

Abstract
The model organism Streptomyces coelicolor represents a genus that produces a vast range of bioactive secondary metabolites. We describe a versatile procedure for systematic and comprehensive mutagenesis of the S. coelicolor genome. The high-throughput process relies on in vitro transposon mutagenesis of an ordered cosmid library; mutagenized cosmids with fully characterized insertions are then transferred by intergeneric conjugation into Streptomyces, where gene replacement is selected. The procedure can yield insertions in upward of 90% of genes, and its application to the entire genome is underway. The methodology could be applied to many other organisms that can receive DNA via RK2/RP4-mediated intergeneric conjugation. The system permits introduction of mutations into different genetic backgrounds and qualitative measurement of the expression of disrupted genes as demonstrated in the analysis of a hybrid histidine kinase and response regulator gene pair, osaAB, involved in osmoadaptation in Streptomyces. The independently transcribed response regulator gene, osaB, is essential for osmoadaptation; when grown with supplementary osmolyte, an osaB mutant cannot erect aerial hyphae and produces up to fivefold greater antibiotic yields than the wild-type strain.
Streptomycetes are complex multicellular saprophytic soil bacteria, perhaps best known for their ability to produce over two-thirds of naturally derived antibiotics currently in medical use (as well as many other valuable pharmaceuticals). Detailed investigations into how differentiation and reproductive growth are coordinated with antibiotic production have focused on the model organism Streptomyces coelicolor A3(2). The onset of secondary metabolism in this species is easily visualized, because two of the antibiotics it produces, undecylprodigiosin and actinorhodin, are highly pigmented. The sequence of the 8.7 Mb linear chromosome of S. coelicolor A3(2) has recently been completed (Bentley et al. 2002), revealing synteny between the central core of the chromosome and the whole chromosomes of actinomycete pathogens such as Mycobacterium tuberculosis. Outside of this ancestral core, the chromosome arms are comprised of largely non-essential contingency genes. Insight into actinomycete gene function can be achieved by mutational analysis. Here, we describe a procedure for systematic insertional mutagenesis that provides both greater versatility and higher throughput than previous methods used to disrupt Streptomyces genes. Gene disruption is achieved by adapting in vitro transposon mutagenesis (Goryshin and Reznikoff 1998), using a derivative of Tn5, to obtain insertions in representatives of the ordered cosmid library that was previously used to sequence the S. coelicolor genome (Redenbach et al. 1996). A transposon insertion library of a cosmid is recovered initially in Escherichia coli, and the location of each insertion then determined by DNA sequencing. The inclusion of an origin of transfer (oriT; RK2) in the transposon allows each cosmid to be subsequently mobilized into S. coelicolor by conjugal transfer, yielding exconjugants with the desired gene disruption with high efficiency.
To demonstrate the utility of this procedure, we have compared how mutations in a predicted response regulator have been obtained by both in vivo and in vitro transposon mutagenesis. Two component regulatory systems, each consisting of a sensor histidine kinase and response regulator, are known to regulate aspects of growth and metabolism, including antibiotic production, in S. coelicolor (Ishizuka et al. 1992; Paget et al. 1999a). More complex multicomponent phosphorelays have been described in other bacteria—one extensively studied system regulates the initiation of sporulation in Bacillus subtilis (Hoch 2002). Multicomponent phosphorelays are structurally analogous to less complicated two-component systems; initiating with a hybrid histidine kinase (a sensory kinase with an additional receiver domain), a change in the environment results in autophosphorylation of the kinase domain. Phosphorylation is relayed from histidine onto the aspartate in the receiver domain and then via an intermediate Histidine Phospho Transfer (HPT) module. Ultimately, a response regulator is phosphorylated, thereby modulating its activity, typically as a transcription factor.
Growth of Streptomyces in submerged liquid medium or on agar plates, is often discontinuous (Granozzi et al. 1990; Susstrunk et al. 1998; Vohradsky et al. 2000). On solid medium, an initial phase of rapid growth generates the filamentous substrate mycelium; a second growth phase follows with the erection of aerial hyphae and the production of secondary metabolites (e.g., antibiotics). One important adaptation for this second round of growth is the generation of turgor pressure to support aerial hyphal growth. Consequently, osmoadaptation is critical for streptomycete differentiation. Genes involved in osmosensing and differentiation include those encoding alternative σ factors belonging to the SigB family (Cho et al. 2001; Viollier et al. 2003), believed to coordinate expression of a network of developmental regulons. However, little is yet known about how changes in environmental osmolarity are relayed to trigger expression of these σ factors. S. coelicolor has two annotated hybrid histidine kinases (SCO5748 and an orphan SCO7327) that are similar to a family of fungal hybrid histidine kinases, which have been implicated primarily to function in osmoadaptation and virulence (Brinkman et al. 2001). Using transposon mutagenesis, we have investigated the function of the hybrid histidine kinase SCO5748 and the adjacent response regulator SCO5749, designated here osaAB, and shown that the latter is critical for osmoadaptation, differentiation, and the coordination of antibiotic production.
RESULTS
Disruption of the S. lividans Ortholog of osaB by In Vivo Transposon Mutagenesis
An S. lividans 1326 mutant library was prepared using the Tn5 derivative Tn5493 (Volff and Altenbuchner 1997). One mutant, X0236, exhibiting a bald (lack of aerial mycelium) phenotype on R2YE medium (Fig. 1), was investigated further. To locate the site of insertion, we sequenced inverse PCR products obtained with transposon-specific primers. This revealed insertion of Tn5493 between the putative ribosome-binding site and start codon (at a position equivalent to base 6285218 in the S. coelicolor genome) of a response regulator gene, osaB (accession no. AJ565873), that shares 99% sequence identity with the equivalent gene of the closely related S. coelicolor. In vivo transposon mutagenesis of Streptomyces can lead to the occurrence of mutations unlinked to the site of insertion (Nodwell et al. 1996). We confirmed linkage by crossing X0236 with S. lividans TK64 and selecting for thiostrepton- (conferred by Tn5493) and streptomycin-resistant (counterselecting the donor X0236 strain) exconjugants; all 95 exconjugants tested possessed the same bald phenotype as X0236 when grown on R2YE.
Figure 1.
