive to the overall data, while this would be challenging to identify in a pooling approach using the same collection. A high-density colony approach is not without its drawbacks, but it does offer a unique means of identifying both lethal and nonlethal interactions between bioactive chemicals and genomic libraries. The choice of growth medium will further impact the interactions, especially in the case of minimal salt or cell culture media (such as Roswell Park Memorial Institute [RPMI] medium), with the latter identifying connections that may be more specific in vivo. Indeed, this emphasizes the plasticity of the platform, which is highly customizable to most growth media and strain collections.
The lethal and slow-growth phenotypes described above were classified as interactions in chemical-genetic network maps. Highly populated (nonchemical) nodes in the maps identify genes with deep involvement in E. coli antimicrobial resistance strategies. Many of these lethal interactions are already well characterized, such as the AcrAB-TolC efflux system (Nikaido, 1996; Okusu et al., 1996) and other generic efflux pumps. Combined with network edges arising from growth rate, however, new connections are seen with previously uncharacterized genes and between processes such as cell division and DNA replication. Bacterial division processes have been proposed as targets for new antimicrobials in the past (Lock and Harry, 2008), and here we further show the value of nonessential division processes in antimicrobial potentiation. The platform we present here is highly amendable and can accommodate many types of genomics libraries as they are created, such as plasmid-based overexpression (Kitagawa et al., 2005) or CRISPR-based (Peters et al., 2015) knockdown libraries.
MATERIALS AND METHODS
Bacterial culture conditions and MIC determinations
The Keio collection (Baba et al., 2006) was grown from frozen stocks onto Luria–Bertani (LB) agar medium in 96-density. Colonies were grown overnight at 37°C, then upscaled to 384-density using a Singer Rotor (Singer Instruments, Somerset, UK), and again grown overnight. From the 384-density plates, plates were upscaled to 1536-density, grown overnight, and duplicated in LB medium to make master plates. Master plates were maintained at this density at 4°C for 2–3 wk. Keio plates older than this were recultured using the above steps before screening, to limit the emergence of suppressor strains and/or passaging effects. All plates used in this study contained 25 ml of agar medium and were dried on a completely level surface.
Solid media MICs of drug were determined in a unique manner. First, the liquid MIC was established, which is used as the basis for finding the solid MIC. An initial bed of 25 ml of 2% agarose was then poured into an empty Singer PlusPlate and allowed to dry. A size 16 test tube was used to cut circular holes out of the agarose, and each individual hole was filled with LB agar containing different concentrations of drug until perfectly level with the agarose (∼440 μl). The concentrations spanned from 2× the liquid MIC at the lower end all the way up to 256× the liquid MIC. These plugs were allowed to dry, and the agarose template was removed with a sterile razor, leaving pads of medium containing a concentration gradient of drug (Supplemental Figure S3). Preparing plugs in this manner generates circles of agar that are then the same height as a conventional 25 ml agar pad, with no concave or convex edge curvature. Escherichia coli K-12 BW25113 was pinned in 1536-density from a master plate of the same density onto the agar plugs, using the same Singer Rotor settings throughout this entire study, and was grown for 18 h at 37°C. This ensures that the inoculum for the MIC determination will be equivalent to the inoculum during the chemical-genetic interaction screening.
Chemical-genetic screens were performed by pinning the Keio collection in 1536-density onto antibiotic-supplemented LB agar in duplicate for each respective drug in the experiment. Ranges of concentrations were used for each chemical, typically ranging from 1/16 to 1/4 MIC. It was found that the “sweet spot” for screening concentration was ∼1/4 MIC. This concentration contained the most hits without enriching for pumps and was the concentration used to prepare the network maps. The same inoculation settings for these experiments were used when determining the solid media MICs. Negative controls were run with each Keio collection master plate to reduce batch effects.
