

Abstract
Leukocyte adhesion deficiency Type I (LAD-I)–associated periodontitis is an aggressive form of inflammatory bone loss that has been historically attributed to lack of neutrophil surveillance of the periodontal infection. However, this form of periodontitis has proven unresponsive to antibiotics and/or mechanical removal of the tooth-associated biofilm. Recent studies in LAD-I patients and relevant animal models have shown that the fundamental cause of LAD-I periodontitis involves dysregulation of a granulopoietic cytokine cascade. This cascade includes interleukin IL-23 (IL-23) and IL-17 that drive inflammatory bone loss in LAD-I patients and animal models and, moreover, foster a nutritionally favorable environment for bacterial growth and development of a compositionally unique microbiome. Although the lack of neutrophil surveillance in the periodontal pockets might be expected to lead to uncontrolled bacterial invasion of the underlying connective tissue, microbiological analyses of gingival biopsies from LAD-I patients did not reveal tissue-invasive infection. However, bacterial lipopolysaccharide was shown to translocate into the lesions of LAD-I periodontitis. It is concluded that the bacteria serve as initial triggers for local immunopathology through translocation of bacterial products into the underlying tissues where they unleash the dysregulated IL-23–IL-17 axis. Subsequently, the IL-23/IL-17 inflammatory response sustains and shapes a unique local microbiome which, in turn, can further exacerbate inflammation and bone loss in the susceptible host.
1. Introduction
Periodontitis is an inflammatory disease that typically affects adults [1]. The disease causes destruction of the periodontium (i.e., tooth-supporting tissues such as gingiva and alveolar bone) and constitutes a potential risk factor for certain systemic diseases [2-4]. However, individuals with disorders affecting neutrophil recruitment to the periodontium, such as the rare condition leukocyte adhesion deficiency (LAD), rapidly develop severe periodontitis early in life affecting both the primary and permanent dentition [5-11] (Fig. 1). In addition to severe periodontal bone loss (Fig. 1A) [11], LAD-I patients display neutrophilia (increased blood neutrophil counts) and are susceptible to persistent infections (e.g., pneumonia)[6-9, 12, 13]. Rare monogenic diseases represent an important medical and social issue in its own right, cumulatively affecting 25 million patients in North America alone [14]. Importantly, however, the study of rare diseases, such as LAD-I, is not only relevant to the treatment of patients with these specific disorders; these diseases constitute real-life models to understand human biology and (patho)physiological mechanisms, thereby providing critical insights into common diseases [15-18].
Fig. 1. Clinical and histological profile of LAD-I periodontitis.
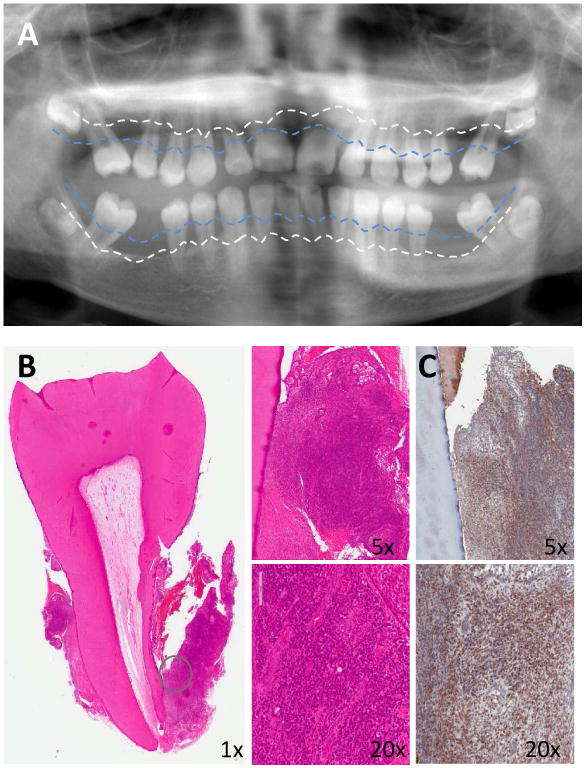
(A) Panoramic radiograph of 11-year-old LAD-I patient with severe bone loss. Blue dotted line represents physiologic bone levels and white dotted line demonstrates current bone levels. (B) H&E staining of extracted tooth and surrounding soft tissues. Encircled soft tissue reveals dense inflammatory infiltrate in the lesion (shown in lower and higher magnification, 5×-20×). (C) Immunohistochemistry for IL-17 in LAD-I tissues. Brown staining indicates IL-17-positive cells (original magnification 5×-20×). Patients were enrolled in an IRB approved protocol and had signed informed consent.
