

Expression of the mitochondrial ATP6 and ATP8 genes of yeast is translationally regulated by F1 ATPase. Dmt1p represses ATP8/ATP6 mRNA translation. Dmt1p prevents the Atp22p translational activator from binding to the mRNA when F1 is limiting. F1 weakens the Dmt1–mRNA interaction, allowing Atp22p to activate translation.
Abstract
Expression of the mitochondrially encoded ATP6 and ATP8 genes is translationally regulated by F1 ATPase. We report a translational repressor (Smt1p) of the ATP6/8 mRNA that, when mutated, restores translation of the encoded Atp6p and Atp8p subunits of the ATP synthase. Heterozygous smt1 mutants fail to rescue the translation defect, indicating that the mutations are recessive. Smt1p is an intrinsic inner membrane protein, which, based on its sedimentation, has a native size twice that of the monomer. Affinity purification of tagged Smt1p followed by reverse transcription of the associated RNA and PCR amplification of the resultant cDNA with gene-specific primers demonstrated the presence in mitochondria of Smt1p-ATP8/ATP6 and Smt1p-COB mRNA complexes. These results indicate that Smt1p is likely to be involved in translational regulation of both mRNAs. Applying Occam’s principle, we favor a mechanistic model in which translation of the ATP8/ATP6 bicistronic mRNA is coupled to the availability of F1 for subsequent assembly of the Atp6p and Atp8p products into the ATP synthase. The mechanism of this regulatory pathway is proposed to entail a displacement of the repressor from the translationally mute Smt1-ATP8/ATP6 complex by F1, thereby permitting the Atp22p activator to interact with and promote translation of the mRNA.
INTRODUCTION
Translation of the bicistronic ATP8/ATP6 mRNA on yeast mitochondrial ribosomes depends on F1 ATPase (Rak et al., 2009) and at least one transcript-specific translation factor encoded by ATP22 (Zeng et al., 2007a). Atp22p is one of a large number of translational activators that target mRNAs for subunits of cytochrome oxidase (Manthey and McEwen, 1995; Costanzo and Fox, 1986; Poutre and Fox, 1987), cytochrome b (Rödel and Fox, 1987), and Atp9p (Ellis et al., 1999). These factors are likely to have similar functions related to some general feature of mitochondrial translation. The requirement of F1 for translation of ATP8/ATP6 mRNA achieves two important results. First, it provides a means by which expression of the mitochondrial ATP6 and ATP8 genes is coordinated with the availability of F1, a product of the nucleocytoplasmic genetic system. Second, translational down-regulation of the ATP8/ATP6 transcript in the absence of F1 prevents accumulation of an Atp6p-Atp9p ring complex capable of equilibrating protons across the inner membrane and discharging the mitochondrial membrane potential (Godard et al., 2011).
In an earlier study (Rak et al., 2009), we reported that ATP22, when present in high copy number, was able to rescue expression in F1 mutants of the reporter gene ARG8m when the latter is substituted for ATP6 in mitochondrial DNA (mtDNA; Steele et al., 1996; Bonnefoy and Fox, 2000). Suppression was attributed to enhanced translation of ARG8m/ATP8 mRNA as a result of the high concentration of the messenger-specific translational activator Atp22p. This approach was used in the present study to screen for and characterize other suppressors. The screen enabled us to identify the product of reading frame YJL147c, here named SMT1 (Suppression of Mitochondrial Translation), as an important component of the regulatory pathway responsible for the F1-dependent translation of the ATP8/ATP6 mRNA. On the basis of the phenotype of an smt1-null mutant and the properties of Smt1p, we propose that it functions to maintain the ATP8/ATP6 mRNA in a translation- mute form under F1 limiting conditions.
RESULTS
Suppression of ATP6 and ATP8 expression in F1 mutants by extragenic mutations
The translation block of the ATP8/ATP6 mRNA in strains of yeast with mutations in either the α and β subunits of F1 or the Atp11p and Atp12p factors that chaperone assembly of F1 was previously corroborated in an arg8 mutants in which either the ATP6 or ATP8 coding sequence was replaced by the ARG8m, a recoded version of the gene for acetylornithine aminotransferase (Bonnefoy et al., 2000). The arginine requirement for growth in the absence of F1 provided a simple means for isolating and identifying extragenic suppressors of the block in ATP8/ATP6 mRNA translation (Rak and Tzagoloff, 2009). As shown previously, overexpression of ATP22, coding for the translational activator of the ATP8/ATP6 mRNA, was found to partially rescue the arginine auxotrophy of MR6ΔATP12ΔATP6, an arg8 and atp12 double mutant with an ARG8m substitution for ATP6 in mitochondrial DNA (Rak et al., 2009). The double mutant was also used to screen for extragenic suppressors by selecting for arginine-independent growth on glucose as the carbon source. MR6ΔATP12ΔATP6 spread at high density (∼108 cells) on minimal glucose medium containing all the growth requirements except for arginine yielded 30–50 revertants with different growth properties after incubation of the plates for 1 wk (Figure 1A). The fastest-growing revertants grew almost as well as the parental strain (Figure 1B). Similar results were obtained with a double arg8, atp12 mutant harboring a mitochondrial genome in which ATP8 was replaced with ARG8m (unpublished data).
FIGURE 1:

Expression of the mitochondrial atp6::ARG8m allele in F1 mutants and revertants. (A) aMRSΔATP12ΔATP6 (200 μl) grown to early stationary phase in YPD was spread on minimal glucose medium supplemented with adenine, uracil, and tryptophan. The photograph of the arginine-independent revertant colonies was taken after incubation of the plate at 30°C for 7 d. (B) Serial dilutions of MRS-3A, an arg8 mutant (top), and arg8 mutants and revertants with additional mutations in ATP12 and the mitochondrial ATP6 genes. The different strains were spotted on minimal glucose medium containing adenine, uracil, tryptophan, with or without arginine. The plates were incubated at 30°C for 2 d. (C) Top, mutant and revertant strains with the indicated genotypes were grown as in B. The ρ0 derivative of the revertant aMRSΔATP12/R1 was crossed to the kar1 strain DFSΔATP6 containing the mitochondrial atp6::ARG8m mutation (middle) and to the kar1 DFSΔATP8 containing the atp8::ARG8m mutation (bottom). Four independent haploid transductants obtained in each transformation were checked for growth in the absence of arginine. (D) The atp2 and atp12 mutants aMRSΔATP2 and aMRSΔATP12, respectively, and aMRSΔATP12/R1, the atp12 mutant with the smt1-1 suppressor allele of R1, were labeled with [35S]methionine in vivo as described previously (Rak and Tzagoloff, 2009). Total cellular proteins were separated by SDS–PAGE on 12% (top) and 17% polyacrylamide gels (bottom), transferred to nitrocellulose, and exposed to x-ray film. (E) The Δarg8 mutant MR6 and the aMRSΔATP12/R1 revertant (Δarg8 Δatp12 smt1-1) were spotted on minimal glucose plus the indicated supplements and on rich ethanol/glycerol (YEPG) medium. The plates were incubated for 2 d at 30°C.