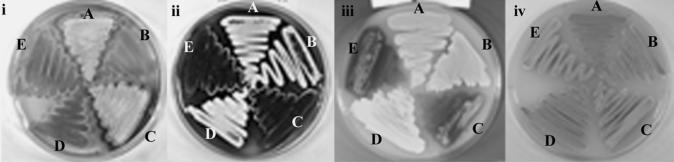
osaB is essential for osmoadaptation. Cultures were incubated for 7 d at 30°C. (i) S. lividans strains grown on R2YE containing 10.3% sucrose: (A) 1326, (B) AB101 containing pPM927, (C) AB101 containing pAB04, (D) AB101, (E) X0236; (ii) S. coelicolor strains grown on R2YE containing 10.3% sucrose: (A) M145, (B) AB201, (C) AB001 containing pPM927, (D) AB001 containing pAB04, (E) AB001; (iii) the same S. coelicolor strains grown on MS containing 25 mM KCl; and (iv) grown on MS with no supplementary osmolyte.
In Vitro Transposon Mutagenesis of Cosmid SC 7C 7
To disrupt genes cloned in Supercos-1 [which itself contains resistance to kanamycin (KanR) and ampicillin (AmpR)] and permit their subsequent transfer into S. coelicolor, a Tn5 derivative, Tn5062, was constructed; the structure of this transposon is illustrated in Figure 2A. An in vitro transposition reaction using Tn5062 and, as target, SC7C7, a representative of the ordered S. coelicolor cosmid library that contains the osaAB genes, yielded in excess of 103 AprR KanR AmpR colonies after electroporation of E. coli JM109. Cosmid DNA was isolated from 192 randomly picked colonies and sequenced using Tn5062-specific primers, revealing that 92% of insertions occurred in Streptomyces DNA. Of these, 90% were within predicted ORFs and 10% in intergenic regions, reflecting the overall proportion of coding DNA (Bentley et al. 2002). The mean distance between independent insertions was 274 bp, and 90% of predicted genes within SC7C7 were represented by at least one insertion cosmid. Details of Tn5062 insertions in this and other cosmids are listed at the S. coelicolor genome database, http://jiio16.jic.bbsrc.ac.uk/S.coelicolor/.
Figure 2.
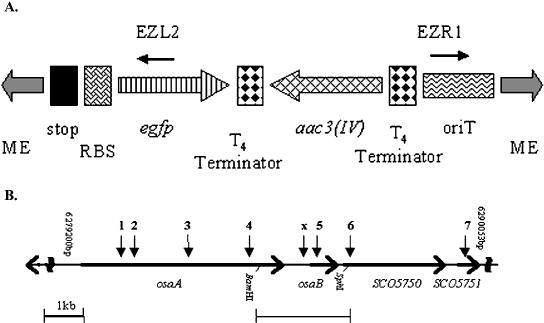
Structure of Tn5062 and the osaAB gene region. (A) Tn5062 is comprised of the following elements (not drawn to scale): two Tn5 mosaic ends (ME) flanking a three-frame translational stop and a consensus streptomycete ribosome-binding site (rbs) preceding a promoterless copy of eGFP, the apramycin resistance gene, aac3(IV), itself flanked by two T4 transcriptional terminators, and oriT(RK2). The locations of the two sequencing primers, EZL2 and EZR1, are indicated. (B) Arrows above the osaAB gene region indicate the location of representative Tn5062 insertions (1–7) and the Tn5493 insertion (x). Tn5062 insertions 1–6 in osaAB are all oriented to place eGFP under transcriptional control of osaAB promoters. S. coelicolor strain AB201 contains an insertion at position 1 (position 6281495 in the chromsome), and strain AB001 contains an insertion at position 5 (position 6285442). The extent of the fragment and the restriction sites used for cloning osaB and its promoter for complementation are indicated beneath the gene. This gene was initially cloned linked to aac3(IV) of the Tn5062 insertion in SCO5751 (insertion 7).
To introduce the mutations into S. coelicolor, which restricts methylated DNA from dam+ dcm+ E. coli (MacNeil 1988), cosmids were passed through the nonmethylating host ET12567/pUZ8002. The plasmid in this strain supplies trans-acting functions for the mobilization of oriT-containing cosmids (Paget et al. 1999b). Once in Streptomyces, Supercos-1 is unable to replicate autonomously, but the long regions of sequence identity in the S. coelicolor cosmids shared with the chromosome promote efficient integration by homologous recombination. A typical intergeneric conjugation yielded between 104 and 105 AprR S. coelicolor colonies. Of these, between 70% and 90% were also KanR, presumably due to integration of the entire cosmid by a single cross-over. The remaining proportion were KanS, indicative of allelic replacement and loss of the Supercos-1 vector. Four independent insertions (of a total of 10) in osaA and one insertion (of three) in osaB (Fig. 2B) were introduced into S. coelicolor in this manner. For each mutation, six independent AprR KanS exconjugants were checked by Southern blot analysis, verifying that the expected double cross-over and allelic replacement had occurred in each case (results not shown).
osaB Is Required for Osmoadaptation Necessary for Differentiation
As in the case of the S. lividans mutant XO236, all six independent osaB mutants of S. coelicolor failed to develop aerial mycelia when grown on R2YE medium that contains 10.3% sucrose, but produced an abundance of the pigmented antibiotics. This phenotype was not due to polar effects on the downstream gene SCO5750; disruption of this gene (insertion 6, Fig. 2B), an ftsK ortholog, resulted in no evident macroscopic phenotype (P. Dyson and P. Herron, unpubl.). A representative osaB mutant, strain AB001, was used for further analyses. Independent of the carbon source (glucose or mannitol), AB001 failed to erect aerial hyphae when grown on a variety of medium in the presence of either 10% sucrose or 250 mM KCl, but produced pigmented antibiotic that appeared qualitatively in excess of the wild-type strain (Fig. 1). Growth and differentiation of AB001 on medium containing no additional osmolytes was similar to the wild-type strain. Introduction of the osaB complementing plasmid, pAB04, into AB001, restored development and antibiotic production to the wild-type pattern under all conditions investigated (Fig. 1). The Tn5062 disruption of osaB was also transferred into S. lividans by intergeneric conjugation from E. coli and selecting for gene replacement. The expected double cross-over in representative exconjugants was confirmed by Southern hybridization (data not shown). The exconjugants exhibited a similar bald phenotype to X0236 carrying the Tn5493 insertion and, like X0236, were more pigmented than the wild-type strain (Fig. 1). Complementation of one representative mutant, AB101, with pAB04, restored the wild-type phenotype. The phenotypes of the four independent osaA mutants of S. coelicolor contrasted to that of the osaB mutant AB001. When grown on medium containing either 10% sucrose or 250 mM KCl, the osaA mutants, represented by strain AB201, exhibited a delay of 24 h, compared with the wild type, before normal aerial growth commenced (reflected in all six AprR KanS exconjugants examined for each of the four mutations; Fig. 2). In the absence of additional osmolyte, no delay in the development of these mutants was observed.
