

The RTK/Ras/Raf cascade is overactive in cancers. Its targets are the MAP kinases Erks, but Erks are not mutated in cancers. An active Erk, Erk1(R84S), is an oncoprotein. Further, Erk1(R84S) and Erk2(R65S) autophosphorylate the TEY motif and Thr-207/Thr-188. Erk2(R65S) autophosphorylates Thr-188 when dually mutated in the TEY.
Abstract
The receptor-tyrosine kinase (RTK)/Ras/Raf pathway is an essential cascade for mediating growth factor signaling. It is abnormally overactive in almost all human cancers. The downstream targets of the pathway are members of the extracellular regulated kinases (Erk1/2) family, suggesting that this family is a mediator of the oncogenic capability of the cascade. Although all oncogenic mutations in the pathway result in strong activation of Erks, activating mutations in Erks themselves were not reported in cancers. Here we used spontaneously active Erk variants to check whether Erk’s activity per se is sufficient for oncogenic transformation. We show that Erk1(R84S) is an oncoprotein, as NIH3T3 cells that express it form foci in tissue culture plates, colonies in soft agar, and tumors in nude mice. We further show that Erk1(R84S) and Erk2(R65S) are intrinsically active due to an unusual autophosphorylation activity they acquire. They autophosphorylate the activatory TEY motif and also other residues, including the critical residue Thr-207 (in Erk1)/Thr-188 (in Erk2). Strikingly, Erk2(R65S) efficiently autophosphorylates its Thr-188 even when dually mutated in the TEY motif. Thus this study shows that Erk1 can be considered a proto-oncogene and that Erk molecules possess unusual autoregulatory properties, some of them independent of TEY phosphorylation.
INTRODUCTION
Mammalian extracellular regulated kinases (Erks), which include two isoforms, Erk1 and Erk2, and several splicing variants form a subgroup within the family of mitogen-activated protein kinases (MAPKs; Boulton et al., 1991; Marshall, 1995; Avruch et al., 2001; Pearson et al., 2001; Shaul and Seger, 2007; Hynes and MacDonald, 2009; Wortzel and Seger, 2011; Cseh et al., 2014). Erk proteins play a pivotal role in numerous critical processes in cell life and are abnormally overactive in most human cancers (Blume-Jensen and Hunter, 2001; Kohno and Pouyssegur, 2006; Roberts and Der, 2007; Shaul and Seger, 2007; Samatar and Poulikakos, 2014). Nonetheless, Erks have not been reported as proto-oncogenes (see later discussion).
Like most MAPKs, Erks manifest full catalytic activity only when phosphorylated on two neighboring residues, a Thr and a Tyr (part of a TEY motif) located within the protein’s activation loop (Robbins et al., 1993; Canagarajah et al., 1997; Cobb and Goldsmith, 2000). This mode of activation is unique to MAPKs, as most other eukaryotic protein kinases (ePKs) are activated by phosphorylation of a single Thr at their activation loop (Huse and Kuriyan, 2002; Nolen et al., 2004; Taylor et al., 2012). In addition, whereas many ePKs are capable of autophosphorylating their activation-loop threonine, Erk proteins have very poor autophosphorylation activity, which is directed primarily toward the Tyr residue and does not stabilize the active conformation to induce efficient catalysis (Seger et al., 1991; Emrick et al., 2001; Levin-Salomon et al., 2008). Phosphorylation of the Erk TEY motif is mediated by specific kinases known as MAPK kinases (MEKs; Marshall, 1994; Pearson et al., 2001; Catalanotti et al., 2009). Dual phosphorylation of Erk2 imposes dramatic conformational changes that are similar in some aspects to the effects of activation-loop phosphorylation on other kinases, for example, the catalytic subunit of protein kinase A. The phospho-Thr at the activation loop of Erk2, T183, forms a network of interactions, including key interactions with helix C residues R68 and R65, thereby stabilizing the “closed” conformation between the N- and C-lobes (Adams et al., 1995; Canagarajah et al., 1997; Huse and Kuriyan, 2002; Nolen et al., 2004).
Although much is known about the structure–function properties of kinases, prediction of point mutations that would generate the kinase spontaneously active and allow study of its specific effect on cells and organisms is not currently feasible. Some kinases, but not all (Adams et al., 1995), can be made intrinsically active by replacing the activation-loop threonine with Asp or Glu (Gould et al., 1991; Huang and Erikson, 1994; Orr and Newton, 1994). The requirement of dual phosphorylation for activation renders Erks less permissive than other kinases to intrinsic activation by mutations (Askari et al., 2006). However, some bona fide active variants of Erk were reported (Emrick et al., 2006; Levin-Salomon et al., 2008). Erk1(R84S) and Erk2(R65S) were constructed on the basis of a mutation, R68S, discovered in the yeast Erk orthologue Mpk1/Slt2. This point mutation leads to an increase in intrinsic catalytic activity (Levin-Salomon et al., 2008). Other active variants include Mpk1(Y268C) (Y261C in mammalian Erk2), which seems to increase biological but not catalytic activity (in yeast; Levin-Salomon et al., 2008) and Erk2(I84A) that was constructed on the basis of the involvement of I84 in transferring structural information from the gatekeeper residue (Emrick et al., 2006). Another mutant, Erk2(D319N), was constructed on the basis of a gain-of-function mutation known as sevenmaker found in the Drosophila Erk orthologue, Rolled (Bott et al., 1994; Brunner et al., 1994). Despite their apparent potential, it is not clear whether these variants are spontaneously active in living mammalian cells and might serve as tools with which to reveal the physiological and pathological consequences of Erks activity per se.
In a large number of human cancers, the high and constitutive activity of Erks is a result of genetic alterations in components of the RTK/Ras/Raf/MEK pathway that function upstream to Erks (Kohno and Pouyssegur, 2006; Roberts and Der, 2007; Samatar and Poulikakos, 2014). For example, the gene encoding the RTK neu/HER2 is amplified in ∼30% of breast cancer cases, and the RTK EGFR is mutated in ∼25% of non–small cell lung carcinoma patients (Hynes and MacDonald, 2009; Mitri et al., 2012). Activating mutations in Ras are prevalent in pancreatic (∼95%), thyroid (55%), colorectal (35%), and lung (35%) carcinomas and in myeloid leukemia (30%; Bos, 1989; Kranenburg, 2005). The gene encoding the B-Raf kinase is mutated in ∼40% (in some reports 80%) of melanoma cases (Pollock and Meltzer, 2002; Pollock et al., 2003). Mutations in MEK have been detected in ∼2% of melanomas and colon carcinomas (Murugan et al., 2009). The RTK/Ras/Raf/MEK pathway may thus suffer deregulation in most, if not all, human tumors (Hanahan and Weinberg, 2000, 2011). Of interest, a fusion MEK-Erk2 protein was shown to be capable of transforming NIH3T3 cells, further supporting the idea that Erks are important mediators of the oncogenic cascade (Robinson et al., 1998). Of note, however, RTKs and Ras activate other pathways in addition to the Raf/MEK cascade. Furthermore, Raf seems to have important effects on the cell not via Erk (Hindley and Kolch, 2002).
The notion that Erk1/2 play critical roles in executing the biological and pathological functions of the Ras/Raf/MEK cascade in cancer is thus based on a large amount of circumstantial evidence. Direct examination of the matter was not performed because it was hitherto not possible to activate the Erk family itself in the cell, let alone activate each isoform individually, in order to ask whether the pathological consequences of such activation would be similar to the consequences of activation of Raf or MEK.
Here we studied the biochemical properties of intrinsically active Erk molecules and addressed aspects of their mechanism of action. We then tested whether they can be used for asking the critical question of whether active Erk alone could execute some functions of the entire RTK/Ras/Raf/MEK pathway. We found that Erk1(R84S) and Erk2(R65S) are efficiently autophosphorylated in cell culture and are capable of spontaneous activation of the downstream pathway. Moreover, we report that NIH3T3 cells expressing Erk1(R84S) form foci on plastic plates, colonies in soft agar, and tumors in nude mice.
Analysis of the biochemical properties of Erk1(R84S) and Erk2(R65S) revealed that they efficiently autophosphorylate their TEY motif, which explains their intrinsic, MEK-independent activity. Unexpectedly, liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis revealed that Erk1(R84S) and Erk2(R65S) autophosphorylate additional residues. One of those is Thr-207/Thr-188 (in Erk1/Erk2), which was reported to be abnormally phosphorylated in the hearts of patients with cardiac hypertrophy (Lorenz et al., 2009; Ruppert et al., 2013). Strikingly, Erk2(R65S) efficiently autophosphorylates its Thr-188 residue even when mutated in the TEY motif, presenting the first case of an Erk molecule that manifests catalysis in the total absence of TEY phosphorylation.