Time-lapse imaging of agar plates
The method of Takeuchi et al. (2014) was used to analyze plates in time-lapse, with several modifications (Supplemental Figure S8). Scanning was performed using Scanner Access Now Easy drivers for Linux; scanimage commands were run as parallel system processes, focusing 2.5 mm off the bed of the scanner using the arguments built in for the Epson Perfection V750 scanner. This was driven with an interface written in the R statistical programming language (Ihaka and Gentleman, 1996). In addition, the power to the scanners was remotely controlled using a Web Power Switch 7 (Digital Loggers, Santa Clara, CA), with Python bindings integrated via the rPython library. The power to the scanners was remotely cut after each scan to reduce the heat on the scanning beds, which can reach at least 50°C due to the scanner lamp staying on for 15 min after each scan (Supplemental Figure S8). We found that this heat dries out agar medium substantially and increases edge effects on Singer plates.
All images were quantified using an in-house script written in ImageJ (Schneider et al., 2012). The full pipeline is described in Supplemental Figure S1. Briefly, images were converted to 8-bit grayscale and background subtracted, and a threshold mask was generated. The margin coordinates of these black colonies were then reapplied to the grayscale image. This allows us to get the exact margins of each colony in the original image, without looking for a colony using a predefined grid. By doing so, we allow for variations in plate orientation, and Singer pinning offsets. Gray-value pixel densities were generated from each plate; a single 1536-density image can be quantified in <10 s using this method. The outputs also describe morphological characteristics of each colony, such as radius, marginal undulations, and circularity. The outputs from ImageJ do not have a microwell-plate grid-coordinate system, so we built a grid system in R, using the centers of each colony to predict the best fit for a 1536-density grid of 32 rows and 48 columns, in a manner similar to Wagih and Parts (2014). This extensive reconstruction and gene annotation pipeline is also described in detail in Supplemental Figure S1.
Analyses of screening data
Data outputs from ImageJ were compiled in R. The mu coefficient from the Gompertz growth-rate model (Zwietering et al., 1990) was used as a measure of growth rate, as shown by Takeuchi et al. (2014). We have slightly modified the Takeuchi method, using the entire colony biomass rather than the inner biomass. This was done after observing the growth kinetics of individual colonies (Supplemental Videos 1–3), where, after compensating for the massive initial (t = 0) biomass, the colonies grow outward preferentially. Calculations were done in batch, using an eight-core cluster with the libraries parallel, foreach, and doParallel, with the Gompertz model fit using the grofit library (Kahm and Hasenbrink, 2010). All data from different antibiotics were compiled and then normalized using an in-house normalization function (Figure 2). The normalization utilizes a two-pass system that normalizes rows, then columns, to the interquartile median of the row or column respectively, set at 1 by definition. Full normalization code is provided in the Supplemental Material; it is highly customizable for kinetic or endpoint reads of plates containing 1536 or 6144 colonies. Normalizing in this way is intuitive, rapid, and does not take an iterative approach; it can even be run in a two-step Microsoft Excel formula.
A multiplicative approach was used to identify strains sensitive, or resistant, to the various probes in this study. The approach is visualized in Figure 3, and plots such as the one seen in Figure 5 are based on the deviation of drug-treated deletion strains from their expected value based on the multiplicative rule. Chemical-genetic interaction profiles that are produced in this manner are typically symmetrical ∼1 (Figure 2, d and f), and the hit cutoff was 2.5σ (from the median), excluding statistical outliers. Hits for large data sets were compiled in R, exported to BioLayout3D 3.3 (Theocharidis et al., 2009), and displayed in an edge-weighted force-directed Fruchterman-Reingold layout. Class files were generated for node color based on antibiotic classification and whether genes were interactions in endpoint biomass, growth rate, or both. For individual drug class analyses, networks were created in Cytoscape 3.2.0 (Shannon et al., 2003) with a spring-embedded force-directed layout. All gene annotations and descriptions were determined using Pathway Tools (version 18.5) software with the E. coli K-12 database (Keseler et al., 2013). The database is for E. coli K-12 MG1655 rather than the Keio collection parent strain BW25113, but the gene ontology is not expected to differ.