LAD represents a group of distinct inherited disorders, which inhibit the normal extravasation of neutrophils and their recruitment to sites of infection or inflammation [6, 8, 10, 11, 19, 20]. LAD patients have defects in the expression or function of the leukocyte-restricted β2 integrins (heterodimeric molecules, each with a distinct CD11 subunit and a common CD18 subunit), or other adhesion molecules. Consequently, their circulating neutrophils cannot adhere to vascular endothelial cells, a function that is required for extravasation [21-23]. LAD type I (LAD-I) is caused by deficiency in β2 integrins, LAD-II is due to defective glycosylation of selectin ligands, and LAD-III involves dysfunction of signaling intermediates affecting integrin activation [13].
The most common type of LAD is LAD-I, an autosomal recessive immunodeficiency caused by mutations in the CD18-encoding ITGB2 gene; therefore, LAD-I patients have defective expression in all β2 integrins [8, 11, 12, 20]. The LFA-1 integrin (CD11a/CD18) plays a crucial role in firm adhesion by interacting with endothelial cell counter-receptors (e.g., intercellular adhesion molecule-1) and is thus required for extravasation of the neutrophils to peripheral tissues [21-23]. In contrast to neutrophils, other types of leukocytes use different or additional adhesion molecules (e.g., VLA-4; very late antigen-4) for firm adhesion and extravasation [24-29]. Consistent with this, the heavy inflammatory infiltrate (Fig. 1B) in the periodontium of LAD-I patients is specifically devoid of neutrophils (which are confined in vessels), whereas lymphocytes and other cells of hematopoietic origin are found in abundance in the periodontium [11].
LAD-I–associated periodontitis (hereafter “LAD-I periodontitis”) has been historically attributed to lack of neutrophil surveillance of the periodontal infection; yet, this form of periodontitis has proven unresponsive to antibiotics and/or mechanical removal of the tooth-associated biofilm [5-7, 11, 19, 30-32]. A recent study in LAD-I patients and relevant animal models has shown that the fundamental cause of LAD-I periodontitis involves dysregulated overproduction of interleukin (IL)-23 and hence IL-17 (Fig. 1C) [11], a pro-inflammatory and pro-osteoclastogenic cytokine implicated in inflammatory bone loss in humans and animal models of arthritis or periodontitis [33-35]. The dysregulation of the IL-23/IL-17 response is consistent with the disruption of a major neutrophil homeostatic mechanism, known as the ‘neutrostat’. This mechanism senses neutrophil recruitment and clearance in peripheral tissues and regulates neutrophil production through a granulopoietic cytokine cascade involving IL-23, IL-17, and granulocyte colony-stimulating factor (G-CSF) [36]. When neutrophils cannot transmigrate to peripheral tissues, as in LAD-I, the neutrostat breaks down leading to unrestrained expression of IL-23 and downstream cytokines including IL-17 and G-CSF. Whereas the upregulation of G-CSF explains the increased granulopoiesis and blood neutrophilia in LAD-I patients, the local overproduction of IL-17 in the periodontium drives inflammation and bone loss [11]. This study [11] conferred clinical relevance to the neutrostat concept established earlier in mice [36] but also provided for the first time a human (and animal) disease correlate of this mechanism. These recent developments beg the question as to whether there is still a role for the bacteria in the pathogenesis of LAD-I periodontitis. This review aims to clarify the precise involvement of the LAD-I-associated periodontal microbiota in the pathogenesis of this aggressive form of periodontitis.