Typical bald mutants of S. coelicolor produce reduced levels of antibiotics (Horinouchi 1996). In contrast, fermentation of AB001 in the presence of supplementary osmolyte resulted in from three- to fivefold greater yields of undecylprodigiosin and actinorhodin compared with the wild-type and complemented strains (Fig. 3). The growth curves of the mutant were similar to those of the wild-type strain. In the absence of additional osmolyte, antibiotic production by AB001 was not significantly elevated.
Figure 3.
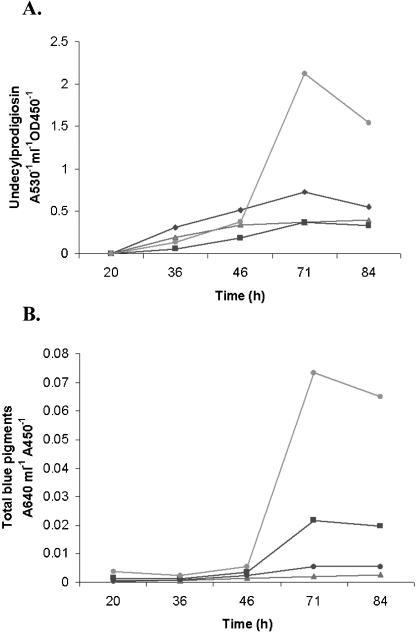
Antibiotic overproduction by the S. coelicolor osaB mutant AB001. Graphs of yields (the average of three independent experiments; standard deviations were <5% of the plotted values) at indicated time-points of (A) actinorhodin and γ-actinorhodin, and (B) undecylprodigiosin relative to cell density (OD450) produced by submerged cultures grown in liquid R5 with or without 10.3% sucrose (S); (•) AB001 (+S); (▪) M145 (+S); (♦) AB001 (-S); (▴) M145 (-S).
osaB Has Its Own Promoter
The majority of sensory histidine kinase and response regulator gene pairs are transcriptionally coupled (West and Stock 2001). By analogy with other transposons, we expected Tn5062 insertions to produce polar mutations. The severity of the osaB mutant phenotype compared with that of the osaA mutants suggested that the mutations in osaA were not polar with respect to osaB and, consequently, that the response regulator is independently regulated. An initial qualitative measure of expression of the two genes was made by monitoring levels of green fluorescent protein in hyphae of the mutants, revealing up-regulation in the substrate hyphae grown in medium with supplementary osmolyte (Fig. 4A). In the case of the differentiating osaA mutant eGFP, up-regulation was also observed in aerial hyphae. To investigate and map the osaB promoter, S1 nuclease protection assays were used (Fig. 4B). The transcriptional start of osaB was mapped to 120 bp upstream of the predicted translational start. Immediately upstream, we identified -10 and -35 sequences that compare well with consensus streptomycete promoter sequences (Strohl 1992). The promoter was active over an extended period of growth on R2YE medium.
Figure 4.
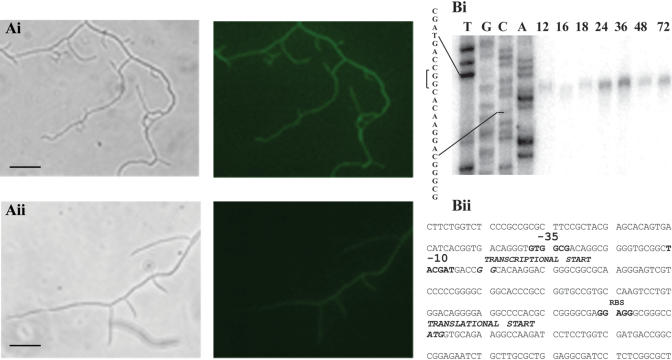
Transcription of osaB. (A) Fluorescence and phase-contrast images of substrate mycelia after 24 h growth of S. coelicolor AB001 on R2YE medium (i) with or (ii) without 10.3% sucrose (Bar, 10 μm). (B) High resolution SI nuclease mapping of the osaB transcription start site. (i) RNA was isolated at the time points indicated from cultures of S. coelicolor M145 grown on R2YE medium containing 10.3% sucrose. (ii) The corresponding nucleotide sequence of the promoter region of osaB, indicating -35 and -10 sequences, transcriptional start (arrow), the probable ribosome-binding site (rbs), and translational start (TS).
DISCUSSION
As more and more genome sequences have become available, procedures for efficiently disrupting defined genes have assumed increasing importance. The system we describe here, based on in vitro transposition, has an inherent throughput and versatility well suited for this new era of functional genomic analysis. Traditional in vivo transposon mutagenesis of a genome is used to generate a library of mutants each containing, ideally, a single random insertion. As illustrated here, with the example of Tn5493 mutagenesis of S. lividans, a mutant with a phenotype of interest, in this case a developmental defect, can then be examined to determine the identity of the disrupted gene by, for example, sequencing inverse PCR products. This highlights one of several technical limitations associated with in vivo systems that also include the requirement to express transposase in the target host, the need to introduce the transposon on a suicide vector and ensure the vector is subsequently lost, and the opportunities for more than a single transposition event that can contribute to genetic instability. An in vitro methodology, based on electro-poration of released Tn5 transposition complexes, transposomes, into the target host has been applied in other bacteria to over-come some of these limitations (Goryshin et al. 2000). Random S. coelicolor mutant libraries have also been constructed by in vitro transposon mutagenesis of DNA shotgun cloned into E. coli vectors (Gehring et al. 2000); mutations were subsequently introduced into S. coelicolor by selecting for marker replacement. Linkage between the transposon insertion and the mutation responsible for a given phenotype was confirmed by use of a procedure of transformation with denatured linear DNA (Oh and Chater 1997), and the identity of the disrupted gene in each mutant was determined by cloning and sequencing DNA linked to the antibiotic resistance marker of the transposon. The in vitro transposition methodology we have developed obviates the need for time-consuming linkage analysis and cloning, and is designed for systematic and comprehensive mutagenesis of the genome, rather than the creation of random insertion libraries.