One of the fast-growing revertants (MR6ΔATP12ΔATP6/R1) was analyzed genetically to ascertain whether the suppressor is nuclear or mitochondrial and whether it is dominant or recessive. A cross of a ρ0 derivative, MR6ΔATP12ΔATP6ρ0, to the revertant produced diploid cells that failed to grow in the absence of arginine, indicative of a recessive nuclear suppressor (Figure 1C). This was confirmed by crosses of the kar1 strains with either an atp6- or an atp8-null mutation in mtDNA (DFSΔATP6 or DFKΔATP8, respectively) to the ρ0 derivative of the R1 revertant. Haploid transductants issued from the two crosses were verified to grow in the absence of arginine (Figure 1C). The suppressor of the R1 revertant was also tested directly for restoration of translation of the ATP8/ATP6 mRNA in an F1 mutant background. Translation of the two proteins was assayed in vivo in the atp12-null mutant and in the ρ0 derivative of the R1 revertant repopulated with wild-type mtDNA. The translational defect of the ATP8/ATP6 mRNA, previously shown to be elicited by the atp12 mutation (Rak and Tzagoloff, 2009), was largely restored in the revertant containing wild-type mtDNA (Figure 1D). As expected, revertants with a normal mitochondrial genome were auxotrophic for arginine (Figure 1E) and did not grow on rich ethanol/glycerol (YEPG) because of impaired F1 assembly in the atp12 mutant (Ackerman and Tzagoloff, 1990).
Cloning of SMT1
To clone the recessive suppressor, we transformed MR6ΔATP12ΔATP6/R1 with a library of yeast genomic DNA in the episomal plasmid YEp24 (Botstein and Davis, 1982). Approximately 1500 transformants were plated on 10 minimal glucose plates containing all the growth requirements of this strain plus arginine. After replication of the transformants on minimal glucose medium lacking arginine, those found to be auxotrophic for arginine were further checked for plasmid-dependent loss of growth in arginine-deficient medium. Of the several dozen putative arginine auxotrophs, only a few survived this second screen. The plasmid DNA isolated from the R1 revertant strain was determined to have a nuclear DNA insert of ∼8.8 kb from chromosome X (Figure 2A). Suppression of growth in arginine-less medium was mapped to a region of the insert between the PstI and NheI sites and (pSMT1/ST9 in Figure 2A). This region contained reading frame YJL147c, reported to code for a nonessential protein associated with mitochondrial ribosomes (Kehrein et al., 2015b). A null allele of the YJL147c reading frame, henceforth referred to as SMT1 (Suppression of mitochondrial translation), was constructed by replacing the 492 nucleotides between the internal EcoRI and HindIII sites with a 1-kb fragment containing the yeast URA3 gene. This plasmid was used to replace wild-type SMT1 of W303 with the smt1::URA3 null allele by homologous recombination (Rothstein, 1983). Uracil-independent clones were verified by PCR amplification of the SMT1 locus to have the null allele. The smt1-null mutants grew as well as the parental wild-type strains on rich ethanol/glycerol medium (YEPG; Figure 2B, top). Deletion of SMT1 restored translation of the bicistronic ATP8/ATP6 mRNA in the F1 mutant (Figure 2, B and C). It is interesting that the smt1-null mutants displayed an increased synthesis of Atp6p, as evidenced by the accumulation of the unprocessed protein with its presequence (pre-Atp6p in Figure 2C). The identity of this band was confirmed by transformation of the smt1 mutant with a high-copy plasmid containing ATP23, the gene for the protease that processes the precursor (Zeng et al., 2007b; Osman et al., 2007). The resultant transformants displayed only mature Atp6p (Figure 2C, right).
FIGURE 2:

Suppression of the ATP6 mRNA translation defect by an smt1-null mutation. (A) Restriction map of the plasmid isolated from the R1 revertant (pSMT1/T1). Only the locations of the EcoRI (E), HindIII (H), BclI (Bc), NheI (Nh), and PstI (P) sites in the inset are shown. The bars labeled ST1 and ST2 correspond to the regions of T1 subcloned in YIp351. The bar labeled ST9 represent the region isolated by colony hybridization of the plasmid library constructed from nuclear DNA of the R1 revertant. All three subclones contained wild-type SMT1 (clear bar). The map of the smt1-null allele is shown above the subclones. (B) Suppression of the ARG8m expression by an smt1-null mutation. Top, the parental W303-1A and the smt1-null mutants in opposite mating types were serially diluted, spotted on rich glucose (YPD) and glycerol/ethanol (YEPG) media, and incubated for 2 d at 30°C. Middle and bottom, MRS-3B (Δarg8) and the arg8 mutant with the indicated additional mutations were tested for growth on YPD and YEPG as in the top. (C) The parental wild-type strain W303-1A, the smt1-null mutants in each mating type, and the atp1-null mutant with and without the smt1-null mutation were labeled and separated by SDS–PAGE, and the Western lot was exposed to x-rays as in Figure 1D. (D) The revertant aMRSΔATP2ΔATP6/R1 (Δatp2 atp6::ARG8m) without and with ectopic SMT1 was grown in the absence and presence of arginine. All the aMRS strains had the arg8::HIS3 allele.
The selection used to clone SMT1 was based on the recessiveness of the mutation in the revertant. This was confirmed by transforming the R1 revertant with SMT1 on a high-copy plasmid. Randomly picked transformants harboring wild-type SMT1 displayed the reappearance of the arginine auxotrophy (Figure 2D).
Is Smt1p the suppressor in the R1 revertant?
Because R1 suppressor and the smt1-null mutant are recessive, expression of ARG8m in a diploid mutant issued from a cross of both strains would be expected only if the two suppressors are allelic. In the complementation test, the diploid cells homozygous for the atp12 and homothallic atp6::ARG8m and heterozygous for the smt1 deletion and R1 suppressor (MRSΔATP12,ATP6,SMT1/R1) grew in the absence of arginine, indicating that the R1 suppressor is allelic with the smt1-null mutation (Figure 3A). Sequencing of PCR-amplified smt1 in the revertant showed a single base change of a G to a T, converting the glutamic acid codon 251 into a stop codon (Figure 3B). This allele will be referred to as smt1-1. No difference was noted in the growth properties of the smt1-null mutant, the R1 revertant, and the wild type on rich glucose or ethanol/glycerol medium.