Thus we report a unique autophosphorylation activity of Erk molecules that does not require activation-loop phosphorylation. We further report that Erk1(R84S) transforms NIH3T3 cells, indicating that Erks are critical mediators of the oncogenic proficiency of the RTK/Ras/Raf/MEK cascade.
RESULTS
Erk1(R84S) and Erk2(R65S) are spontaneously active in HEK293 and NIH3T3 cells
Several mutants of the Erk family were reported for their intrinsic catalytic activity or gain-of-function property (Brunner et al., 1994; Emrick et al., 2001, 2006; Levin-Salomon et al., 2008). The prominent ones are 1) mammalian Erk1(R84S), Erk2(R65S), Erk1(I103A), and Erk2(I84A), which are catalytically intrinsically active as purified recombinant proteins (Emrick et al., 2006; Levin-Salomon et al., 2008), 2) Drosophila Rolled(D334N) (D338N/D319N in Erk1/Erk2 numeration), which carries the gain-of-function mutation known as sevenmaker (Brunner et al., 1994), and 3) yeast Mpk1(Y268C) (Y280C/Y261C in Erk1/Erk2 numeration; Levin-Salomon et al., 2008). To test whether the intrinsic activity or gain-of-function properties of the mutants could also be manifested in mammalian cells, we transiently expressed the Erk1 and Erk2 mutants in HEK293 cells and monitored the level of phosphorylation of their TEY motif (Figure 1A). Of the mutants tested, Erk1(R84S), Erk1(D338N), Erk2(R65S), and Erk2(I84A) were found to be spontaneously phosphorylated, namely, in serum-starved cells, not exposed to any stimulus, at levels higher than the respective Erk1(WT) and Erk2(WT) proteins (Figure 1A). Erk2 molecules carrying the mutation equivalent to Y268C of Mpk1 (Erk1(280C) and Erk2(Y261C)) or the sevenmaker mutation in Erk2 (D319N) or the I103A mutation in Erk1 were not spontaneously phosphorylated at high levels (Figure 1A).
FIGURE 1:

Some Erk1 and Erk2 variants are spontaneously phosphorylated when expressed in HEK293T and NIH3T3 cells. (A) pCEFL vectors carrying cDNAs encoding the indicated Erk1/2 molecules or a control empty vector were introduced into HEK293 cells. At 48 h posttransfection, cells were serum starved for 24 h and harvested. Protein lysates prepared from these cells were analyzed by Western blotting, using antibodies that specifically recognize phospho-Erk or the relevant tags (6xHis-Erk1, HA-Erk2, or GAPDH). (B) The same pCEFL vectors were introduced to NIH3T3 cells. At 48 h posttransfection, the cells were harvested and subjected to Western blot analysis with the indicated antibodies.
The same Erk mutants were also transiently expressed in NIH3T3 cells. In this case serum starvation was not applied because this procedure had a hazardous effect on the cells. Nevertheless, in these cells too, Erk1(R84S) and Erk2(R65S) were spontaneously phosphorylated at higher levels than the ectopically expressed Erk1(WT) and Erk2(WT) proteins (Figure 1B). Of note, for an unknown reason, Erk2(D319N) and Erk2(Y261C), which were expressed in HEK293 cells, were not expressed at all in NIH3T3 cells (Figure 1B).
The foregoing experiments show that Erk1(R84S) and Erk2(R65S) are spontaneously phosphorylated/activated in HEK293 and NIH3T3 cells. To check whether those Erk proteins can activate their downstream targets, we monitored their effect on relevant reporter genes of the pathway. We observed that in HEK293T cells, Erk1(R84S), but not Erk2(R65S), induced transcription of an AP-1–mediated promoter (AP-1 luciferase; Askari et al., 2007; Figure 2A). We also tested activation of a promoter known to respond to active Ras, containing a Ras-responsive element (a combination of AP-1 and Ets elements; Foos et al., 1998). This reporter gene (pY2) was efficiently activated (14-fold) by transiently expressed Erk1(R84S) in HEK293 cells and also, but less efficiently (fourfold), by Erk2(R65S) (Figure 2B). Thus, Erk2(R65S), although seemingly strongly activated based on its phosphorylation status (Figure 1A), was not efficiently activating the downstream cascade. Erk1(R84S), on the other hand, was spontaneously active in all assays. Table 1 summarizes the properties of the mutants tested.
FIGURE 2:
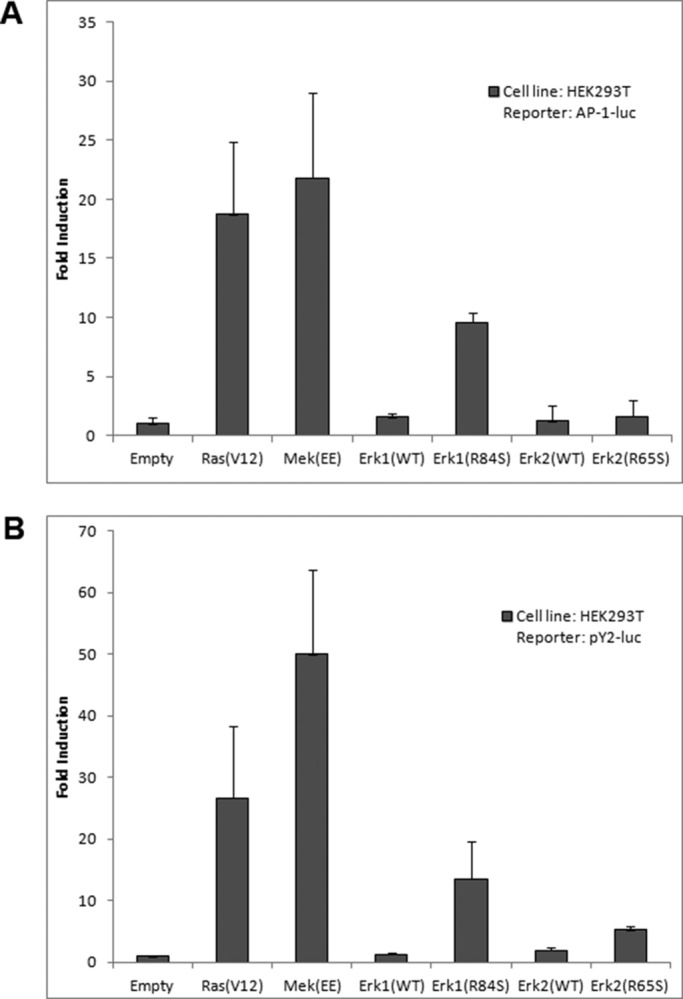
Erk1(R84S) induces AP-1– and (AP-1+Ets)–mediated transcriptional activity in HEK293T cells. HEK293T cells were cotransfected with the indicated pCEFL vectors together with (A) AP-1-luc or (B) (AP-1+Ets)–luciferase plasmids. A plasmid encoding Renilla luciferase (pRL-TK) was also added to each transfection mixture. At 48 h posttransfection, cells were harvested, and the lysates were subjected to the luciferase assay as described in Materials and Methods.
TABLE 1:
Biochemical and physiological properties acquired by Erk molecules.
| Spontaneous phosphorylation in HEK293 cells | Spontaneous phosphorylation in NIH3T3 cells | Spontaneous phosphorylation in stable NIH3T3 cultures | Activation of AP-1–luciferase in HEK293 cells | Activation of pY-2–luciferase in HEK293 cells | |
|---|---|---|---|---|---|
| Erk1(WT) | + | – | – | 2-fold | 1-fold |
| Erk1(R84S) | +++ | ++ | +++ | 10-fold | 14-fold |
| Erk1(I103A) | – | – | – | Not tested | Not tested |
| Erk1(D338N) | ++ | – | Not tested | Not tested | Not tested |
| Erk1(Y280C) | + | – | Not tested | Not tested | Not tested |
| Erk2(WT) | – | – | – | 0-fold | 2-fold |
| Erk2(R65S) | ++ | + | – | 2-fold | 5-fold |
| Erk2(I84A) | + | – | – | Not tested | Not tested |
| Erk2(D319N) | +/– | – | Not tested | Not tested | Not tested |
| Erk2(Y261C) | – | – | Not tested | Not tested | Not tested |
Erk1(R84S) transforms NIH3T3 cells
A key question regarding the function of Erks as downstream targets of the RTK/Ras/Raf/MEK pathway is whether the oncogenic proficiency of the pathway could be mediated solely via the activation of Erk molecules. This issue could be addressed by asking whether activation of Erks is sufficient to impose oncogenic transformation. We thus tested whether expression of some of the intrinsically active mutants transform NIH3T3 cells. By selecting G418-resistant cultures, after transfection of the relevant expression vectors to NIH3T3 cells, we established cultures that stably express hexahistidine (6xHis)-tagged Erk1(WT), Erk1(R84S), or Erk1(I103A) or hemagglutinin (HA)-tagged Erk2(WT), Erk2(R65S), or Erk2(I84A). As controls, we prepared G418-resistant cells constitutively expressing Ras(G12V) or MEK(EE) and cells harboring an “empty” vector. Spontaneous phosphorylation of Erk was observed in cells stably expressing Ras(G12V) or 6xHis-tagged Erk1(R84S) (Figure 3A). Phosphorylation of Erk1(R84S) was unusually strong. However, this high phosphorylation stems not only from the intrinsic activity of this mutant, but also from the high steady-state level of the protein, as reflected in a Western blot using anti-polyhistidine antibodies (Figure 3A). It seems that the R84S mutation affects the proteins at two levels; first, it increases its intrinsic (MEK-independent) catalytic properties (Levin-Salomon et al., 2008), and second, it endows it with a degree of stability that results in high steady-state levels. Unexpectedly, Erk2(R65S) was not spontaneously phosphorylated in the stable NIH3T3 cell culture (Figure 3A). In transient expression experiments with the same cell line, Erk2(R65S) was phosphorylated at higher levels than Erk2(WT), although these levels were not as high as in HEK293 cells (Figure 1, A and B).