In vitro transposon mutagenesis of a cosmid is efficient and allows the recovery of several thousand clones in E. coli. Cosmid extraction from E. coli and subsequent sequencing are procedures suitable for automation, offering a high throughput to determine the site of insertion in each cosmid. We chose to characterize 192 clones from each in vitro reaction, yielding insertions in, on average, 87% of genes. As more cosmids are systematically mutagenized, the redundant nature of the overlapping cosmid library will inevitably result in a higher percentage of genes containing insertions For example, mutagenesis of three contiguous and overlapping cosmids has provided insertions in 31 of the 32 predicted ORFs (97% coverage) present in the central cosmid. Moreover, even more comprehensive coverage could evidently be achieved by sequencing more clones. We have achieved saturation mutagenesis, with at least one insertion per predicted ORF, by analysis of 384 clones derived from mutagenesis of one representative cosmid. In practice, as part of an ongoing program of saturation mutagenesis, we are analyzing 192 clones per reaction, as a compromise to economize on time and costs. After mutagenesis of overlapping cosmids, any remaining ORFs requiring mutagenesis will be targeted using PCR-generated cassettes (Gust et al. 2003).
The inclusion of oriT from an IncP-group plasmid in the transposon allows conjugation to be used for intergeneric transfer of cosmid DNA from E. coli to Streptomyces. In comparison with transformation protocols described for Streptomyces (Kieser et al. 2000), conjugation is much more efficient. Allelic replacement in exconjugants is efficient, with the implication that phenotypes of several replicate exconjugants can be compared to confirm an association with a specific gene disruption. With independent insertions distributed on average every 274 bp, a typical gene can be disrupted in multiple positions. In the case of osaA, a similar phenotype resulted from four independent insertions, providing strong evidence as to the consequence of disruption of this gene. Although not apparent in the case of osaA, multiple transposon insertions in a coding sequence may provide additional information on domain functionality. Another versatile aspect of the system is that mutations can be moved into different genetic backgrounds or even into related species, as demonstrated in this study.
We have used the procedure to disrupt osaB of S. coelicolor and S. lividans. In both species, the mutations resulted in the inability of the mutants to erect aerial hyphae on medium supplemented with osmolyte. We explored this defect in more detail in S. coelicolor, demonstrating that the inability to adapt results in from three to five times greater yields of the two pigmented secondary metabolites, actinorhodin and undecylprodigiosin. These antibiotics are structurally diverse and derived from quite different precursors, indicating a general effect on the regulation of antibiotic biosynthesis. The overproduction of antibiotics is likely to be a response to the disruption of the complex regulatory network, involving stress adaptation, which coordinates morphological and physiological differentiation.
The much less severe phenotype of delayed osmoadaptation and development due to mutation in the upstream hybrid histidine kinase gene, osaA, was unexpected. This implied that, unlike osaB, this gene is not essential for osmoadaptation. Whether the second hybrid histidine kinase, SCO7327, an orphan, works in conjunction with osaA, remains a possibility, and such redundancy could explain the mild phenotype. Moreover, we could also infer that osaAB are not necessarily transcriptionally coupled. This was confirmed both by mapping the osaB promoter and demonstrating genetic complementation in the osaB mutant with the gene and its promoter alone. Construction of the complementing plasmid was facilitated by cloning osaB linked to the antibiotic resistance gene of a Tn5062 insertion in a neighboring gene, illustrating another use of the mutant cosmid library. Due to the inclusion of a promoterless reporter gene, eGFP, Tn5062 insertions also permit analysis of expression of disrupted genes. In this study, fluorescence microscopy was used to reveal up-regulation of osaAB in the presence of supplementary osmolyte. The reporter gene also permits analysis of any tissue specificity of expression.
The build up of osmotic pressure to drive aerial growth requires coordinated changes in global gene expression. We are proceeding with more detailed investigations into osmoadaptation in S. coelicolor by, for example, defining the function of OsaB. This response regulator has an atypical output domain; rather than a DNA-binding motif, it has a predicted coiled-coil structure. This implies an interaction with one or more coiled-coil proteins that mediate the response, thereby coordinating maintenance of turgor and synchronizing progression of secondary metabolism. Insight into the response will utilize a variety of proteomic and functional genomic techniques, underpinned by further studies of mutants. Tn5062 mutagenesis of all of the S. coelicolor cosmids is at present underway, and the resulting libraries will be an invaluable resource for the analysis and manipulation of this model organism; to date, more than 250 mutated cosmids have been distributed to over 15 researchers worldwide. Information on the status of the systematic mutagenesis of the S. coelicolor genome is available at http://jiio16.jic.bbsrc.ac.uk/S.coelicolor/. Moreover, this mutagenesis procedure could be beneficially applied to many other organisms, including eukaryotic cells that can receive DNA via RK2/RP4-mediated transkingdom conjugation (Inomata et al. 1994; Waters 2001).
METHODS
Bacterial Strains, Growth Conditions, and Southern Hybridizations
Bacterial strains and plasmids used in this study are listed in Table 1. E. coli strains were grown on L agar plates and L broth medium containing ampicillin (50 μg/mL), apramycin (100 μg/mL), tetracycline (12.5 μg/mL), kanamycin (25 μg/mL), chloramphenicol (25 μg/mL), spectinomycin (100 μg/mL), or streptomycin (25 μg/mL) where appropriate. Streptomyces strains were grown on R2YE, MS agar, minimal medium [containing either 10% (w/v) glucose or 5% (w/v) mannitol], and nutrient agar at 30°C (Kieser et al. 2000) as circumstances required. Where stated, medium was supplemented with either 10% sucrose or 250 mM KCl. Medium for growth of streptomycete strains was supplemented with apramycin (100 μg/mL), kanamycin (25 μg/mL), spectinomycin (200 μg/mL), thiostrepton (25 μg/mL), or nalidixic acid (20 μg/mL) where appropriate. Unless stated otherwise, plasmids and mutated cosmids were introduced into Streptomyces by intergeneric conjugation from E. coli ET12567/pUZ8002 (Paget et al. 1999b). Streptomyces genomic DNA was isolated using a Fast Prep Soil DNA isolation kit (Bio101). Briefly, streptomycete strains were grown on nutrient agar for 24 h, after which 100 mg of mycelia was scraped off and DNA extracted according to the manufacturers' instructions. Southern hybridization was by established procedures using the digoxigenin-labeled 3442-bp Tn5062 PvuII fragment as a probe.