FIGURE 3:

Allelism test and sequence of the R1 suppressor allele. (A) The indicated strains 1) MRSΔATP12ΔATP6, 2) aMRSΔATP12ΔATP6/R1, 3) aMRSΔATP12ΔATP6/R1ρ0, and 4) a/αMRSΔATP12,ATP6,SMT1/R1 were streaked on YPD medium and after 2 d replicated on minimal glucose plus adenine, uracil, and tryptophan with and without arginine. The replicas were incubated for 2 d at 30°C. The plate lacking arginine was copied twice because of some residual growth of MRSΔATP12ΔATP6 after the first replication due to the endogenous arginine from growth in YPD. Right, the pertinent genotypes. (B) Sequence of the smt1 mutation in the cDNA copied from nuclear DNA of the R1 revertant. The single-base G751T mutation creates an ochre terminator at the glutamic codon 251.
Localization of Smt1p
The SMT1 gene was modified by in-frame fusion of the terminal codon to a sequence coding for the hemagglutinin tag (HA). This construct was inserted at the LEU2 locus of the smt1-null mutant aW303ΔSMT1. Both the mutant and transformant aW303/SMT1-HA with the SMT1-HA allele were respiratory competent. Mitochondria but not the postmitochondrial supernatant fraction of the transformant with the HA-tagged Smt1p, contained a 44-kDa protein detectable with an antibody against the HA tag (Figure 4A). This protein, corresponding to Smt1p-HA, was associated with the membrane fraction after sonic disruption of mitochondria and was not extracted from the membrane with alkaline carbonate, indicating that it is an intrinsic membrane protein (Figure 4B). Smt1p-HA was resistant to digestion with proteinase K in mitoplasts lacking an intact outer membrane. Based on the proteinase K susceptibility of known markers for the intermembrane space (cytochrome b2), the inner membrane (Sco1p), and the matrix (α-ketoglutarate dehydrogenase complex), Smt1p is an inner membrane protein facing the matrix.
FIGURE 4:
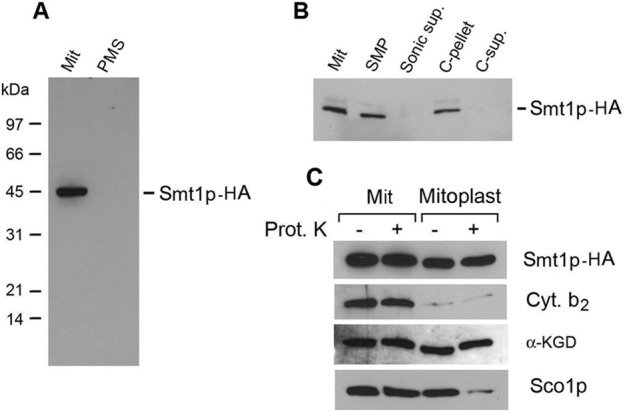
Localization of Smt1p. (A) aW303/SMT1-HA, containing Smt1p with a C-terminal HA tag, was used to isolate mitochondria (Mit) and the postmitochondrial supernatant fraction (PMS) containing soluble cytosolic proteins. Equivalent amounts of mitochondria (50 μg protein) and the soluble cytosolic protein fraction were separated by SDS–PAGE, transferred to nitrocellulose, and probed first with a monoclonal antibody against the HA and then by a secondary peroxidase-coupled anti-mouse antibody. The antigen–antibody complexes were visualized with the Super Signal Chemiluminescent Substrate Kit (Pierce, Rockford, IL). (B) Mitochondria (Mit), 0.5 ml, at a protein concentration of 10 mg/ml were disrupted by sonic oscillation and centrifuged for 20 min at 70,000 × gav into submitochondrial membrane vesicles (SMPs) and the supernatant (Sonic sup.) consisting of soluble proteins of the intermembrane space and matrix. The submitochondrial membranes were suspended in a final volume of 0.5 ml of 0.1 M sodium carbonate and 1 mM PMSF and incubated at 4°C for 30 min. The suspension was centrifuged at 100,000 × gav to separate the insoluble intrinsic (C-pellet) from soluble extrinsic proteins (C-sup). Equivalent volumes of each fraction were separated by SDS–PAGE and the Western blot treated as in A. (C) Mitochondria (Mit) were converted into mitoplasts under hypotonic conditions. Both were incubated with and without protease K (Prot. K) as described previously (Zeng et al., 2007b). Samples normalized to 50 μg of starting mitochondrial protein were separated by SDS–PAGE and transferred to nitrocellulose. The Western blot was probed with a primary monoclonal antibody against the HA tag to detect Smt1p-HA and with rabbit polyclonal antibodies against the indicated mitochondrial protein markers. Antigen–antibody complexes were visualized as in A, except that the mitochondrial makers were detected with a peroxidase-coupled anti-rabbit antibody.
Purification and sedimentation properties of Smt1p-CH
Our failure to purify Smt1p from cells containing the protein with a single polyhistidine tag prompted us to further modify SMT1 by inserting between the end of the gene and the histidine codons a short sequence coding for the protein C epitope (Lychty et al., 2005). This new construct was integrated at the LEU2 locus of the smt1-null mutant. The SMT1-CH gene in the resultant strain aW303/SMT1-CH was verified to retain its ability to suppress translation of the ATP8/ATP6 mRNA in F1 mutants. Transformation of the Arg+ R1 revertant with integrative plasmids containing either the wild-type (pSMT1/ST19) or the modified (pSMT1/ST18) gene in both cases conferred an arginine-dependent growth phenotype on the revertant (Figure 5A).
FIGURE 5:

(A) Suppression by SMT1-CH of Atp6p translational in the R1 revertant. 1) R1 aMRSΔATP12ΔATP6/R1, 2) aMRSΔATP12ATP6, 3) aMRSΔATP12ATP6/R1/ST18, and 4) aMRSΔATP12ATP6/R1/ST19. The YPD master plate of each strain was replicated and grown on minimal glucose medium plus and minus arginine as in Figure 3A. Left, the pertinent genotypes. (B) Smt1p-CH was purified by the two-step affinity protocol described in Materials and Methods. The different fractions at the indicated concentration relative to the starting volume of mitochondria were separated by SDS–PAGE on a 12% polyacrylamide gel and probed as in Figure 4A, except that the primary monoclonal antibody was against the polyhistidine tag. Mitochondria (M), lauryl maltoside extract (Ex), pellet after lauryl maltoside extraction (P), the protein fraction of nonadsorbed proteins on Ni-NTA (Ni-NTA FT), high-imidazole washes of Ni-NTA beads (E1, E2), nonadsorbed proteins on the protein C antibody beads (P-C FT), and three sequential EDTA eluates of the P-C beads (C1, C2, C3). Most of the Smt1-CH was eluted in the first high-imidazole wash and in the first two EDTA eluates from the protein C beads. (C) Fractions E1 and E2 (Ni-NTA) and C1 and C2 (P-C) were combined and separated as in A. The amount of each fraction loaded relative to the starting volume of mitochondria is indicated at the top of each lane. The gel was stained with Coomassie blue. (D) The combined EDTA eluates (C1 and C2) from the P-C beads were separated as in A and either silver stained (Ag) or transferred to nitrocellulose and visualized with a primary antibody against the polyhistidine tag. (E) Top, the EDTA eluate from the P-C beads was mixed with lactate dehydrogenase (LD) and hemoglobin (HGB) and applied to a linear 10–25% sucrose gradient containing in 10 mM Tris-Cl, pH 7.5, 0.1 M NaCl, and 0.1% lauryl maltoside. The gradient was centrifuged at 60,000 rpm in a Beckman 60SW rotor at 4°C for 6 h, and 13 fractions were collected from the bottom of the tube. Each fraction was assayed for lactate dehydrogenase (solid dots) and hemoglobin (open dots). Each fraction was also probed for Smt1p-CH as described in B. Bottom, same as before, except that a strain expressing HA-tagged Smt1p was used and the gradient was loaded with a lauryl maltoside extract of mitochondria without affinity purification. The primary antibody was a monoclonal directed against the HA tag. The peak fractions of lactate dehydrogenase (140 kDa) and hemoglobin (65 kDa) are indicated by the arrows.