FIGURE 3:

Erk1(R84S) is highly expressed and strongly phosphorylated and induces oncogenic transformation of NIH3T3 cells. (A) NIH3T3 cells were transfected with the indicated expression vectors and selected for stable presence of the plasmid using the antibiotic G-418. Four weeks after transfection, cells were harvested. Protein lysates prepared from these cells were subjected to Western blotting, using antibodies that specifically recognize the indicated proteins. (B) NIH3T3 cells were transfected with the indicated vectors and grown in the presence of G-418. Cells were harvested at the indicated time points, and protein lysates prepared were analyzed by Western blot. Asterisk indicates the transfected HA-Erk2. (C) NIH3T3 cells transfected with the indicated vectors were allowed to grow to high density and reach confluence. Continuation of proliferation and appearance of foci was monitored. Cells were fixed and stained with crystal violet 4 wk after transfection. (D) Cells expressing Erk1(R84S) form colonies in soft agar. Cultures of NIH3T3 cells stably expressing the indicated proteins were seeded in triplicate in 96-well plates containing soft agar. Two weeks after plating, cells were stained with MTT and photographed.
In an attempt to understand why Erk2(R65S) is not phosphorylated when it is constitutively expressed, we transfected NIH3T3 cells with the expression vectors, applied G418 selection, and monitored expression levels and phosphorylation status of the ectopically expressed Erk proteins (Figure 3B). We observed that in the first days posttransfection, both Erk1(R84S) and Erk2(R65S) were phosphorylated. During the course of selection, however, in which cells that did not integrate the expression vectors to the genome were dying, phosphorylation of Erk2(R65S) was not detected (Figure 3B). This explains the observation that Erk2(R65S) is not phosphorylated in NIH3T3 cultures that stably express it (Figure 3A). We also noted that expression levels of Erk2 molecules, particularly Erk2(R65S), were also very low, as long exposure time of the Western blot film were required to observe it. Perhaps active Erk2 harms the well being of cells, enforcing a selective pressure that selects cells in which Erk2(R65S) is not phosphorylated/activated. By contrast, expression and phosphorylation levels of Erk1(R84S) remained high.
To test whether the cultures that stably express the Erk variants may be oncogenically transformed, we allowed cells to reach 100% confluence and checked whether any of the cultures lost the capability of contact inhibition of growth. Unregulated growth and appearance of foci was observed in cultures harboring vectors expressing Ras(G12V), MEK(EE), or Erk1(R84S) (Figure 3C). No foci were developed by cells harboring vectors expressing Erk1(I103A), Erk2(I84A), or Erk2(R65S) (Figure 3C and unpublished data). We further tested the capability of the NIH3T3 cultures to form colonies in soft agar. In agreement with their ability to form foci, cells of cultures expressing MEK(EE) or Erk1(R84S) developed colonies in soft agar, whereas cells harboring an empty vector or a vector carrying Erk1(WT), Erk1(I103A), Erk2(WT), Erk2(I84A), or Erk2(R65S) did not (Figure 3D). Finally, we tested whether cells of the various NIH3T3 cultures form tumors when subcutaneously injected to nude mice. Within 8 d postinjection, we observed that mice injected with cells expressing MEK(EE) or Erk1(R84S) developed large tumors, whereas mice injected with cells expressing Erk1(WT) or harboring an empty vector formed almost undetectable tumors (Table 2). Two weeks postinjection, mice injected with cells expressing Erk1(R84S) manifested tumors 2.5 times larger than those injected with cells expressing MEK(EE) (Table 2). Thus, based on the criteria of forming foci, colonies in soft agar, and tumors in nude mice, the cells expressing Erk1(R84S) could be regarded as transformed. This result strongly suggests that Erk1 molecules can become oncogenic.
TABLE 2:
Volume of subcutaneous tumors that evolved in nude mice after injection of cells.
| Tumor volume (mm3) at given time after injection of cells | |||
|---|---|---|---|
| Plasmid | At 0 d | At 8 d | At 15 d |
| Empty | 0 | 0 | 0 |
| MekEE | 0 | 308 ± 96.2 | 1009 ± 394.7 |
| ERK1(WT) | 0 | 59 ± 63.9 | 535 ± 472.9 |
| ERK1(R84S) | 0 | 333 ± 165.7 | Killed |
Each value represents an average volume of tumors measured in 10 mice.
It is not clear whether Erk2 could also become oncogenic. Because Erk2(R65S) carries a mutation equivalent to R84S of Erk1 and is similarly active catalytically (Levin-Salomon et al., 2008), it would be expected to be an oncoprotein too. The results of Robinson et al. (1998) with the MEK-Erk2 fusion protein also suggest that Erk2 could become an oncoprotein. Erk2(R65S) is a less efficient activator of AP-1– and (AP-1+Ets)–mediated transcription than Erk1(R84S) (Figure 2), suggesting that perhaps active Erk2 might not be as oncogenic as active Erk1. In any case, because in the stable NIH3T3 culture, Erk2(R65S) is not phosphorylated and is expressed at low level (Figure 3A), we cannot tell what would be the phenotype of cells in which Erk2 is highly active.
The intrinsic activity of Erk1(R84S) and Erk2(R65S) is associated with efficient autophosphorylation of their TEY motif
The described experiments show that Erk1(R84S) and Erk2(R65S) are catalytically active spontaneously in mammalian cells and that Erk1(R84S) is an oncoprotein. It is important therefore to reveal their biochemical properties, particularly the mechanism underlying their MEK-independent activity.
The R84S and R65S mutations in Erk1 and Erk2, respectively, were generated based on a corresponding mutation, R68S, in the yeast Mpk1/Slt2, which rendered the protein independent of its MEKs (Levin-Salomon et al., 2008), namely, the Mpk1(R68S) protein was active in the total absence of its MEKs Mkk1 and Mkk2 and allowed mkk1∆mkk2∆ cells to proliferate in the presence of caffeine (Levin-Salomon et al., 2008; Figure 4A, row 4). This yeast system thus provides a clear-cut assay for monitoring whether a given mutant is truly independent of upstream signaling. In an attempt to reveal the mechanism that rendered Erk1(R84S) and Erk2(R65S) intrinsically active, we thus also studied the properties of the yeast Mpk1(R68S) protein, taking advantage of the biological output it enables. We first tested whether Mkk1/2-independent ability of Mpk1(R68S) requires phosphorylation of the TEY motif, as this phosphorylation is normally catalyzed by Mkk1/2. We therefore looked at the phosphorylation status of Mpk1(R68S) in mkk1∆mkk2∆ cells. We found that this mutant, unlike Mpk1(WT), is phosphorylated on its TEY motif, although to a low level, in mkk1∆mkk2∆ cells (Figure 4B, lane 4). This phosphorylation could be a result of either phosphorylation by a MEK other than Mkk1 or Mkk2 or autophosphorylation activity. Two approaches were taken to distinguish between these possibilities. First, we expressed Mpk1(R68S) in cells of the mkk1∆mkk2∆pbs2∆ste7∆ strain (Levin-Salomon et al., 2009), which lacks all four yeast MEKs, and tested the ability of the transformants to proliferate on medium supplemented with caffeine. We observed that mkk1∆mkk2∆pbs2∆ste7∆ cells expressing Mpk1(R68S) did proliferate to a certain degree under these conditions (Figure 4C, row 4). In addition, the Mpk1(R68S) protein was phosphorylated, although to a low level, in these cells (Figure 4D, lane 4). It seems therefore that the mechanism underlying the Mkk1/2-independent activity of Mpk1(R68S) does not involve activation by another MEK. Second, we prepared a kinase-dead version of Mpk1(R68S) in order to check whether its phosphorylation is dependent on its own catalytic activity. For that, we replaced the Lys-54, critical for ATP binding, with Arg. As expected (Robinson et al., 1996), the K54R mutation is hazardous to the enzyme, as it rendered Mpk1(WT) incapable of rescuing mpk1∆ cells (Figure 4E, row 4). Similarly, the Mpk1(R68S+K54R) protein did not allow growth on caffeine of mkk1∆mkk2∆ cells (Figure 4A, bottom row) or mkk1∆mkk2∆pbs2∆ste7∆ cells (Figure 4C, bottom row) and was not phosphorylated in these cells (Figure 4, B and D). These results combined indicate that Mpk1(R68S) acquired its intrinsic activity via autophosphorylation of its activation loop.