Table 1.
Bacterial Strains and Plasmids
| Strain or plasmid | Characteristics | Reference or source |
|---|---|---|
| S. coelicolor M145 | prototrophic, SCP1- SCP2- PgI+ | Kieser et al. 2000 |
| S. lividans 1326 | SLP2+ SLP3+ | Kieser et al. 2000 |
| S. lividans TK64 | pro-2 str-6 SLP2- SLP3- | Kieser et al. 2000 |
| S. lividans X0236 | osaB::Tn5493 | This work |
| S. coelicolor AB001 | osaB::Tn5062 | This work |
| S. lividans AB101 | osaB::Tn5062 | This work |
| S. coelicolor AB201 | osaA::Tn5062 | This work |
| E. coli JM109 | F′ traD36 proA+B+ lac1q Δ(lacZ)M15/Δ(lac-proAB) glnV44 e14- gyrA96 recA1 relA1 endA1 thi hsdR17 | Yanisch-Perron et al. 1985 |
| E. coli ET12567 (pUZ8002) | dam13::Tn9 dcm6 hsdM hsdR recF143 16 zjj201::Tn10 galK2 galT22 ara14 lacY1 xyl5 leuB6 thi1 tonA31 rpsL136 hisG4 tsx78 mtli glnV44, containing the nontransmissible oriT mobilizing plasmid, pUZ8002 | |
| SC7C7 | Supercos-1 containing chromosomal DNA from S. coelicolor; bla kan | Redenbach et al. 1996 |
| pET26B+ | E. coli cloning vector; kan | Novagen |
| pALTER1 | E. coli cloning vector; bla tet | Promega |
| pEGFP-N1 | E. coli mammalian eGFP shuttle vector; kan | Clontech |
| pHP45Ωacc | E. coli vector; aac3(IV) | Blondelet-Rouault et al. 1997 |
| pMOD〈MCS〉 | E. coli vector with Tn5 inverted repeats; bla | Epicentre |
| pIJ8660 | integrative vector for Steptomyces; oriT (RK2), aac3(IV) | Sun et al. 1999 |
| pFP11 | pALTER1 containing eGFP | This work |
| pFP12 | pFP11, eGFP with 5′ Ndel site | This work |
| pVC101 | pET26B+ containing three-frame translational stop | This work |
| pVC102 | pVC101 containing streptomycete rbs | This work |
| pVC107 | pVC102 containing eGFP | This work |
| pQM501 | pALTER1 containing aac3(IV) | This work |
| pQM504 | pMOD〈MCS〉 containing aac3(IV) | This work |
| pQM5052 | pQM504 containing oriT(RK2) | This work |
| pQM5062 | pQM5052 containing eGFP; Tn5062 | This work |
| pUC18 | E. coli cloning vector; bla | Yanisch-Perron et al. 1985 |
| pD81 | pALTER1 containing osaB, SCO5750, aac3(IV), tet | This work |
| pD04 | pUC18 containing osaB | This work |
| pAD04 | pD04 ligated to pALTER1 | This work |
| pPM927 | integrative vector for Streptomyces; oriT(RK2) int attP (pSAM2) aadA, tsr | Smokvina et al. 1990 |
| pPM04 | pPM927 containing osaB | This work |
Isolation and Characterization of X0236
In vivo transposon mutagenesis of S. lividans using Tn5493 (Volff and Altenbuchner 1997) was carried out after transformation with pJOE2577 as previously described (Herron et al. 1999). Linkage of the developmental mutation in X0236 with Tn5493 was confirmed by mating the mutant with the plasmid-free strain S. lividans TK64. Exconjugants were isolated on medium supplemented with thiostrepton and streptomycin (10 μg/mL), the latter to counterselect the donor. The location of the insertion in X0236 was determined by inverse PCR (Martin and Mohn 1999). X0236 genomic DNA (2.5 μg) was digested with SalI, which cuts once within Tn5493. The digested DNA was extracted with phenolchloroform, ethanol precipitated, and resuspended in 1 mL of 1×T4 DNA ligase reaction mix containing 15 Weiss units of T4 DNA ligase (Life Technologies) and incubated overnight at 14°C. Subsequently, the DNA was extracted with phenolchloroform, ethanol precipitated, and resuspended in 25 μL of sterile distilled water. Two, 50-μL PCR reactions were then set up containing 5 μL of DNA, 1 mM of MgSO4, 200 μM of dNTPS, 0.5 units of exo- Vent DNA polymerase (New England Biolabs), 1× reaction buffer, and 0.5 μM of each primer. Different primer pairs were used to generate PCR products from each of the two circular molecules generated after ligation, firstly, Tsr3and Tsr4, and secondly, Tsr7and Tsr8 (Table 2). The PCR was carried out in a MJ Research PTC-200 DNA engine by initially heating to 95°C for 3 min, followed by five cycles of amplification (95°C, 1 min; 55°C, 1 min; 72°C, 2 min); subsequently, a further 30 cycles of amplification were carried out (95°C, 1 min; 60°C, 1 min; 72°C, 2 min), followed by a final extension at 72°C for 7 min. Agarose gel electrophoresis of the products revealed single bands in each reaction. The two PCR products corresponding to the two flanking regions of the insertion were then further amplified with pairs of nested primers (Tsr1/Tsr2 for the Tsr3/Tsr4 product and Tsr5/Tsr6 for the Tsr7/Tsr8 product) using the same conditions as before. These PCR products were then sequenced, using the nested primer pairs, with a Beckman-Coulter CEQ 2000XL sequencer.
Table 2.