Mitochondria of aW303/SMT1-CH were used to purify the protein from a lauryl maltoside extract by two sequential affinity steps. Most of the protein was recovered in the lauryl maltoside extract and was adsorbed on both the nickel–nitriloacetic acid (Ni-NTA) and the protein C antibody–coated beads from which it was eluted under nondenaturing conditions in the presence of EDTA (Figure 5B). The elution fraction obtained by this procedure consisted predominantly of a major product that migrated as a 44-kDa protein and of a less abundant product slightly smaller in size (Figure 5, C and D). Mass spectrophotometric analysis of the two bands confirmed both to be Smt1p-CH. The shorter and less abundant protein may be the product of alternate processing of the N-terminal extension in the precursor during its import into mitochondria or of a proteinase-sensitive site cleaved during the purification.
The size of purified Smt1p was assessed by sedimentation of the fraction eluted from the P-C antibody beads in sucrose gradients. The protein peaked at a position of the gradient intermediate between the hemoglobin and lactate dehydrogenase size standards, with an estimated mass of 80–90 kDa corresponding an Smt1p dimer (Figure 5E, top). Similar results were obtained with a crude lauryl maltoside extract from a strain expressing HA-tagged Smt1p analyzed under similar conditions (Figure 5E, bottom).
Detection of Smt1p-ATP8/ATP6 mRNA complex
Restoration of the ATP6 and ATP8 expression in F1 mutants lacking Smt1p suggested that the latter might be a repressor that modulates translation of the ATP8/ATP6 mRNA (Figure 6A) in response to the demand of Atp8p and Atp6p for assembly with F1 and other protein constituents of the ATP synthase. The proposed function of Smt1p as a translational repressor implies its interaction with the target RNA. The existence of such a complex was tested in aW303ΔNUC1/SMT1-CH, a strain containing Smt1p double tagged with a protein C and polyhistidine and having a null mutation in NUC1, which codes for an abundant nuclease of yeast mitochondria (Vincent et al., 1988). The nuc1 mutation did not abolish but did substantially reduce the loss of mitochondrial RNA after their extraction with digitonin (Figure 6B). The procedure used to assess association of ATP8/ATP6 mRNA with Smt1p-CH (Figure 6C) entailed extraction of mitochondria with digitonin followed by affinity purification of Smt1p in the extract on protein C antibody beads and reverse transcription PCR (RT-PCR) amplification with primers specific for ATP8/ATP6 mRNA (Figure 6D).
FIGURE 6:

Detection of Smt1p-ATP8/ATP6 transcript in mitochondria. (A) Map of the ATP8/ATP6 mRNA. The ATP8 and ATP6 genes are denoted by the solid bars. The locations of the RT and PCR primers are shown by the arrows. (B) Stabilization of mitochondria RNAs in digitonin extracts of mitochondria from yeast with a Δnuc1 mutation. Mitochondria (Mito) from the Δnuc1 mutant W303ΔNUC1 were extracted with phenol and nucleic acids, precipitated with ethanol, and separated on a 1% agarose gel, without treatment and after digestion with MseI restriction endonuclease to digest DNA and with RNase I to digest RNA. Mitochondria were extracted with 2% digitonin and treated identically. The two mitochondrial ribosomal RNAs and DNA are identified in the margin. (C) Outline of protocol used to purify the Smt1p-CH-ATP8/ATP6 mRNA complex. (D) Smt1p was purified from 25 μg of starting mitochondria, separated by SDS–PAGE, and probed as in Figure 4A, except that the primary antibody was against the polyhistidine tag. The fractions shown are mitochondria (M), digitonin extract (Ex), the fraction that did not adsorb to the protein C antibody beads (FT), and eluate from the beads (EL). The samples loaded were normalized to the starting volume of mitochondria. (E) Detection of an ATP8/ATP6 cDNA after RT-PCR amplification of the fraction enriched on protein C antibody beads. Left, each lane of the gel was loaded with the PCR product obtained from the equivalent of 25 μg of starting mitochondrial protein from aW303ΔNUC1 (SMT1 Δnuc1) and aW303ΔNUC1/SMT1-CH (SMT1-CH Δnuc1). The mitochondria were extracted with 2% digitonin, and the extract was purified on beads as described in Materials and Methods. One-half of the nucleic acids extracted from protein C eluate was either reverse transcribed (+RT) prior to PCR amplification or directly PCR amplified (–RT). The product shown in the extreme right lane was obtained with Atp6-29 and Atp6-24 PCR primers using purified mitochondrial DNA as the template. Right, same as the left, except that the mitochondria were aW303ΔNUC1/SMT1-CH (SMT1-CH Δnuc1) and aW303ΔNUC1/LAT1-CH (LAT1-CH Δnuc1), a strain that expresses the dihydrolipoamide acetyltransferase component (E2) of pyruvate dehydrogenase with a CH tag. (F) Effect on cDNA synthesis of addition of mitochondrial RNA from MR10 to the protein C eluate. The conditions for purification and RT-PCR amplification were the same as in E. The indicated amounts of purified total RNA from MR10 (atp6::ARG8m) were added to the nucleic acids extracted from the protein C antibody beads. The purified RNA (10 μg) was also used directly as the template for PCR amplification either without or with prior reverse transcription. A faint product was obtained in both conditions. The origin of this product is not known.