FIGURE 4:

The intrinsic activity of Mpk1(R68S) is dependent on the autophosphorylation of the TEY motif. mkk1∆mkk2∆ cells (A), mkk1∆mkk2∆pbs2∆ste7∆ cells (C), or Mpk1∆ cells (E) expressing the indicated Mpk1 molecules were plated in five dilutions on plates containing YPD supplemented with 15 mM caffeine (left) or on plates not containing caffeine (YNB –URA; right). Western blot analysis with the indicated antibodies was performed on protein lysates prepared from mkk1∆mkk2∆ cells (B), mkk1∆mkk2∆pbs2∆ste7∆ cells (D), or Mpk1 cells (F) expressing the indicated Mpk1 molecules.
We could not test directly, in vitro, the autophosphorylation activity of Mpk1(R68S) because the protein was insoluble during various purification attempts. Unlike Mpk1, the mammalian Erk proteins carrying the equivalent mutation, Erk1(R84S) and Erk2(R65S), could be readily purified as recombinant proteins (Levin-Salomon et al., 2008), and we were able to test their capability of autophosphorylation in a kinase assay containing [γ-32P]ATP and no other substrate. The mutants manifested efficient autophosphorylation capability in this assay (Figure 5A). Western blot analysis of the autophosphorylated proteins, using anti–phospho-Erk antibodies, showed that autophosphorylation events occurred at the TEY motif (Figure 5B). This was also confirmed by capillary LC/MS/MS analysis; see later description). We then tested the activity of Erk2(R65S) bearing mutations at T183 and Y185 of the TEY motif. All proteins tested—Erk2(R65S+T183A), Erk2(R65S+Y185F), and Erk2(R65S+T183A+Y185F)—failed to phosphorylate the substrate provided (myelin basic protein [MBP]; Figure 5C), indicating that the spontaneous activity of Erk2(R65S) toward MBP was absolutely dependent on TEY phosphorylation. Thus autophosphorylation of both T183 and Y185 is essential for the intrinsic activity of the intrinsically active Erks.
FIGURE 5:

Erk1(R84S) and Erk2(R65S) are capable of efficient autophosphorylation at the TEY motif. (A) Autophosphorylation capabilities of the purified Erks were assessed by incubating the proteins in a kinase assay mixture with [γ-32P]ATP and no other substrate. Reactions were terminated at the indicated time points, separated via SDS–PAGE, stained with Coomassie brilliant blue, and exposed to x-ray film. (B) Autophosphorylation of the active variants occurs at the TEY motif. The indicated proteins were subjected to Western blot analysis using antibodies that react with Erk1/2 proteins phosphorylated at their TEY motif (αpErk) or with antibodies that react with the polyhistidine tag (αHis). (C) Intact TEY motif is essential for catalytic activity of Erk2(R65S) toward the substrate MBP. Catalytic activity of purified recombinant Erks carrying mutations at the TEY motif was analyzed with or without preincubation with active MEK1, using [γ-32p]ATP and MBP as substrates. Reaction mixtures were spotted on filter papers and quantified. Activity of MEK1-activated Erk2(WT) was defined as 100%. In parallel, a sample from each reaction was subjected to SDS–PAGE, stained with Coomassie brilliant blue, and exposed to x-ray film.
In addition to autophosphorylating their TEY motif, Erk1(R84S) and Erk2(R65S) also autophosphorylate their Thr-207/Thr-188 residues
Although Erk1(R84S) and Erk2(R65S) are active catalytically and biologically independent of MEK, they still require phosphorylation of their TEY motif for their activity (Figure 5C). Western blot analysis with anti–phospho-TEY antibodies (anti–phospho-Erk) indeed verified that the strong autophosphorylation activity of these mutants, measured by radioactivity (Figure 5A), is directed toward the TEY motif both in vitro and in cell cultures (Figures 1A and 5B). To unequivocally confirm this conclusion, we further determined the phosphorylated sites on Erk1(R84S) and Erk2(R65S) after autophosphorylation reactions, via capillary LC-MS/MS analysis. This analysis confirmed that Erk1(R84S) and Erk2(R65S) are phosphorylated on their TEY motif but disclosed unexpected phosphorylation events on additional phosphorylation sites. Only one of these sites, T207 in Erk1/T188 in Erk2, was found to be autophosphorylated in both Erk1(R84S) and Erk2(R65S). With antibodies generated specifically against phospho–Thr-207-Erk1, which cross-react with phospho–Thr-188-Erk2 (a gift of Steven Pelech and Kinexus Bioinformatics Corporation, Vancouver, Canada) or with a commercial antibody raised against phospho–Thr-188-Erk2 (Badrilla, Leeds, United Kingdom), we observed that Erk1(R84S) and Erk2(R65S) were spontaneously phosphorylated on Thr-207/Thr-188 not only in vitro (Figure 6A) but also when transiently expressed in HEK293 cells (Figure 6B). Phosphorylation of Thr-207/Thr-188 did not change when the cells were exposed to epidermal growth factor (EGF) (Figure 6B). Of importance, not only the active variant, but also Erk2(WT) was phosphorylated to some degree on T188 when expressed in HEK293 cells (Figure 6B), indicating that this phosphorylation event is not peculiar to the active mutants. We thus further tested the status of T207 phosphorylation in the NIH3T3 cells transformed by Erk1(R84S) and found that Erk1(R84S) is constantly phosphorylated on T207 (Figure 6C). Of note, phosphorylation of T188 was previously reported to occur in the hearts of patients with cardiac hypertrophy (Lorenz et al., 2009; Ruppert et al., 2013). T188 phosphorylation in Erk2 was proposed to have an activating effect on the protein because in Erk2(T188D) knock-in mice, Erk’s substrates were hyperphosphorylated, and the mice were prone to cardiac hypertrophy. The effect of T188 phosphorylation on the catalytic properties of the protein was not reported, however (Lorenz et al., 2009; Ruppert et al., 2013). Structural analysis revealed that the hydroxyl of the T188 side chain forms hydrogen bond interactions with the catalytic base, D147, which is responsible for activating Erk’s substrates. The T188-D147 interaction was proposed to contribute to the stabilization of the activation-loop conformation, constraining activation of T183 and Y185 by D147 and thereby preventing their autophosphorylation (Emrick et al., 2006). In an attempt to test whether in Erk2(R65S) the spontaneous T188 phosphorylation enhances catalysis, we mutated T188 to Ala or to Asp in both Erk2(WT) and Erk2(R65S). Erk2(T188A), but not Erk2(T188D), manifested a dramatic increase in autophosphorylation activity as monitored in a radioactive autophosphorylation assay (Figure 6D). Mutating T188 to either Asp or Ala in Erk2(R65S) had no effect on its autophosphorylation activity (Figure 6D). Capillary LC-MS/MS analysis showed that in both Erk2(T188A) and Erk2(R65S+T188A), the efficient autophosphorylation is directed exclusively toward Y185, whereas T183 remains unphosphorylated. As a result, these proteins are not expected to be catalytically active toward external substrates. This was indeed verified in a kinase assay with MBP as a substrate (Figure 6E, right). Furthermore, Erk molecules carrying mutations in T188 were inactive even after MEK phosphorylation (Figure 6E, left), indicating that intact T188 is critical for catalysis. Perhaps phosphorylation of T188 is an inhibitor of Y185 phosphorylation, yet the exact consequence of T207/T188 phosphorylation is yet to be determined (see Discussion).