Oligonucleotides Used. The Nonhomologous Tail in the S1 Mapping Primer S1-04nht Is Underlined
| Tn5062 linker oligonucleotides | |
|---|---|
| VC1 | 5′-GATCTGAATTCGGATCCTAATTAATTAATCTAGAAAGGAGGTGATCA |
| VC2 | 5′-TATGATCACCTCCTTTCTAGATTAATTAATTAGGATCCGAATTCA |
| VC3 | 5′-TATGGACGGAGCTCGGCCGCTTAAGGTACCGAATTCC |
| VC4 | 5′-TCGAGGAATTCGGTACCTTAAGCGGCCGAGCTCCGTCCA |
| Tn5062 sequencing primers | |
|---|---|
| EZR1 | 5′-ATGCGCTCCATCAAGAAGAG |
| EZL2 | 5′-TCCAGCTCGACCAGGATG |
| IPCR primers | |
|---|---|
| Tsr1 | 5′-GCAAGGGGAAGTCGTCGCTCTCTGG |
| Tsr2 | 5′-CGGCGTGGGGACGTCGTCGTTCTCG |
| Tsr3 | 5′-GGATTTGCGATGGTGTCCAACTCAG |
| Tsr4 | 5′-GGACGTCGAGCCCCTCATGCACAGC |
| Tsr5 | 5′-GGAGAGAACGACGGGAAGGGAGAAG |
| Tsr6 | 5′-GACCTCGATTCGTCAGTGATGATCC |
| Tsr7 | 5′-CGGATTGTCCCCGAGTTCCTTCACG |
| Tsr8 | 5′-GCGTCTTCCGCCTCGGTTTCCATCC |
| S1 mapping primers | |
|---|---|
| S1-04nht | 5′-GCGCAAGCTTCCCGAAGCCGTGTAGAAGTA |
| S1-04r | 5′-AGTGCTTTGAGCGCTTCCT |
Construction of Tn5062
DNA manipulations were carried out according to established procedures (Sambrook et al. 1989). eGFP from pEGFP-N1 was subcloned into pALTER1 as a 787-bp HindIII–XbaI fragment creating pFP11. An NdeI site was introduced at the start codon of eGFP in pFP11by site-directed mutagenesis using an Altered Sites Kit from Promega according to the manufacturers' instructions, resulting in pFP12; ACCATG (pFP11) was changed to CATATG (pFP12), where ATG is the first codon of the eGFP gene. The three-frame translational stop was constructed as a linker made from the oligonucleotides VC1 and VC2 (Table 2) and cloned into BglII–NdeI-digested pET26B+, creating pVC101. This was digested with NdeI and XhoI and ligated to a second linker synthesized as the oligonucleotides VC3 and VC4 (Table 2) carrying a Streptomyces consensus ribosome-binding site, creating pVC102. eGFP was cloned from pFP12 into pVC102 as a 725-bp NdeI–EagI fragment giving pVC107. aac3(IV) was first moved to pALTER1 from pHP45Ωaac as a 1783-bp EcoRI fragment, creating pQM501. This plasmid was digested with SmaI and the 1794-bp aac3(IV) fragment introduced between the Tn5 inverted repeats of pMOD〈MCS〉 by blunt-ended ligation with EcoICRI and HincII-digested pMOD〈MCS〉, resulting in pQM504. oriT (RK2) was introduced into pQM504 as a 786-bp PstI fragment from pIJ8660, giving pQM5052. Finally, eGFP was added to pQM5052 as a 782-bp EcoRI fragment from pVC107. This plasmid was named pQM5062 and allowed Tn5062 to be liberated from the plasmid backbone by digestion with PvuII as a 3442-bp fragment (Fig. 2A). The sequence of the transposon (accession no. AJ566337) was verified using a Beckman-Coulter CEQ 2000XL sequencer.
In Vitro Transposition
The 3442-bp Tn5062 PvuII fragment was isolated from an agarose gel using a QIAEX II gel extraction system (Qiagen) and further purified using a QIAquick PCR purification kit (Qiagen) before being resuspended in 10 μL of sterile distilled water. The following reaction mix was prepared according to the manufacturers' instructions (Epicentre) and incubated for 2 h at 37°C: 1 μL of EZ::Tn 10× Reaction Buffer; 200 ng (ca. 7.6 × 10-9 μMoles) SC7C7 DNA; 17.5 ng (7.6 × 10-9 μMoles) Tn5062 DNA; sterile distilled water to give a final volume of 10 and 1 μL of EZ::TN Transposase. After completion, the reaction was stopped by adding 1 μL of EZ::TN stop solution and incubated at 70°C for 10 min. A total of 1 μL of the transposition reaction was then transformed into electrocompetent E. coli JM109 cells and plated on L agar supplemented with ampicillin, kanamycin, and apramycin to select for colonies containing transposed cosmids. A total of 96 ampicillin, kanamycin, and apramycin resistant colonies were picked and inoculated to a 96-square deep well growth block (ABgene), each well containing 1 mL of L broth supplemented with ampicillin, kanamycin, and apramycin. Following overnight incubation (225 rpm), 1.3 μL of each of the 96 cultures was then transferred to a second growth block, each well containing 1.3 mL of 2 × YT supplemented with apramycin and incubated for 18 h at 225 rpm. A total of 330 μL of 60% (w/v) glycerol was then added to each of the 96 wells from the first growth block, mixed, and stored at -70°C. Cosmid DNA was isolated from the cultures in the second growth block using a Wizard SV 96 kit (Promega) and stored at -70°C. For sequencing, ∼500 ng of DNA from each of the 96 samples was transferred to a 96-well PCR plate (ABgene) and heated to 86°C for 5 min in a PTC-200 DNA engine (MJ Research). We then added to each sample 2 μL of transposon-specific sequencing primer EZR1 or EZL2 (10 pmole/μL; Table 2; Fig. 2A) and 8 μL of CEQ DTCS quick-start sequencing kit (Beckman-Coulter). The sequencing reactions were then carried out in a PTC-200 DNA engine by heating to 96°C (20 sec), 55°C (20 sec), and 60°C (4 min) for 50 cycles. The samples were then analyzed on a CEQ 2000XL sequencer.
Identification of Tn5062 Insertion Sites
Flanking sequences to Tn5062 insertions were identified by analysis of raw sequence files using DNAMAN (Lynnon Biosoft). The location and orientation of each insertion was established by comparison of the flanking sequence (BLASTn) with the S. coelicolor genome sequence (http://www.sanger.ac.uk/Projects/S_coelicolor/). Information on the coding sequences present on the SC7C7 real cosmid was determined from the S. coelicolor genome database (http://jiio16.jic.bbsrc.ac.uk/S.coelicolor/).