Supplementary Material
Acknowledgments
This research was supported in part by an operating grant from the Canadian Institutes for Health Research (CIHR; MOP-81330), a Leaders Opportunity Fund grant from the Canada Foundation for Innovation, and a Tier I Canada Research Chair award to E.D.B. S.F. was supported by the CIHR Drug Safety and Effectiveness Cross-Disciplinary Training program (DSECT), and J.-P.C. was supported by Fonds de Recherche en Santé du Québec.
Abbreviations used:
- CFUs
colony-forming units
- ECA
enterobacterial common antigen
- LB
Luria–Bertani
- LPS
lipopolysaccharide
- MCL
Markov cluster
- MIC
minimum inhibitory concentration.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-08-0573) on January 20, 2016.
REFERENCES
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, Youn J-Y, Ou J, Luis B-JS, Bandyopadhyay S, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi AA, Baneyx F. Hyperosmotic shock induces the sigma32 and sigmaE stress regulons of Escherichia coli. Mol Microbiol. 1999;34:1029–1038. doi: 10.1046/j.1365-2958.1999.01664.x. [DOI] [PubMed] [Google Scholar]
- Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J Biomol Screen. 2003;8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- Brochado AR, Typas A. High-throughput approaches to understanding gene function and mapping network architecture in bacteria. Curr Opin Microbiol. 2013;16:199–206. doi: 10.1016/j.mib.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Cascales E, Gavioli M, Sturgis JN, Lloubès R. Proton motive force drives the interaction of the inner membrane TolA and outer membrane pal proteins in Escherichia coli. Mol Microbiol. 2000;38:904–915. doi: 10.1046/j.1365-2958.2000.02190.x. [DOI] [PubMed] [Google Scholar]
- Chase JW, Richardson CC. Escherichia coli mutants deficient in exonuclease VII. J Bacteriol. 1977;129:934–947. doi: 10.1128/jb.129.2.934-947.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour AH. Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu Rev Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- Ejim L, Farha MA, Falconer SB, Wildenhain J, Coombes BK, Tyers M, Brown ED, Wright GD. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol. 2011;7:348–350. doi: 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- Grishin AM, Ajamian E, Tao L, Zhang L, Menard R, Cygler M. Structural and functional studies of the Escherichia coli phenylacetyl-CoA monooxygenase complex. J Biol Chem. 2011;286:10735–10743. doi: 10.1074/jbc.M110.194423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JJ, Wade WD, Akierman S, Vacchi-Suzzi C, Stremick CA, Turner RJ, Ceri H. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob Agents Chemother. 2009;53:2253–2258. doi: 10.1128/AAC.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Aoyama H, Irikura T, Iyobe S, Mitsuhashi S. Differences in susceptibility to quinolones of outer membrane mutants of Salmonella typhimurium and Escherichia coli. Antimicrob Agents Chemother. 1986;29:535–538. doi: 10.1128/aac.29.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- Kahm M, Hasenbrink G. grofit: fitting biological growth curves with R. J Stat Softw. 2010;33 [Google Scholar]
- Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Lee AY, St Onge RP, Proctor MJ, Wallace IM, Nile AH, Spagnuolo PA, Jitkova Y, Gronda M, Wu Y, Kim MK, et al. Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science. 2014;344:208–211. doi: 10.1126/science.1250217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother. 2010;54:1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock RL, Harry EJ. Cell-division inhibitors: new insights for future antibiotics. Nat Rev Drug Discov. 2008;7:324–338. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- Mangat CS, Bharat A, Gehrke SS, Brown ED. Rank ordering plate data facilitates data visualization and normalization in high-throughput screening. J Biomol Screen. 2014;19:1314–1320. doi: 10.1177/1087057114534298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Dieter U, Barr K, Starman R, Hatch L, Rick PD. Nucleotide sequence of the Escherichia coli rfe gene involved in the synthesis of enterobacterial common antigen. Molecular cloning of the rfe-rff gene cluster. J Biol Chem. 1992;267:746–753. [PubMed] [Google Scholar]