FIGURE 6:

The active variants Erk1(R84S) and Erk2(R65S) are autophosphorylated at a novel phosphoacceptor, T207 (in Erk1) and T188 (in Erk2), which is critical for catalytic activity. (A) The indicated Erk proteins (recombinant purified) were subjected to Western blot analysis, using antibodies specifically raised against phospho-Thr-207/Thr-188 (αpT207/T188) and anti-polyhistidine antibody (αHis). (B) The indicated ERKs were introduced into HEK293T cells. At 48 h posttransfection, cells were serum starved for 24 h. Then cells were exposed or not exposed to 50 ng/ml EGF for 10 min, harvested, and subjected to Western blot analysis using the indicated antibodies. (C) Phosphorylation of T207 in Erk1(R84S) is not affected by EGF. NIH3T3 cells stably expressing Erk1(WT) or Erk1(R84S) were collected, and protein lysates were prepared at the indicated time point after EGF addition. Cell lysates were subjected to Western blot analysis using the indicated antibodies. (D) Erk2(T188A), but not Erk2(T188D), manifested dramatic increase in autophosphorylation activity. Autophosphorylation of purified Erk2(R65S), Erk2(T188A), and Erk2(T188D) was tested by incubating the proteins in a kinase assay mixture with [γ-32P]ATP and no other substrate. Samples were removed from the assay at the indicated time points and subjected to SDS–PAGE. Gels were stained with Coomassie brilliant blue, dried, and exposed to x-ray film. (E) Mutating T188 of Erk2 to either Asp or Ala diminished its catalytic activity. Catalytic activity of the indicated protein was assayed as described in the legend to Figure 5.
Because any catalytic activity of MAPKs is dependent on phosphorylation of the TXY motif, we sought to verify that this also accounts for the spontaneous autophosphorylation of Erk2(R65S) toward T188. We thus measured T188 phosphorylation in the Erk2(R65S+T183A+Y185F) protein. As a control, we tested the autophosphorylation of Erk2(WT) mutated in its TEY motif (Figure 7B). Totally unexpectedly, Erk2(R65S+T183A+Y185F) was found to be a very efficient autophosphorylating enzyme, even more efficient than Erk2(R65S) (Figure 7A, bottom). In addition, intriguingly, Erk2(T183A) manifested an efficient autophosphorylation activity, significantly higher than that of Erk2(WT) (Figure 7A, top). Thus activation-loop phosphoacceptors may be inhibitors of an autophosphorylation reaction. A Western blot analysis confirmed these findings and showed that all proteins carrying the R65S mutation, including Erk2(R65S+T183A+Y185F), were phosphorylated on T188 (Figure 7B). LC/MS/MS analysis further confirmed that Erk2(R65S+T183A+Y185F) is phosphorylated on T188 and perhaps on several more residues. Autophosphorylation of Erk2(T183A) occurs mainly on Tyr-185 as the protein reacts with the common anti–phospho-Erk antibodies (Figure 7B). To our best knowledge, this is the first case of a MAPK molecule that manifests any type of catalysis in the total absence of TXY phosphorylation. The R65S mutation may enforce a conformation that particularly supports phosphorylation of T188.
FIGURE 7:

T188 is phosphorylated in Erk2(R65S) independently of TEY phosphorylation. (A) Autophosphorylation capabilities of purified recombinant Erks carrying mutations at the TEY motif were tested in the presence of [γ-32P]ATP as described in the legend to Figure 5A. (B) Purified recombinant Erks carrying mutations at the TEY motif were subjected to Western blot analysis using antibodies specifically raised against phospho–Thr-207/Thr-188 (αpT207/T188), as well as αpErk and αErk antibodies.
The R65S mutation induces just subtle changes in Erk2’s crystal structure
Changing the residue at the 84 position of Erk1 and the 65 position of Erk2 results in dramatic changes in the properties of the proteins (Figures 1–3 and 5). The structural function of R65, as revealed from the crystal structures of Erk2 and phospho-Erk2 (Zhang et al., 1994; Canagarajah et al., 1997), is to assist in stabilizing the domain closure and promote the geometry of the phosphate-binding ribbon in the active conformation. These effects are a consequence of R65 interaction with phospho-T183, bridged by a solvent molecule. In an effort to reveal the effect of the R65S mutation on Erk2’s conformation, we elucidated the crystal structure of Erk2(R65S) at a resolution of 1.53 Å. (The protein DataBank numbers of the described structures are ERK2(WT) 4S31; ERK2(R65S) 4S2Z, and the data collection is shown in Supplemental Table S1a.) Comparing the structure of Erk2(R65S) and that of Erk2(WT) (including that of Erk2(WT), which we crystallized) revealed no significant differences, global or local, between the proteins. We further established the crystal structure of Erk2(WT) and Erk2(R65S) bound to β,γ imido-adenosine 5′-triphosphate (AMP-PNP) and again could not observe significant differences between the proteins. (The protein DataBank numbers of the described structures are ERK2-ANP(WT) 4S32; ERK2(R65S)-ANP 4S33, and the data collection is shown in Supplemental Table S1b.) A single intriguing observation was the presence of hydrogen bond interaction between S65 (in Erk2(R65S) and Y34 (Figure 8). Because Y34 resides in the P-loop, this new interaction may somehow affect ATP binding. Nonetheless, the fact that the dramatic differences in the catalytic properties of the protein are not reflected in the crystal structure is enigmatic.
FIGURE 8:
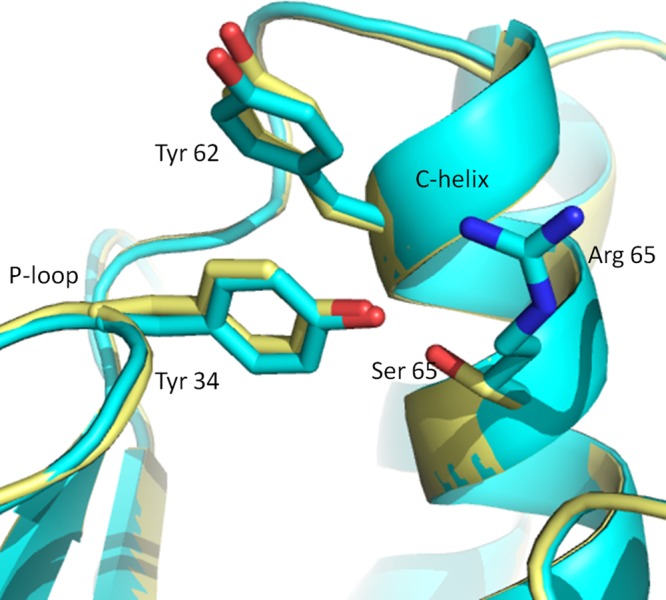
Minor conformational changes, apparent only at the site of mutation, are observed when the crystal structure of Erk2(R65S) is compared with that of Erk2(WT). Superimposition of the structures of Erk2(R65S) (yellow) and Erk2(WT) (cyan) at the region of the mutation. The side-chain hydroxyl of S65 in Erk2(R65S) is directed toward Y34 from the P-loop, forming a polar interaction, whereas R65 in Erk2(WT) does not form any interaction with this region. Another hydrophobic interaction, between Y34 and Y62 from the C-helix, is similar in both proteins.
In summary, the yeast Mpk1(R68S) and the mammalian Erk1(R84S) and Erk2(R65S) mutants are able to bypass the need of MEK-mediated phosphorylation because they have the capability to autophosphorylate the TEY motif, perhaps via reorientation of the phosphate- binding ribbon motif. Erk1(R84S) and Erk2(R65S) further acquired the ability to spontaneously autophosphorylate other regulatory sites, primarily T207/T188, a site that is essential for catalysis. Not only is phosphorylation of T207/T188 independent of TEY phosphorylation, but also the TEY motif seems to inhibit this autophosphorylation reaction.
DISCUSSION
In most cell types, activation of the RTK/Ras/Raf/MEK pathway leads to dramatic physiological effects (Hynes and MacDonald, 2009; Lemmon and Schlessinger, 2010). Constitutive activity of the pathway is measured in almost all cancer cases (Kohno and Pouyssegur, 2006; Roberts and Der, 2007; Hanahan and Weinberg, 2011), and expression of constitutively active (oncogenic) variants of RTKs, Ras, MEK (Cowley et al., 1994; Mansour et al., 1994), or a MEK-Erk2 fusion protein (Robinson et al., 1998), transforms cells in culture (Voisin et al., 2008). Current understanding is therefore that Erk proteins are the only targets of this pathway, implying that activation of Erks alone would be oncogenic, but perhaps not as oncogenic as are upstream components, because RTKs, Ras, and Raf activate more pathways that also contribute to oncogenesis (Marshall, 1991; Campbell et al., 1998; Yamamoto et al., 1999; Lemmon and Schlessinger, 2010). The observation that Erk1(R84S) acquired oncogenic properties supports the notion that activation of Erk is a very important element in the oncogenic effect of the cascade. Nonetheless, active RTKs or Ras seem to be more efficient and more aggressive oncoproteins than Raf, which in turn is more aggressive than active MEKs, which are quite rare in cancer (Murugan et al., 2009). Perhaps oncogenic transformation by active Erk is even less efficient and less severe, explaining why active Erk has not been identified in diseases. It is curious, however, that in a single cancer case, reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) database, a patient with endometrium carcinoma was carrying an R84H mutation in her ERK1 gene (mutations ID, COSM4875436). Furthermore, a screen for mutations that render cells resistant to several chemotherapeutic agents also identified the R84H mutation in ERK1 (Goetz et al., 2014). It could be, therefore, that activating mutations in ERKs, particularly mutations in R84, will be proven to have clinical significance. In any case, the finding of this study pointing to ERK1 as a proto-oncogene suggests that Erk1 is a critical downstream target of RTKs, Ras, and Raf as a mediator of their oncogenicity.