PCR amplification of mitochondrial DNA produced a product of ∼1.2 kb, consistent with the expected size of the ATP8/ATP6 mRNA based on the locations of the two primers. A cDNA product of the same size was also detected after RT-PCR amplification of nucleic acids in the protein fraction purified on the protein C beads (Figure 6E). This cDNA was confirmed by sequencing to span the sequence of the ATP8/ATP6 locus between the two primers. The product with the ATP8/ATP6 sequence depended on a prior treatment of the fraction with reverse transcriptase, indicating that the cDNA was copied from RNA. The 1.2-kb product was not observed with mitochondria from a strain with the nuc1 mutation but with untagged Smt1p (Figure 6E, left) or mitochondria from a strain expressing C-terminal CH tag fused to dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase complex, encoded by LAT1, used as a negative control (Figure 6E, middle).
Additional evidence excluding the presence of the ATP8/ATP6 transcript due to nonspecific binding of RNA to the beads was excluded by lack of competition when excess yeast RNA purified from the atp6-null mutant MR10 was added to the extract (Figure 6F). Addition of this RNA increased the amount of final ATP8/ATP6 cDNA as a result of more efficient precipitation of the cDNA in the presence of the MR10 RNA.
The specificity of Smt1p for mitochondrial transcripts was also examined by testing its ability to bind the mRNA for the COB, COX3, and ATP9 for the cytochrome b subunit of the bc1 complex, subunit 3 of cytochrome oxidase, and subunit 9 of the ATP synthase, respectively. The primers internal to the COB coding sequence (Figure 7A) were verified to amplify the gene from mitochondrial DNA (Figure 7B). The COB primers also amplified a product of the right size and sequence from the RNA in the Smt1p-CH purified fraction. This product depended on the inclusion of a reverse transcriptase step before PCR amplification (Figure 7C) and was not seen in the nuc1-null mutant with the normal SMT1 gene (Figure 7C). The results obtained with PCR primers spanning the COX3 and ATP9 coding sequences could not be interpreted. Even though these primers amplified single bands of the right size with mitochondrial DNA as template, the smeared products obtained with the nuc1-negative control and with purified Smt1p-CH precluded an answer as to whether they are also associated with Smt1p.
FIGURE 7:
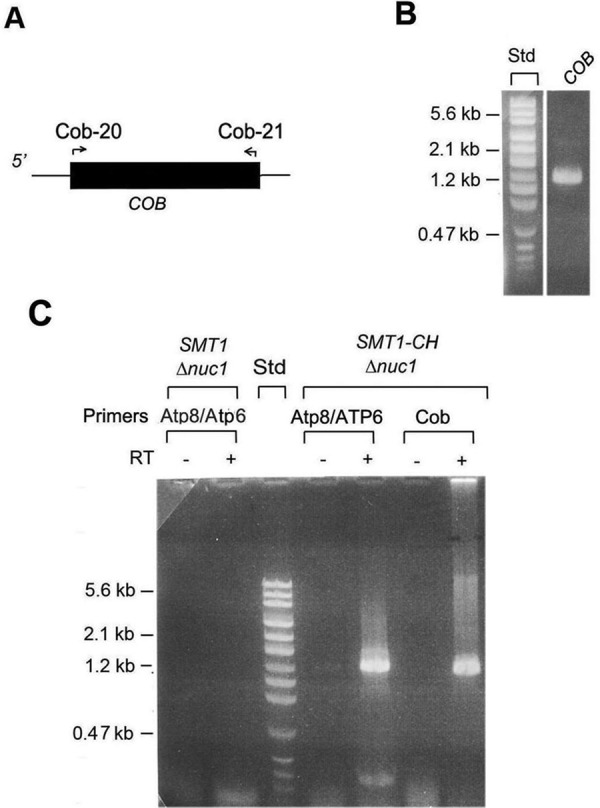
Presence of COB mRNA in purified Smt1p-CH. (A) The locations of primers used for RT-PCR amplification of COB mRNA are shown by the arrows. (B) PCR amplification of COB from mtDNA of the intronless strain MRSIo. (C) Smt1p was purified from digitonin extracts of mitochondria from the control strain W303ΔNUC1 with unmodified SMT1 (SMT1 Δnuc1) and from aW303ΔNUC1/SMT1-CH (SMT1-CH Δnuc1) as described in the legend to Figure 6, C and E. The eluate from the protein C antibody beads was further processed as in Figure 6E.
The ATP8/ATP6 and COB mRNAs were also detected in Smt1p purified by the procedure using lauryl maltoside instead of digitonin and including purification on Ni-NTA before the protein C antibody beads (unpublished data).
DISCUSSION
In this study, we used genetic and biochemical means to gain additional mechanistic details underlying the previously observed F1-dependent translation of the ATP8/ATP6 mRNA (Rak and Tzagoloff, 2009). Using a double arg8 and atp12 (F1 chaperone) mutant with a substitution of the mitochondrial ATP6 gene by recoded ARG8m for acetylornithine transaminase synthesis on mitochondrial ribosomes (Steele et al., 1996), we obtained a number of arginine-independent revertants. The R1 revertant reported here grew as well as wild type in the absence of arginine and was ascertained to have a recessive mutation in a nuclear gene. The recessive nature of the suppressor was taken advantage of to isolate SMT1, which, when mutated, confers arginine- independent growth and, like the suppressor in R1, is also recessive. Sequencing of the SMT1 in the R1 revertant indicated that the suppressor mutation creates a premature termination codon in the gene. Allelism of the R1 suppressor with the smt1-null mutation was also confirmed genetically.
Smt1p sedimented approximately midway between lactate dehydrogenase and hemoglobin, suggesting a size two times larger than the monomer estimated by SDS–PAGE. Smt1p behaves as an intrinsic inner membrane protein facing the matrix compartment.
A requisite for the proposed function of Smt1p as a translational repressor is that it interacts with the ATP8/ATP6 mRNA. The presence of the ATP8/ATP6 mRNA was confirmed by PCR amplification of a reverse transcriptase–dependent cDNA product obtained with ATP8 and ATP6 primers when the template was extracted from Smt1p-CH purified on protein C antibody beads in the presence of digitonin or lauryl maltoside. On the basis of our genetic and biochemical results, we propose that the previously observed F1-dependent regulation of Atp6p and Atp8p synthesis involves a mechanism at the core of which activation of ATP8/ATP6 mRNA translation is prevented by repressor proteins. A second important feature of this model is the labilization of the repressor-ATP8/ATP6 mRNA complex by F1, allowing Atp22p to exercise its function as a translational activator (Figure 8). This would imply a common or overlapping binding site on the mRNA for Atp22p and Smt1p.