- Michaut M, Bader GD. Multiple genetic interaction experiments provide complementary information useful for gene function prediction. PLoS Comput Biol. 2012;8:e1002559. doi: 10.1371/journal.pcbi.1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganathan A, Wood MP, Moore SD. The large ribosomal subunit protein L9 enables the growth of EF-P deficient cells and enhances small subunit maturation. PLoS One. 2015;10:e0120060. doi: 10.1371/journal.pone.0120060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H, Basina M, Nguyen V, Rosenberg EY. Multidrug efflux pump AcrAB of Salmonella typhimurium excretes only those beta-lactam antibiotics containing lipophilic side chains. J Bacteriol. 1998;180:4686–4692. doi: 10.1128/jb.180.17.4686-4692.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara M, Wu HC, Sankaran K, Rick PD. Identification and characterization of a new lipoprotein, NlpI, in Escherichia coli K-12. J Bacteriol. 1999;181:4318–4325. doi: 10.1128/jb.181.14.4318-4325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol. 1996;178:306–308. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Silvis MR, Zhao D, Hawkins JS, Gross CA, Qi LS. Bacterial CRISPR: accomplishments and prospects. Curr Opin Microbiol. 2015;27:121–126. doi: 10.1016/j.mib.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddock LJ, Walters RN. Bactericidal activities of five quinolones for Escherichia coli strains with mutations in genes encoding the SOS response or cell division. Antimicrob Agents Chemother. 1992;36:819–825. doi: 10.1128/aac.36.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruusala T, Andersson D, Ehrenberg M, Kurland CG. Hyper-accurate ribosomes inhibit growth. EMBO J. 1984;3:2575–2580. doi: 10.1002/j.1460-2075.1984.tb02176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JM, Davis JH, Mangat CS, Williamson JR, Brown ED. Discovery of a small molecule that inhibits bacterial ribosome biogenesis. Elife. 2014;3:e03574. doi: 10.7554/eLife.03574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi R, Tamura T, Nakayashiki T, Tanaka Y, Muto A, Wanner BL, Mori H. Colony-Live—a high-throughput method for measuring microbial colony growth kinetics—reveals diverse growth effects of gene knockouts in Escherichia coli. BMC Microbiol. 2014;14:171. doi: 10.1186/1471-2180-14-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamae C, Liu A, Kim K, Daniel S, Hong J, Becket E, Bui A, Solaimani P, Tran KP, Yang H, et al. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J Bacteriol. 2008;190:5981–5988. doi: 10.1128/JB.01982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharidis A, van Dongen S, Enright AJ, Freeman TC. Network visualization and analysis of gene expression data using BioLayout Express3D. Nat Protocols. 2009;4:1535–1550. doi: 10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- Tong A, Evangelista M, Parsons AB, Xu H. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M, et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Nichols RJ, Siegele DA, Shales M. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vales LD, Rabin BA, Chase JW. Subunit structure of Escherichia coli exonuclease VII. J Biol Chem. 1982;257:8799–8805. [PubMed] [Google Scholar]
- Vales LD, Rabin BA, Chase JW. Isolation and preliminary characterization of Escherichia coli mutants deficient in exonuclease VII. J Bacteriol. 1983;155:1116–1122. doi: 10.1128/jb.155.3.1116-1122.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S. Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal App. 2008;30:121–141. [Google Scholar]
- Wagih O, Parts L. gitter: a robust and accurate method for quantification of colony sizes from plate images. G3 (Bethesda) 2014;4:547–552. doi: 10.1534/g3.113.009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwietering MH, Jongenburger I, Rombouts FM, van ‘t Riet K. Modeling of the bacterial growth curve. Appl Environ Microbiol. 1990;56:1875–1881. doi: 10.1128/aem.56.6.1875-1881.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.