Cloning of osaB
The mutant cosmid library facilitates the cloning of genes for complementation. By selecting a Tn5062 insertion in a gene flanking a region of interest and choosing suitable restriction sites, the gene can be excised and cloned together with aac3(IV), permitting positive selection. SC7C7.D08 contains a Tn5062 insertion in SCO5751 at position 19059 bp (with respect to the cloned S. coelicolor DNA). osaB was initially subcloned from this cosmid together with aac3(IV) on a 9439-bp BglII–EcoRI fragment into pALTERI, digested with BamHI and EcoRI to create pD81. A 2332-bp BamHI–SphI fragment containing osaB was moved into pUC18, giving pD04. pD04 was then linearized by HindIII and ligated to pALTER1 to generate a chimera, pAD04. Finally, osaB and its promoter region were introduced into pPM927 as a 2362-bp BamHI–XbaI fragment, resulting in the complementing plasmid pAB04.
Fluorescence Microscopy
Samples for light and fluorescence microscopy were prepared by taking surface impressions of mature plates and from cultures grown against sterile coverslips. Hyphae were fixed in methanol and mounted in 40% glycerol (Kieser et al. 2000). Samples were studied using a Nikon Eclipse E600 epifluorescent microscope (with a standard FITC filter set), and images were captured using a RS Photometrics digital camera. Appropriate controls were used to establish levels of background fluorescence and determine suitable exposure times.
RNA Isolation
Mycelia were harvested from cellophane discs overlaid onto R2YE medium, and then resuspended in RNAprotect Bacteria Reagent (Qiagen) prior to RNA isolation using a Qiagen RNeasy Midi kit. An additional step was incorporated to remove any contaminating DNA, via an on-column DNase treatment using a Qiagen RNase free DNase set. The quality and concentration of the RNA were assayed using gel electrophoresis and spectrophotometrically.
S1 Nuclease Protection Assay
Amplification of a 385-bp fragment from the template pD04 using S1-04nht and S1-04r (Table 2) generated the probe for S1 nuclease mapping of the osaB promoter. The primers associated 244 bp before the predicted translational start of osaB and 131 bp into the coding sequence. A nonhomologous tail was incorporated on the end of the S1-04nht primer. The PCR product was purified using a Qiagen QiaQuick PCR clean up kit and end labeled with [γ32P]ATP using T4 polynucleotide kinase. The probe was radioactively labeled on both ends, but subsequent to hybridization, the labeled nonhomologous tail was digested with S1 nuclease, permitting clear distinction between the true protected fragment and DNA–DNA hybrids. A total of 20 μg of RNA was hybridized to 100–200 cpm of probe in NaTCA at 70°C for 10 min, then 45°C for 15 h. The reaction was treated with S1 nuclease (100 units), denatured, and separated out on a 6% acrylamide gel alongside of a sequencing ladder (Kieser et al. 2000). The sequencing ladder was generated using pD04 as a template, S1-04r primer and a Sequenase quick denature plasmid sequencing kit (Amersham Pharmacia Biotech).
Antibiotic Analysis
Pregerminated spores were inoculated in 25 mL of liquid R5 medium (with 5% PEG 8000), in the presence and absence of 10.3% sucrose, to give an initial OD450 of between 0.03 and 0.05. Three independent fermentations for each strain and condition were in baffled flasks at 30°C, 230 rpm. During the time course, samples were assayed for growth rate (OD450) alongside of antibiotic production. Undecylprodigiosin was extracted overnight from pelleted mycelia with acidified methanol (Strauch et al. 1991) and quantified by measuring absorbance at 530 nm. γ-actinorhodin and actinorhodin were extracted by adding KOH to a final concentration of 1N (Bystrykh et al. 1996). The extracted pigments were then quantified by measuring absorbance at 640 nm, and absorption spectra from 400 to 700 nm confirmed the authenticity of the blue pigments.
Acknowledgments
This work was supported by a BBSRC Investigating Gene Function grant (IGF 58/12431). A.B. was supported by a BBSRC studentship. We thank Tobias Kieser and Govind Chandra, John Innes Centre, Norwich, UK, for establishing the mutagenesis Web site.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
[The sequence data from this study have been submitted to EMBL under accession nos. AJ565873 and AJ566337.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1710304. Article published online before print in April 2004.
References
- Bentley, S.D., Chater, K.F., Cerdeno-Tarraga, A.M., Challis, G.L., Thomson, N.R., James, K.D., Harris, D.E., Quail, M.A., Kieser, H., Harper, D., et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417: 141-147. [DOI] [PubMed] [Google Scholar]
- Blondelet-Rouault, M.H., Weiser, J., Lebrihi, A., Branny, P., and Pernodet, J.L. 1997. Antibiotic resistance gene cassettes derived from the Ω interposon for use in E. coli and Streptomyces. Gene 190: 315-317. [DOI] [PubMed] [Google Scholar]
- Brinkman, F.S., Macfarlane, E.L., Warrener, P., and Hancock, R.E. 2001. Evolutionary relationships among virulence-associated histidine kinases. Infect. Immun. 69: 5207-5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh, L.V., Fernandez-Moreno, M.A., Herrema, J.K., Malpartida, F., Hopwood, D.A., and Dijkhuizen, L. 1996. Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J. Bacteriol. 178: 2238-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, Y.H., Lee, E.J., Ahn, B.E., and Roe, J.H. 2001. SigB, an RNA polymerase σ factor required for osmoprotection and proper differentiation of Streptomyces coelicolor. Mol. Microbiol. 42: 205-214. [DOI] [PubMed] [Google Scholar]
- Gehring, A.M., Nodwell, J.R., Beverley, S.M., and Losick R. 2000. Genomewide insertional mutagenesis in Streptomyces coelicolor reveals additional genes involved in morphological differentiation. Proc. Natl. Acad. Sci. 97: 9642-9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryshin, I.Y. and Reznikoff, W.S. 1998. Tn5 in vitro transposition. J. Biol. Chem. 273: 7367-7374. [DOI] [PubMed] [Google Scholar]
- Goryshin, I.Y., Jendrisak, J., Hoffman, L.M., Meis, R., and Reznikoff, W.S. 2000. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat. Biotechnol. 18: 97-100. [DOI] [PubMed] [Google Scholar]
- Granozzi, C., Billetta, R., Passantino, R., Sollazo, M., and Puglia, A.M. 1990. A breakdown in macromolecular synthesis preceding differentiation in Streptomyces coelicolor A3(2). J. Gen. Microbiol. 136: 713-716. [DOI] [PubMed] [Google Scholar]
- Gust, B., Challis, G.L., Fowler, K., Kieser, T., and Chater, K.F. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. 100: 1541-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron, P.R., Evans, M.C., and Dyson, P.J. 1999. Low target site specificity of an IS6100-based mini-transposon, Tn1792, developed for transposon mutagenesis of antibiotic-producing Streptomyces. FEMS Microbiol. Lett. 171: 215-221. [DOI] [PubMed] [Google Scholar]
- Hoch, J.A. 2002. Control of signal transduction in the sporulation phosphorelay. In Signals, switches, regulons and cascades: Control of bacterial gene expression (eds. D.A. Hodgson and C.M. Thomas), pp. 199-212. Cambridge University Press, Cambridge, UK.