FIGURE 8:
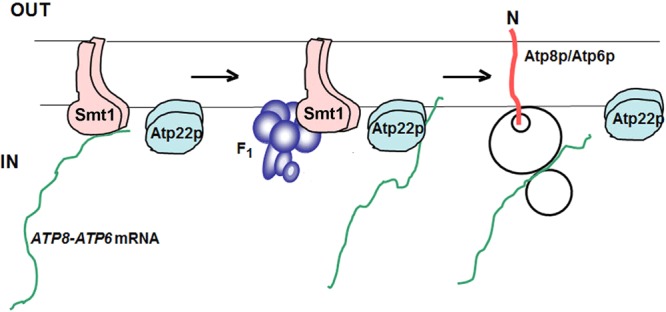
Model of ATP8/ATP6 mRNA translational regulation by F1. In the absence of F1, the ATP8/ATP6 mRNA is associated with Smt1p. When bound to the repressor, the mRNA is prevented from interacting with Atp22p to initiate translation. F1 is proposed to weaken the interaction of the repressor with the ATP8/ATP6 mRNA, thereby allowing Atp22p to bind to the mRNA and activate translation. Based on its sedimentation, native Atp22p, like Smt1p, is a dimer (unpublished data). When overexpressed from ATP22 on a high-copy plasmid, Atp22p is able to interact with a limited amount of ATP8/ATP6 mRNA and partially suppress the translational block in the absence of F1.
Of interest, RNA binding by Smt1p is not confined to the ATP8/ATP6 mRNA. Primers internal to COB yielded a PCR product of the right sequence when RNA extracted from purified Smt1p-CH was reverse transcribed and PCR amplified. Smt1p (Mrx5p) was recently reported to be associated with ribosomes in a large complex termed Miorex (Kehrein et al., 2015a, b). Purified Smt1p is a single dimeric protein, suggesting that the purification procedure may have led to its dissociation from the Miorex complex. The presence of the COB mRNA cannot be explained by the presence of the Miorex complex with both ATP8/ATP6 and COB mRNA, as purified Smt1p-CH consists of a single protein. Instead, it suggests a possible involvement of Smt1p in translational regulation of at least one other mitochondrial gene product.
MATERIALS AND METHODS
Yeast strains and growth media
The yeast strains used in this study and their genotypes are listed in Table 1. The compositions of yeast extract/peptone/glucose (YPD), YP plus ethanol/glycerol, YP plus galactose, and minimal glucose media have been described (Myers et al., 1985).
TABLE 1:
Genotypes and sources of Saccharomyces cerevisiae strains. Continued
| Strain | Genotype | mtDNA | Source |
|---|---|---|---|
| W303-1A | MATa ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ | R. Rothstein (Columbia University, New York, NY) |
| W303-1B | MATα ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ | R. Rothstein |
| DFSΔATP6 | MATα kar1 ade2 leu2 ura3 arg8::URA3 | ρ+ atp6::ARG8m | This study |
| DFKΔATP8 | MATα kar1 ade2 leu2 lys2 ura3 arg8::URA3 | ρ+ atp8::ARG8m | This study |
| MR6ΔATP6 | MATa ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 arg8::HIS3 | ρ+ atp6::ARG8m | Rak and Tzagoloff (2009) |
| MR6ΔATP8 | MATa ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 arg8::HIS3 | ρ+ atp8::ARG8m | Rak and Tzagoloff (2009) |
| aW303ΔSMT1 | MATa ade2-1 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 | ρ+ | This study |
| W303ΔSMT1 | MATα ade2-1 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 | ρ+ | This study |
| aW303/SMT1-HA | MATa ade2-1 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 leu2::pSMT1/ST5 | ρ+ | This study |
| aW303/SMT1-CH | MATa ade2-1 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 leu2::pSMT1/ST13 | ρ+ | This study |
| aW303/SMT1-CHρ0 | MATa ade2-1 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 leu2::pSMT1/ST13 | ρ0 | This study |
| aW303ΔNUC1 | MATa ade2-1 his3-1,15 leu2-3,112 nuc1::TRP1 trp1-1 ura3-1 | ρ+ | This study |
| aW303ΔNUC1ΔSMT1 | MATa ade2-1 his3-1,15 leu2-3,112 nuc1::TRP1 smt1::URA3 trp1-1 ura3-1 | ρ+ | This study |
| aW303ΔNUC1/SMT1-CH | MATa ade2-1 his3-1,15 leu2-3,112 nuc1::TRP1 smt1::URA3trp1-1 ura3-1 leu2::pSMT1/ST13 | ρ+ | This study |
| aW303ΔNUC1/LAT1-CH | MATa ade2-1 his3-1,15 lat1::HIS3 leu2-3,112 nuc1::TRP1 trp1-1 ura3-1 leu2::pLAT1/ST6 | ρ+ | This study |
| MR6 | MATa ade2-1 arg8::HIS3 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ | Rak et al. (2007) |
| MR10 | MATa ade2-1 arg8::HIS3 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ atp6::ARG8m | Rak et al. (2007) |
| MRSI0 | MATa ade2-1 arg8::HIS3 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+, intronless | Rak and Tzagoloff (2009) |
| aW303ΔATP1/SMT1-CH | MATa ade2-1 atp1::HIS3 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 leu2::pSMT1/ST13 | ρ+ | This study |
| aW303ΔCOX2/SMT1-CH | MATa ade2-1 atp1::HIS3 his3-1,15 leu2-3,112 smt1::URA3 trp1-1 ura3-1 leu2::pSMT1/ST13 | ρ+ cox2::ARG8m | This study |
| MRS-3A | MATa ade2-1 arg8::HIS3 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ | Rak and Tzagoloff (2009) |
| aMRSΔATP2ΔATP6 | MATa ade2-1arg8::HIS3 atp2::LEU2 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ atp6::ARG8m | Rak and Tzagoloff (2009) |
| aMRSΔATP2ΔATP6/R1 | MATa ade2-1 arg8::HIS3 atp2::LEU2 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 | ρ+ atp6::ARG8m | This study |
| aMRSΔATP12 | MATa ade2-1 arg8::HIS3 atp12::HIS3 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ | Rak and Tzagoloff (2009) |
| aMRSΔATP12/R1 | MATa ade2-1 arg8::HIS3 atp12::HIS3 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 | ρ+ | This study |
| aMRSΔATP12ΔATP6 | MATa ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ atp6::ARG8m | Rak and Tzagoloff (2009) |
| MRSΔATP12ΔATP6 | MATα ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 trp1-1 ura3-1 | ρ+ atp6::ARG8m | This study |
| aMRSΔATP12ΔATP6/R1 | MATa ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 | ρ+ atp6::ARG8m | This study |
| aMRSΔATP12ΔATP6/R1ρ0 | MATa ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 | ρ0 | This study |
| aMRSΔATP12ΔATP6/R2ρ0 | MATa ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 trp1-1 ura3-1 R2sup | ρ0 atp6::ARG8m | This study |
| aMRSΔATP12,ATP6//R1/SMT1-CH | MATa ade2-1 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 arg8::HIS3 atp12::LEU2 trp1::pSMT1/ST18 | ρ+ atp6::ARG8m | This study |
| aMRSΔATP12,ATP6//R1/SMT1 | MATa ade2-1 arg8::HIS3 atp12::LEU2 his3-1,15 leu2-3,112 smt1-1 trp1-1 ura3-1 trp1::pSMT1/ST19 | ρ+ atp6::ARG8m | This study |
| a/αMRSΔATP12,ATP6,SMT1/R1 | MATa/α ade2-1/ade2-1 arg8::HIS3/arg8::HIS3 atp12::LEU2/atp12::LEU2 his3-1,15/his3-1,15 leu2-3,112/leu2-3,112 smt1-1/smt1::HIS3 trp1-1/trp1-1 ura3-1/ura3-1 | ρ+ atp6::ARG8m | This study |
Construction of the smt1-null allele
The 3-kb BclI fragment of pSMT1/T1 containing the entire SMT1 coding sequence plus 5′ and 3′ flanking sequences was cloned into the BamH1 site of YEp352B, a shuttle plasmid identical to YEp352 (Hill et al., 1986) except for the presence of a single BamH1 site instead of the multiple cloning sequence. The resultant plasmid pSMT1/ST2 was digested with EcoR1 plus HindIII to remove 500 base pairs from the middle of the gene. The gapped plasmid was ligated to a 1-kb EcoRI-HindIII fragment containing the yeast URA3 gene to yield pSMT1/ST3. The smt1::URA3 allele plus flanking sequences was removed from pSMT1/ST3 as a BstU1-NheI fragment and was used to transform W303-1A. Several uracil-independent and respiratory-competent clones (aW303ΔSMT1) obtained from the transformation were verified by PCR amplification of the SMT1 locus to carry the smt1-null allele. The alpha mating type smt1-null mutant was obtained from a cross of aW303ΔSMT1 to W303-1B.