We cannot conclude whether Erk2 is also a proto-oncogene because we could not obtain stable clones in which Erk2(R65S) is highly expressed and phosphorylated. Nonetheless, the observation that MEK-Erk2 protein is oncogenic (Robinson et al., 1998) suggests that Erk2 might also be a proto-oncogene.
The fact that MEK is an activator of both Erk1 and Erk2 and is a bona fide proto-oncogene (Cowley et al., 1994; Mansour et al., 1994; Murugan et al., 2009) might imply that simultaneous activation of both Erk1 and Erk2 would give rise to a more efficient transformation. We therefore coexpressed active variants of both Erk1 and Erk2 in NIH3T3 cells but did not observe more foci than those that appeared after expression of Erk1(R84S) alone. It could be, however, that Erk2(R65S) is not the proper molecule with which to ask this question because it seems that the cells (at least NIH3T3 cells) somehow inactivate it.
The mechanism underlying the MEK-independent activity of Mpk1(R68S), Erk1(R84S), and Erk2(R65S) involves acquirement of efficient autophosphorylation capabilities on both the Thr and Tyr of the activation loop. Erk proteins were previously reported to possess weak autophosphorylation capability, primarily toward Tyr residue only (Seger et al., 1991), or autophosphorylation toward T188 (of Erk2) under particular conditions (Lorenz et al., 2009). Here we showed that Erk proteins do possess an efficient autophosphorylation and autoactivation capability, suggesting that Erks are similar to most ePKs in this respect. However, this activatory autophosphorylation ability, which is spontaneous in most ePKs, is occluded in Erks and must be uncovered. It is not known which structural element(s) restrict spontaneous autophosphorylation in Erks and distinguish them from most ePKs. Perhaps the MAPK insert domain is involved (Beenstock et al., 2014). We were able to unmask the autophosphorylation activity of Erks by converting the particular Arg at the αC helix to Ser. We suppose that there are also physiological conditions under which Erk’s autophosphorylation is induced. Activation by induced autophosphorylation rather than by the canonical MAPK cascade was reported for the MAPKs p38α and p38β (Ge et al., 2002; Salvador et al., 2005; De Nicola et al., 2013). Of note, whereas MEK-independent activation of MAP kinases of the p38 family results in molecules that are monophosphorylated on Thr only (Bell and Engelberg, 2003; Askari et al., 2006, 2009; Beenstock et al., 2014), the autoactivation of the Erk molecules described here provides dually phosphorylated molecules. Therefore there must be fundamental mechanistic differences between the autophosphorylation reaction of p38 and Erk molecules.
A striking mechanism is responsible for the autophosphorylation of T207 and T188 in Erk1 and Erk2, respectively. Not only is this mechanism independent of TEY phosphorylation, it seems to be inhibited by T183 and Y185. A possible mechanistic model suggests competition between T183, T185, and T188 on the interaction with the hydroxyl activator Asp-147 (Emrick et al., 2006). Changes in the αC-helix, for example, by the R65S/R84S mutation, make the protein more flexible and motional, allowing several phosphoacceptors to be activated by Asp-147.
The effect of T188 phosphorylation on Erk2 catalytic, biological, and pathological function is not clear and is not trivial to study because mutating Thr-188 to Ala or Asp abolishes catalytic activity altogether. Because Erk1(R84S) and Erk2(R65S) are spontaneously active in vitro and in cells in culture and at the same time are autophosphorylated at T207 and T188, respectively, this phosphorylation could be associated with the mechanism that renders them intrinsically active. Further, when stably expressed in NIH3T3 cells, Erk1(R84S) is constantly active and constantly phosphorylated on T207, supporting the linkage between this phosphorylation and activity. However, given that any change in the 188 position of Erk2 renders the kinase inactive, the possibility remains that T188 phosphorylation reduces Erk2’s phosphotransferase activity. This latter notion has been supported by independent and parallel studies performed with Erk1 with extensive mutations of it T-activation phosphosites (Lai and Pelech, 2016). Whatever the role of T207/T188 may be, given that Erk1(R84S) and Erk2(R65S) are spontaneously phosphorylated at this residue, they provide a convenient experimental system for testing it. T207/T188 of the activation loop of Erks is highly conserved in many ePKs, so that understanding the functionality of phosphorylation of this residue could be important for understanding the regulation of most ePKs. Finally, the mechanism of T188 phosphorylation seems rather unique, because in the context of Erk2(R65S), it is independent of TEY phosphorylation, namely, MAP kinases can acquire some modes of active conformation without being phosphorylated.
MATERIALS AND METHODS
Plasmids
Plasmids carrying the mutants Erk1(R84S), Erk1(I103A), Erk1(D338N), Erk1(Y280C), Erk2(R65S), Erk2(I84A), Erk2(D319N), and Erk2(Y261C) were previously described (Emrick et al., 2006; Levin-Salomon et al., 2008). For the creation of new mutations, pCEFL vector (Invitrogen, Carlsbad, CA) containing either rat-Erk1(WT) or rat-Erk2(WT), with Erk1 N-terminally tagged with 6xHis or Erk2 N-terminally tagged with HA sequence (YPYDVPDYA), served as templates for site-directed mutagenesis for the mutations Erk2(T188A) and Erk2(T188D), using the Stratagene (La Jolla, CA) QuikChange kit according to manufacturer’s instructions. All cDNAs were verified via DNA sequencing. Primers used for the mutagenesis are shown in Table 3. The pET15b or pHIS parallel (Addgene, Cambridge, MA) vectors containing cDNA encoding the Erk variants were used for protein expression and purification. The ERK2 cDNA containing mutations at the TEY motif were transferred to pHIS parallel vectors using the NcoI and NotI restriction enzymes from pBluescript (pBS) vectors containing the indicated mutations. The K54R mutation was introduced into MPK1 via site-directed mutagenesis on the plasmids pAES426-HA-MPK1(WT) and pAES426-HA-MPK1(R68S) (Levin-Salomon et al., 2009) to obtain pAES426-HA-MPK1(K54R) and pAES426-HA-MPK1(K54R+R68S), respectively. Sequences of the primers used for mutagenesis are listed in Table 3.
TABLE 3:
Primers used for site-directed mutagenesis.
| Description | Primer sequence |
|---|---|
| ERK2-T188A-F | 5′-GGGTTCTTGACAGAGTATGTAGCCGCGCGTTGGTACAGAGC-3′ |
| ERK2-T188A-R | 5′-TGTACCAACGCGCGGCTACATACTCTGTCAAGAACCC-3′ |
| ERK2-T188D-F | 5′-GGGTTCTTGACAGAGTATGTAGCCGACCGTTGGTACAGAGC-3′ |
| ERK2-T188D-R | 5′-TGTACCAACGGTCGGCTACATACTCTGTCAAGAACCC-3′ |
| MPK1-K54R-F | 5′-CCACAGTTGCCATCAGGAAAGTGAC-3′ |
| MPK1-K54R-R | 5′-GTCACTTTCCTGATGGCAACTGTGG-3′ |
Cell cultures and transfection procedures
NIH3T3 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 0.044 M sodium hydrogen carbonate, and streptomycin and penicillin (Beit-Haemek, Beit-Haemek, Israel). HEK293T cells were cultured in the same medium supplemented with 10% FBS, 0.017 M sodium hydrogen carbonate, 1 mM sodium pyruvate, and streptomycin and penicillin. All cells were incubated at 37ºC and 5% CO2. HEK293T cells were plated 24 h before transfection at 1.5 × 105 cells per 3-cm plate or 8 × 105 cells per 10-cm plate. Medium was changed to half the volume approximately 2 h before transfection. Cells were transfected with 10 μg of cDNA (pCEFL plasmids) for 24 h using the calcium phosphate method (Kingston et al., 2003), washed with phosphate-buffered saline (PBS), and provided with fresh medium. At 48 h posttransfection, cells were either starved for serum (incubated in medium with no serum) or received fresh medium for an additional 24 h. Then cells were lysed by addition of 80 μl of sample buffer containing 10% glycerol, 3% SDS, 0.2 M Tris, pH 6.8, 5% β-mercaptoethanol, and phenol blue dye, followed by 10 min of boiling at 100ºC. NIH3T3 cells were transfected using TurboFect Transfection Reagent (Thermo Scientific, Waltham, MA) or TransIT-X2 System (Mirus Bio LLC, Madison, WI) according to manufacturer’s protocol.