- Horinouchi, S. 1996. Streptomyces genes involved in aerial mycelium formation. FEMS Microbiol. Lett. 141: 1-9. [DOI] [PubMed] [Google Scholar]
- Inomata, K., Nishikawa, M., and Yoshida, K. 1994. The yeast Saccharomyces kluveryii as a recipient eukaryote in transkingdom conjugation: Behavior of transmitted plasmids in transconjugants. J. Bacteriol. 176: 4770-4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka, H., Horinouchi, S., Kieser, H.M., Hopwood, D.A., and Beppu, T. 1992. A putative 2-component regulatory system involved in secondary metabolism in Streptomyces spp. J. Bacteriol. 174: 7585-7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser, T., Bibb, M.J., Buttner, M.J., Chater, K.F., and Hopwood, D.A. 2000. Practical Streptomyces Genetics. John Innes Foundation, Norwich, UK.
- MacNeil, D.J. 1988. Characterization of a unique methyl-specific restriction system in Streptomyces avermitilis. J. Bacteriol. 170: 5607-5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, V.J. and Mohn, W.W. 1999. An alternative inverse PCR (IPCR) method to amplify DNA sequences flanking Tn5 insertions. J. Microbiol. Meth. 35: 163-166. [DOI] [PubMed] [Google Scholar]
- Nodwell, J.R., McGovern, K., and Losick, R. 1996. An oligopeptide permease responsible for the import of an extracellular signal governing aerial mycelium formation in Streptomyces coelicolor. Mol. Microbiol. 22: 881-893. [DOI] [PubMed] [Google Scholar]
- Oh, S.H. and Chater, K.F. 1997. Denaturation of circular or linear DNA facilitates targeted integrative transformation of Streptomyces coelicolor A3(2): Possible relevance to other organisms. J. Bacteriol. 179: 122-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget, M.S., Leibovitz, E., and Buttner, M.J. 1999a. A putative two-component signal transduction system regulates σE, a σ factor required for normal cell wall integrity in Streptomyces coelicolor A3(2). Mol. Microbiol. 33: 97-107. [DOI] [PubMed] [Google Scholar]
- Paget, M.S.B., Chamberlin, L., Atrih, A., Foster, S.J., and Buttner, M.J. 1999b. Evidence that the extracytoplasmic function σ factor σ(E) is required for normal cell wall structure in Streptomyces coelicolor A3(2). J. Bacteriol. 181: 204-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redenbach, M., Kieser, H.M., Denapaite, D., Eichner, A., Cullum, J., Kinashi, H., and Hopwood, D.A. 1996. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol. Microbiol. 21: 77-96. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Smokvina, T., Mazodier, P., Boccard, F., Thompson, C.J., and Guerineau, M. 1990. Construction of a series of pSAM2-based integrative vectors for use in actinomycetes. Gene 94: 53-59. [DOI] [PubMed] [Google Scholar]
- Strauch, E., Takano, E., Baylis, H.A., and Bibb, M.J. 1991. The stringent response in Streptomyces coelicolor A3(2). Mol. Microbiol. 5: 289-298. [DOI] [PubMed] [Google Scholar]
- Strohl, W.R. 1992. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 20: 961-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J., Kelemen, G.H., Fernández-Abalos, J.M., and Bibb, M.J. 1999. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145: 2221-2227. [DOI] [PubMed] [Google Scholar]
- Susstrunk, U., Pidoux, J., Taubert, S., Ullmann, A., and Thompson, C.J. 1998. Pleiotropic effects of cAMP on germination, antibiotic biosynthesis and morphological development in Streptomyces coelicolor. Mol. Microbiol. 30: 33-46. [DOI] [PubMed] [Google Scholar]
- Viollier, P.H., Kelemen, G.H., Dale, G.E., Nguyen, K.T., Buttner, M.J., and Thompson, C.J. 2003. Specialized osmotic stress response systems involve multiple SigB-like σ factors in Streptomyces coelicolor. Mol. Microbiol. 47: 699-714. [DOI] [PubMed] [Google Scholar]
- Vohradsky, J., Li, X.M., Dale, G., Folcher, M., Nguyen, L., Viollier, P.H., and Thompson, C.J. 2000. Developmental control of stress stimulons in Streptomyces coelicolor revealed by statistical analyses of global gene expression patterns. J. Bacteriol. 182: 4979-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff, J.N. and Altenbuchner, J. 1997. High frequency transposition of the Tn5 derivative Tn5493 in Streptomyces lividans. Gene 194: 81-86. [DOI] [PubMed] [Google Scholar]
- Waters, V. 2001. Conjugation between bacterial and mammalian cells. Nat. Genet. 29: 375-376. [DOI] [PubMed] [Google Scholar]
- West, A.H. and Stock, A.M. 2001. Histidine kinases and response regulator proteins in two-component signalling systems. Trends Biochem. Sci. 26: 369-376. [DOI] [PubMed] [Google Scholar]
- Yanisch-Perron, C., Vieira, J., and Messing, J. 1985. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103-119. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.sanger.ac.uk/Projects/S_coelicolor/; S. coelicolor genome sequence database.
- http://jiio16.jic.bbsrc.ac.uk/S.coelicolor/; S. coelicolor genome sequence and mutagenesis database including cosmid sequences.