Construction of yeast strains that synthesize Smt1p with a C-terminal hemagglutinin and a double protein-C plus polyhistidine tag
Primers Smt1-1 and Smt1-2 (Table 2) were used to amplify SMT1 fused at the 3′ end with a sequence coding for the HA tag. The PCR product consisting of ∼500 nucleotides of the 5′-untranslated region (5′-UTR), the entire SMT1 coding sequence fused to the sequence of the tag, was digested with a combination of SacI and PstI and ligated to the YIp351 (Hill et al., 1986) linearized with the same enzymes, yielding plasmid pSMT1/ST5. Similarly, primers Smt1-1 and Smt1-3 were used to amplify the 5′-UTR and coding sequence of SMT1 without the termination codon. The PCR product was digested with a combination of SacI and Pst1 and ligated to YIp351-CH cut with the same combination of enzymes. The product of this ligation (pSMT1/ST13) resulted in an in-frame ligation of SMT1 to a sequence coding for the protein C tag followed by three glycines followed by six histidine codons and a TAA terminator. The last codon of SMT1 was followed by the Leu and Gln codons contributed by the PstI cloning site before the start of the CH tag (Table 2). Both pSMT1/ST5 and pSMT1/ST13 were linearized at the ClaI site internal to the LEU2 gene of YIp351. The linear plasmid sequence was integrated at the leu2 locus of aW303ΔSMT1 (Rothstein, 1983) to obtain aW303/SMT1-HA and aW303/SMT1-CH, respectively.
TABLE 2:
Sequences of primers and of CH double tag.
| Primer name | Primer sequence |
|---|---|
| Primers used for construction of HA- and CH-tagged Smt1p | |
| Smt1-1 | 5′ ggcgagctctgctatcggccgtacatagtaag |
| Smt1-2 | 5′ ggcctgcagtcaagcgtagtctgggacgtcgtatgggtatatgaggttcaatggtaagtg |
| Smt1-3 | 5′ cctgcagtatgaggttcaatggtaagtg |
| ATP8/ATP6 primers | |
| Atp6-29 | 5′ cctatgatcttaagattatatgtatctag |
| Atp6-24 | 5′ ggtactaatggtaatggtgtaccagcaggtacg |
| COB primers | |
| Cob-20 | 5′ atggcatttagaaaatcaaatgtg |
| Cob-21 | 5′ ctctaccgatatagaataaaacattttc |
| CH double tag | CTGCAGGAAGATCAGGTAGATCCACGGTTAATCGATGGTAAGGGAGGAGGACACCATCACCATCATCACTAA LeuGlnGluAspGlnValAspProArgLeuIleAspGlyLysGlyGlyGlyHisHisHisHisHisHisEnd |
Purification of Smt1p
The procedure of Herrmann et al. (1994) was used to prepare mitochondria from aW303/SMT1-CH grown to early stationary phase in YPGal. In a typical purification, 20 ml of the mitochondrial suspension at a protein concentration of 10 mg/ml was adjusted to 2 mM phenylmethanesulfonate fluoride (PMSF) with a 0.2 M ethanolic solution and 1% lauryl maltoside with a 10% solution. After centrifugation at 70,000 × gav for 10 min, the supernatant was adjusted to pH 8.2 with 1.5 M Tris-Cl, pH 8.8, and added to 1 ml of packed Ni-NTA beads that had been prewashed with Ni-NTA wash buffer (10 mM Tris-Cl, pH 7.5, 10 mM imidazole, pH 8, 100 mM NaCl). The mixture was rotated at 4°C for 2 h, and nonadsorbed proteins were removed by centrifugation at 1000 rpm for 10 s. The beads were washed three times with 5 ml of Ni-NTA wash buffer containing 0.1% lauryl maltoside and eluted with 2 ml of Ni-NTA elution buffer (10 mM Tris-Cl, pH 7.5, 350 mM imidazole, 100 mM NaCl, 0.1% lauryl maltoside). The eluate from the Ni-NTA beads was adjusted to 2 mM CaCl2 and added to 200 ml of packed protein C antibody beads (Anti-Protein C Affinity Matrix; Roche Life Sciences, Indianapolis, IN) that had been prewashed in PC wash buffer (10 mM Tris-Cl, pH 7.5, 2 mM CaCl2, 100 mM NaCl, 0.1% lauryl maltoside). After rotation of the mixture for 1.5 h, unadsorbed proteins were removed by centrifugation at 1000 rpm for 10 s, and the beads were washed four times with 1 ml of PC wash buffer. The washed beads were then eluted twice with 100 μl of PC elution buffer (10 mM Tris-Cl, pH 7.5, 7 mM EDTA, 100 mM NaCl, 0.1% lauryl maltoside).
Pull down of Smt1p and RT-PCR amplification of ATP8/ATP6 cDNA
A mitochondrial suspension representing 250 μg of protein was centrifuged at 10,000 × gav for 10 min and used to purify Smt1p as described, with the following modifications. The pellet was extracted in 50 μl of 2% digitonin in PC wash buffer. The digitonin extract was added to 30 μl of packed protein C antibody beads and rotated for 30 min. After four washes of the beads with PC buffer containing 0.5% digitonin, proteins were eluted with 60 μl of PC elution buffer containing 0.5% digitonin.