Western blotting
A 100-ng amount of recombinant proteins, 30 μg of protein lysates prepared from mammalian cells, or 45 μg of lysates prepared from yeast cells was separated by SDS–PAGE and subsequently transferred to a nitrocellulose membrane. After incubation of the membrane with the appropriate antibodies (see Table 4), specific proteins were visualized using an enhanced chemiluminescence detection reagent.
TABLE 4:
Antibodies used in this study.
| Antibody | Manufacturer |
|---|---|
| Anti–phospho-Erk | Cell Signaling (#4377) |
| Anti-Erk | Cell Signaling (#4370) |
| Anti-Mpk1 | Santa Cruz Biotechnology (SC6803) |
| Anti-polyhistidine | Sigma-Aldrich (H1029) |
| Anti-HA | Santa Cruz Biotechnology (12CA5) |
| Anti–phospho-Thr-188 | Badrilla (A010-40AP) |
| Anti–phospho-Thr-207 | Kinexus Bioinformatics (PK865) |
Soft agar assay
Approximately 5000 cells of each NIH3T3 culture that stably expressed Erk variants or contained a control vector were seeded in triplicate in a 96-well plate. The assay was performed using a CytoSelect 96-well Transformation Assay kit (Cell Biolabs, San Diego, CA) according to manufacturer’s protocol. After a 2-wk incubation period, cells were stained using MTT reagent and incubated for 4 h. All the plates were photographed using an Olympus IX70 microscope.
Foci formation assay
Approximately 1.5 × 105 NIH3T3 cells were plated on a 3-cm plate, transfected with the relevant plasmid, and selected for the presence of the plasmid by incubation with 400 μg/ml G-418. Four weeks after transfection, cells were fixed with 100% MeOH for 20 min, stained with 4% crystal violet dye for 5 min, and photographed.
Luciferase assay
Approximately 7.5 × 105 HEK293T cells were cotransfected with an ERK expression vector, Renilla luciferase, and either 6×AP-1-luc construct (Askari et al., 2009) or pY2-luc construct (Foos et al., 1998). The assay was preformed 48 h posttransfection. Cells were lysed according to manufacturer’s protocol (Dual Luc system; Promega, Madison, WI), and luciferase was measured with a Tube luminometer (GloMax). The firefly luciferase activity was normalized to the Renilla luciferase activity.
Tumorigenicity assay in nude mice
Female nude mice (Harlan; Rehovot, Israel) at 6–7 wk of age were divided into groups of 10 animals each. NIH3T3 cells stably expressing the different Erk variants were plated in 750-ml flasks and allowed to grow to ∼80% confluence. The cells were washed with PBS and harvested. Cell pellets were resuspended in PBS at a density of 1 × 107/ml, and 200 μl of these cells was injected subcutaneously into the right flank of each animal. Tumor volumes were measured over a period of 2 wk after injection. Volume of the growing tumors was calculated as V = LW2/2, where L is length and W is width.
Yeast strains and media
The Saccharomyces cerevisiae strains used in this study were mkk1Δ/mkk2Δ (BY4741; Mat a; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0, YPL140c::kanMX4), mkk1::LEU2; Levin-Salomon et al., 2008), mpk1Δ (BY4741; Mat a; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; YHR030c::kanMX4; obtained from EUROSCARF, Frankfurt, Germany), and pbs2Δste7Δmkk1Δmkk2Δ (BY4741; Mat a; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; YJL128c::kanMX4; ste7::LEU2; mkk1::HygromycinB; mkk2::HIS3; Levin-Salomon et al., 2009). Yeast cultures were maintained on YPD (1% yeast extract, 2% Bacto Peptone, and 2% glucose, plus, where indicated, 15 mM caffeine) or on the synthetic medium YNB-URA (0.17% yeast nitrogen base without amino acids and NH4(SO4)2, 0.5% ammonium sulfate, 2% glucose, and 40 mg/l adenine, histidine, tryptophan, lysine, leucine, and methionine).
Yeast cell plasmid transformation
Plasmids were introduced into the relevant yeast strain by the lithium acetate method as described by Schiestl and Gietz (1989).
Growth assay of yeast cells on medium supplemented with caffeine
Yeast cultures were grown in liquid medium of YNB –URA to mid logarithmic phase (OD600 = 0.5). Then five serial 10-fold dilutions were prepared, starting at OD600 = 0.4 (approximate concentrations 107, 106, 105, 104, and 103 cells/ml). For each yeast strain, 5 μl of each dilution was spotted on the YPD plates supplemented with 15 mM caffeine or on plates containing YNB –URA medium and grown at 30ºC for 6 d.
Preparation of yeast cell lysates
Cell cultures (12 ml) were grown on YNB –URA to OD600 = 0.4–0.6. Cultures were pelleted and resuspended in 10 ml of 20% trichloroacetic acid (TCA). After the samples were pelleted again, they were resuspended in 200 ml of 20% TCA at room temperature (25°C), and 400 mg of glass beads was added. Each sample was vortex-mixed twice for 8 min. Supernatants were transferred to new Eppendorf tubes, and glass beads were rinsed twice with 200 μl of 5% TCA (the final concentration of TCA was 10%). After centrifugation, pellets were resuspended in 100 μl of 2× Laemmli sample buffer, followed by addition of 50 μl of 1 M Tris base. Samples were vortex-mixed for 30 s and boiled for 3 min before centrifugation. The supernatant was used.
Expression and purification of Erk molecules in E. coli cells
Erk variants were expressed and purified as previously described (Levin-Salomon et al., 2008).
Paper-spotted kinase assay
To activate the recombinant Erk variants, 1 μg of a purified recombinant 6xHis-tagged Erk protein and 10 ng of recombinant active MEK1 (14-429; Upstate Biotechnology, Lake Placid, NY) were used. Final reaction conditions were 100 mM NaCl, 35 mM Tris-Cl, pH 8, 15 mM MgCl2, 5 mM 2-glycerolphosphate, 0.2 mM Na3VO4, 0.02 mM dithiothreitol (DTT), 1 mM ethylene glycol tetraacetic acid, and 100 μM ATP. The activation reactions proceeded for 30 min at 30°C and were terminated by placement on ice.
The kinase assays were initialized by the addition of 45 μl of reaction mixture to 5 μl of Erk enzyme. Final reaction conditions were 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 8, 0.1 mM benzamidine, 10 mM MgCl2, 25 mM 2-glycerolphosphate, 1 mM Na3VO4, 0.1 mM DTT, 0.5 μg/μl MBP (M1891; Sigma-Aldrich, St. Louis, MO), 0.1 mM ATP, and 0.1 μCi of [γ-32P]ATP. The kinase reactions proceeded for 15 min at 30°C and were terminated by the addition of 50 μl of 0.5 M EDTA, pH 8 (250 mM final), and placement on ice. After reaction termination, aliquots of 85 μl from each well were spotted onto 3 × 3–cm paper squares of Whatman No. 3MM and briefly air-dried. Each square was rinsed four times with 10% TCA and 3% sodium pyrophosphate (10 ml/square) for 1.5 h (each time), twice with 100% ethanol (4 ml/square) for 20 min each, and air-dried. The radioactivity of each square was counted using a scintillation counter running a 32P Cherenkov program. Experimental points were in triplicate. In parallel, Laemmli sample buffer was added to 15-μl samples of each reaction. These samples were then boiled at 100°C for 5 min and separated on 15% SDS–PAGE. The gel was dried and exposed to x-ray film.
Autophosphorylation assay
A 1-μg amount of a purified recombinant 6xHis-tagged Erk protein was incubated in Erk-activating buffer lacking MEK1. The autophosphorylation reactions proceeded for the indicated time in each experiment at 30°C and were terminated by placement on ice and addition of Laemmli sample buffer. All samples were boiled at 100°C for 5 min and separated on 10% SDS–PAGE. The gel was exposed to x-ray film.