The eluate was extracted with an equal volume of water-saturated phenol, and nucleic acids were precipitated by addition of 5 M NaCl to a final concentration of 0.25 M and three volumes of ethanol. The pelleted and dried nucleic acid was dissolved in 10 μl of water and reverse transcribed in a final volume of 20 μl with the Atp6-24 primer using the Script cDNA synthesis kit (BioLab). The resultant cDNA was precipitated by addition of salt and ethanol and PCR amplified with primers Atp6-29 and Atp6-24 in a final volume of 50 μl. The cDNA was separated on a 1% agarose gel and visualized by ethidium bromide staining.
Miscellaneous procedures
Yeast was transformed by the LiAc procedure of Schiestl and Gietz (1989). Standard protocols were used for DNA cloning, purification of plasmids from Escherichia coli, and colony hybridization (Sambrook et al., 1989). The conditions for in vivo labeling of yeast mitochondrial gene products with [35S]methionine have been described (Rak et al., 2009). Proteins were separated by SDS–PAGE in the buffer system of Laemmli (1970). Protein concentration was determined by the procedure of Lowry et al. (1951).
Acknowledgments
This research was supported by Grant HL02274 from the National Institutes of Health.
Abbreviations used:
- EDTA
ethylenediaminetetraacetic acid
- ρ− mutant
respiratory-deficient mutant with partially deleted mitochondrial DNA
- ρ+ genome
wild-type mitochondrial DNA
- ρ0 mutant
respiratory-deficient mutant lacking mitochondrial DNA.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0642) on January 28, 2016.
REFERENCES
- Ackerman S, Tzagoloff A. Identification of two nuclear genes (ATP11, ATP12) required for the assembly of yeast F1 ATPase. Proc Natl Acad Sci USA. 1990;87:4986–4990. doi: 10.1073/pnas.87.13.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefoy N, Fox TD. In vivo analysis of mutated initiation codons in the mitochondrial COX2 gene of Saccharomyces cerevisiae fused to the reporter gene ARG8m reveals lack of downstream reinitiation. Mol Gen Genet. 2000;262:1036–1046. doi: 10.1007/pl00008646. [DOI] [PubMed] [Google Scholar]
- Botstein D, Davis RW. Principles and practice of recombinant DNA research in yeast. In: Strathern JN, Jones EW, Broach JR, editors. The Molecular Biology of the Yeast Saccharomyces cerevisiae: Metabolism and Gene Expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1982. pp. 607–636. [Google Scholar]
- Costanzo MC, Fox TD. Product of Saccharomyces cerevisiae nuclear gene PET494 activates translation of a specific mitochondrial mRNA. Mol Cell Biol. 1986;6:3694–3703. doi: 10.1128/mcb.6.11.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis TP, Lukins HB, Nagley P, Corner BE. Suppression of a nuclear aep2 mutation in Saccharomyces cerevisiae by a base substitution in the 5’-untranslated region of the mitochondrial oli1 gene encoding subunit 9 of ATP synthase. Genetics. 1999;151:1353–1363. doi: 10.1093/genetics/151.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godard F, Tetaud E, Duvezin-Caubet S, di Rago JP. A genetic screen targeted on the FO component of mitochondrial ATP synthase in Saccharomyces cerevisiae. J Biol Chem. 2011;286:18181–18189. doi: 10.1074/jbc.M110.214825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Foelsch H, Neupert W, Stuart RA. Isolation of yeast mitochondria and study of mitochondrial protein translation. In: Celis JE, editor. Cell Biology: A Laboratory Handbook. I. San Diego, CA: Academic Press; 1994. pp. 538–544. [Google Scholar]
- Hill JE, Myers AM, Koerner TJ, Tzagoloff A. Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- Kehrein K, Möller-Hergt BV, Ott M. The MIOREX complex: lean management of mitochondrial gene expression. Oncotarget. 2015a;6:16806–16807. doi: 10.18632/oncotarget.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrein K, Schilling R, Möller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M. Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep. 2015b;10:843–853. doi: 10.1016/j.celrep.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lychty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of protein tags for protein purification. Protein Expr Purif. 2005;41:98–105. doi: 10.1016/j.pep.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Manthey GM, McEwen JE. The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J. 1995;14:4031–4043. doi: 10.1002/j.1460-2075.1995.tb00074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AM, Pape KL, Tzagoloff A. Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae. EMBO J. 1985;4:2087–2092. doi: 10.1002/j.1460-2075.1985.tb03896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Wilmes C, Tatsuta T, Langer T. Prohibitins interact genetically with Atp23, a novel processing peptidase and chaperone for the F1Fo-ATP synthase. Mol Biol Cell. 2007;18:627–635. doi: 10.1091/mbc.E06-09-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poutre CG, Fox TD. PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics. 1987;115:637–647. doi: 10.1093/genetics/115.4.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak M, Tetaud E, Godard F, Sagot I, Salin B, Duvezin-Caubet S, Slonimski PP, Rytka J, di Rago JP. Yeast cells lacking the mitochondrial gene encoding the ATP synthase subunit 6 exhibit a selective loss of complex IV and unusual mitochondrial morphology. J Biol Chem. 2007;282:10853–10864. doi: 10.1074/jbc.M608692200. [DOI] [PubMed] [Google Scholar]
- Rak M, Tzagoloff A. F1-dependent translation of mitochondrially encoded Atp6p and Atp8p subunits of yeast ATP synthase. Proc Natl Acad Sci USA. 2009;106:18509–18514. doi: 10.1073/pnas.0910351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Rödel G, Fox TD. The yeast nuclear gene CBS1 is required for translation of mitochondrial mRNAs bearing the cob 5’ untranslated leader. Mol Gen Genet. 1987;206:45–50. doi: 10.1007/BF00326534. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schiestl RH, Gietz RD. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Steele DF, Butler CA, Fox TD. Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc Natl Acad Sci USA. 1996;93:5253–5257. doi: 10.1073/pnas.93.11.5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent RD, Hofmann TJ, Zassenhaus HP. Sequence and expression of NUC1, the gene encoding the mitochondrial nuclease in Saccharomyces cerevisiae. Nucleic Acids Res. 1988;16:3297–3312. doi: 10.1093/nar/16.8.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Hourset A, Tzagoloff A. The Saccharomyces cerevisiae ATP22 gene codes for the mitochondrial ATPase subunit 6-specific translation factor. Genetics. 2007a;175:55–63. doi: 10.1534/genetics.106.065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Neupert W, Tzagoloff A. The metalloprotease encoded by ATP23 has a dual function in processing and assembly of subunit 6 of mitochondrial ATPase. Mol Biol Cell. 2007b;18:617–626. doi: 10.1091/mbc.E06-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]