Capillary LC-MS/MS analysis for identification of phosphorylation sites
A 1-μg amount of protein from each autophosphorylated Erk sample was trypsin digested and desalted on a disposable reversed-phase column (C18 Micro Tip Column; Harvard Apparatus, Holliston, MA). The samples were first brought to a concentration of 8 M urea and 100 mM ammonium bicarbonate. Next 45 mM DTT was added to final concentration of 2.6 mM, and the samples were incubated for 30 min at 60°C with mild agitation. The samples were cooled to room temperature, and 150 mM iodoacetamide was added to final concentration of 8.8 mM for 30 min at room temperature in the dark. After fourfold dilution (to 2 M urea) with water, trypsin was added at a ratio of 1:50 (trypsin to protein), and the samples were incubated overnight at 37°C. A second dose of trypsin was given after the overnight digestion, and the samples were left to continue the proteolysis for an additional 4 h at 37°C. The samples were then acidified with 2 μl of 0.1% trifluoroacetic acid (TFA) to stop the reaction. Peptides desalting was carried out by C18 Micro Tip Columns (Harvard Apparatus). The tips were washed with 80% acetonitrile and 0.1% TFA (elution solution), followed by 0.1% TFA (desalting solution). The samples were loaded in 0.1% TFA on the C18 microcolumns, and the flowthrough fractions were kept. The tryptic peptides were desalted with 0.1% TFA and eluted with 80% acetonitrile with 0.1% TFA. Next the samples were completely dried by vacuum centrifugation and resuspended in 0.1% formic acid for LC-MS/MS on an OrbitrapXL (Thermo Scientific).
Crystallization, data collection, and solution of ERK variants
See the Supplemental Materials.
Supplementary Material
Acknowledgments
We thank Steven Pelech (University of British Columbia, Vancouver, Canada) for sharing unpublished information and for fruitful discussions. We also thank Steven Pelech and the Kinexus Bioinformatics Corporation (Vancouver, Canada) for the gift of anti–phospho-T207 Erk1 antibodies. This study was supported by the Israel Science Foundation (Center of Excellence Grants 180/09 and 1772/13 and personal Grant 593/15), the Binational US–Israel Science Foundation (Grant 2009116), the Israel Cancer Research Fund, and the Singapore National Research Foundation under its HUJ-NUS partnership program at the Campus for Research Excellence and Technology Enterprise (CREATE).
Abbreviations used:
- EGF
epidermal growth factor
- ePKs
eukaryotic protein kinases
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- MBP
myelin basic protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-07-0521) on December 10, 2015.
REFERENCES
- Adams JA, McGlone ML, Gibson R, Taylor SS. Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry. 1995;34:2447–2454. doi: 10.1021/bi00008a007. [DOI] [PubMed] [Google Scholar]
- Askari N, Beenstock J, Livnah O, Engelberg D. P38alpha is active in vitro and in vivo when monophosphorylated at threonine 180. Biochemistry. 2009;48:2497–2504. doi: 10.1021/bi900024v. [DOI] [PubMed] [Google Scholar]
- Askari N, Diskin R, Avitzour M, Capone R, Livnah O, Engelberg D. Hyperactive variants of p38alpha induce, whereas hyperactive variants of p38gamma suppress, activating protein 1-mediated transcription. J Biol Chem. 2007;282:91–99. doi: 10.1074/jbc.M608012200. [DOI] [PubMed] [Google Scholar]
- Askari N, Diskin R, Avitzour M, Yaakov G, Livnah O, Engelberg D. MAP-quest: could we produce constitutively active variants of MAP kinases. Mol Cell Endocrinol. 2006;252:231–240. doi: 10.1016/j.mce.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, Zhang XF. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- Beenstock J, Ben-Yehuda S, Melamed D, Admon A, Livnah O, Ahn NG, Engelberg D. The p38 mitogen-activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the -G helix and MAPK insert. J Biol Chem. 2014;289:23546–23556. doi: 10.1074/jbc.M114.578237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell M, Engelberg D. Phosphorylation of Tyr-176 of the yeast MAPK Hog1/p38 is not vital for Hog1 biological activity. J Biol Chem. 2003;278:14603–14606. doi: 10.1074/jbc.C300006200. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;1:4682–4689. [PubMed] [Google Scholar]
- Bott CM, Thorneycroft SG, Marshall CJ. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 1994;352:201–205. doi: 10.1016/0014-5793(94)00958-9. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Brunner D, Oellers N, Szabad J, Biggs WH, Zipursky SL, Hafen E. A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell. 1994;76:875–888. doi: 10.1016/0092-8674(94)90362-x. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, Baccarini M. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16:294–303. doi: 10.1038/nsmb.1564. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. Dimerization in MAP-kinase signaling. Trends Biochem Sci. 2000;25:7–9. doi: 10.1016/s0968-0004(99)01508-x. [DOI] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein-protein interaction in the Raf/Mek/Erk pathway. FEBS Lett. 2014;588:2398–2406. doi: 10.1016/j.febslet.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nicola GF, Martin ED, Chaikuad A, Bassi R, Clark J, Martino L, Verma S, Sicard P, Tata R, Atkinson RA, et al. Mechanism and consequence of the autoactivation of p38α mitogen-activated protein kinase promoted by {TAB1} Nat Struct Mol Biol. 2013;20:1182–1190. doi: 10.1038/nsmb.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrick MA, Hoofnagle AN, Miller AS, Ten Eyck LF, Ahn NG. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J Biol Chem. 2001;276:46469–46479. doi: 10.1074/jbc.M107708200. [DOI] [PubMed] [Google Scholar]
- Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc Natl Acad Sci USA. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foos G, García-Ramírez JJ, Galang CK, Hauser CA. Elevated expression of Ets2 or distinct portions of Ets2 can reverse Ras-mediated cellular transformation. J Biol Chem. 1998;273:18871–18880. doi: 10.1074/jbc.273.30.18871. [DOI] [PubMed] [Google Scholar]
- Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway Inhibitors. Cancer Res. 2014;74:7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould KL, Moreno S, Owen DJ, Sazer S, Nurse P. Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 1991;10:3297–3309. doi: 10.1002/j.1460-2075.1991.tb04894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hindley A, Kolch W. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J Cell Sci. 2002;115:1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- Huang W, Erikson RL. Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc Natl Acad Sci USA. 1994;91:8960–8963. doi: 10.1073/pnas.91.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Curr Protoc Mol Biol. 2003;Chapter 9:Unit 9.1. doi: 10.1002/0471142727.mb0901s63. [DOI] [PubMed] [Google Scholar]
- Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med. 2006;38:200–211. doi: 10.1080/07853890600551037. [DOI] [PubMed] [Google Scholar]
- Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756:81–82. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Lai S, Pelech S. Regulatory roles of conserved phosphorylation sites in the activation T-loop of the MAP kinase ERK1. Mol Biol Cell. 2016;27:1040–1050. doi: 10.1091/mbc.E15-07-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Salomon V, Kogan K, Ahn NG, Livnah O, Engelberg D. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J Biol Chem. 2008;283:34500–34510. doi: 10.1074/jbc.M806443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Salomon V, Maayan I, Avrahami-Moyal L, Marbach I, Livnah O, Engelberg D. When expressed in yeast, mammalian mitogen-activated protein kinases lose proper regulation and become spontaneously phosphorylated. Biochem J. 2009;417:331–340. doi: 10.1042/BJ20081335. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. How does p21ras transform cells. Trends Genet. 1991;7:91–95. doi: 10.1016/0168-9525(91)90278-X. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Mitri Z, Constantine T, O’Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract 2012. 2012:743193. doi: 10.1155/2012/743193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan AK, Dong J, Xie J, Xing M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009;8:2122–2124. doi: 10.4161/cc.8.13.8710. [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases: controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Orr JW, Newton AC. Requirement for negative charge on “activation loop” of protein kinase C. J Biol Chem. 1994;269:27715–27718. [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Meltzer PS. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell. 2002;2:5–7. doi: 10.1016/s1535-6108(02)00089-2. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, Goldsmith EJ. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5’-triphosphate. Biochemistry. 1996;35:5641–5646. doi: 10.1021/bi952723e. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;8:1141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, Weidemann F, Lohse MJ, Lorenz K. Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci USA. 2013;110:7440–7445. doi: 10.1073/pnas.1221999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, Fornace AJ, Ashwell JD. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6:390–395. doi: 10.1038/ni1177. [DOI] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI. Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Seger R, Ahn NG, Boulton TG, Yancopoulosii GD, Panayotatosii N, Radziejewskaii E, Ericsson L, Bratlien RL, Cobb MH, Krebs EG. Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: implications for their mechanism of activation. Proc Natl Acad Sci USA. 1991;88:6142–6146. doi: 10.1073/pnas.88.14.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Keshwani MM, Steichen JM, Kornev AP. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos Trans R Soc B Biol Sci. 2012;367:2517–2528. doi: 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin L, Julien C, Duhamel S, Gopalbhai K, Claveau I, Saba-El-Leil MK, Rodrigue-Gervais IG, Gaboury L, Lamarre D, Basik M, Meloche S. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer. 2008;8:337. doi: 10.1186/1471-2407-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Taya S, Kaibuchi K. Ras-induced transformation and signaling pathway. J Biochem. 1999;126:799–803. doi: 10.1093/oxfordjournals.jbchem.a022519. [DOI] [PubMed] [Google Scholar]
- Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.