2. LAD-I periodontitis does not involve a tissue-invasive infection
Although the lack of neutrophil surveillance in the periodontal pockets might be expected to lead to uncontrolled bacterial infection and invasion of the underlying connective tissue, microbiological analyses of gingival tissue samples from LAD-I patients did not reveal any unusual tissue-invasive infection within the lesion driving tissue destruction [11]. Specifically, gram staining of extracted teeth and surrounding tissues from LAD-I patients showed microbial colonization of tooth surfaces but not of underlying diseased gingival tissue. Furthermore, the use of real-time PCR of the 16S rRNA gene to quantify bacterial load in gingival tissue sections from LAD-I patients and healthy controls indicated that the bacterial burden within the tissue of LAD-I patients was comparable to that of healthy controls [11]. In stark contrast to healthy tissue, the diseased gingival tissue harbored immunopathological lesions typified by a dense inflammatory infiltrate comprising primarily lymphocytes, including IL-17-expressing subsets (Fig. 1B-C) [11]. This feature was shared by LFA-1–deficient mice, a model mimicking LAD-I.
Both LAD-I patients and LFA-1–deficient mice, nevertheless, display higher tooth-associated bacterial load than their respective healthy controls [11]. Intriguingly, however, antibody-mediated neutralization of IL-17 or of IL-23 in LFA-1–deficient mice not only inhibited inflammatory bone loss but also diminished the total bacterial load to normal levels (similar to those of wild-type healthy mice) [11]. Therefore, the high bacterial load is possibly driven by IL-23/IL-17–dependent inflammation rather than by lack of neutrophil surveillance of the periodontal infection. In this regard, inflammation generates tissue breakdown products (e.g., degraded collagen peptides and heme-containing compounds) which foster the growth and sustenance of periodontitis-associated bacteria [35, 37-40]. Briefly stated, the interventional control of inflammation in LAD-I can suppress the local microbiota, despite the absence of the presumed protective effects of neutrophils in the periodontium. In this context, it is of interest to note that individuals with chronic granulomatous disease (CGD) do not have increased susceptibility to periodontitis (as compared to the general population), even though the defective oxygen-dependent bactericidal activity of their neutrophils renders them susceptible to frequent infections, such as pneumonia and abscesses of the skin [5, 41]. Arguably, therefore, defective immune surveillance by neutrophils is not an overriding factor in susceptibility to periodontitis, whereas the extravasation competence of neutrophils is essential for periodontal tissue homeostasis. The observation that neutrophils migrate normally to the periodontal tissue even in the absence of bacterial colonization as in germ-free mice [42] is consistent with the notion that neutrophil recruitment mediates homeostatic functions that are not necessarily related to infection control. For instance, a recently identified subset of human mature neutrophils was shown to inhibit T cell activation by delivering H2O2 into the immunological synapse in a β2 integrin–dependent manner [43].
3. Bacteria are required to unleash the disinhibited IL-23–IL-17 axis
The above facts and discussion should not be interpreted to mean that the tooth-associated microbiota is not involved in the pathogenic process leading to of LAD-I periodontitis. It was recently shown that subgingival plaque bacteria associated with LAD-I periodontitis readily stimulate IL-23 expression in human macrophages and also in vivo in murine oral tissues [44]. These findings suggest that the bacteria and their products such as lipopolysaccharide (LPS) likely act as stimuli for IL-23 induction by macrophages in the periodontium, thereby unleashing the disinhibited IL-23–IL-17 axis (Fig. 2). In this regard, the bacteria in the LAD-I microbiome (Fig. 3, A and B) do not have to invade the periodontal tissue to stimulate inflammatory cells, as their released bacterial products (e.g., LPS) can readily penetrate through the highly porous gingival junctional epithelium [45]. Indeed, bacterial LPS was shown to translocate into the lesions of LAD-periodontitis (Fig. 3C). Therefore, periodontitis is a manifestation of LAD-I due, in part, to its particular mucosal environment, where the presence of IL-23–inducing bacteria [44, 46] can act as the initial stimulus to unleash the dysregulated IL-23–IL-17 axis (Fig. 2). This notion is substantiated by findings of increased IL-23 and IL-17 production in both LAD-I patients and mouse models [11]. In contrast, once teeth are removed or lost in LAD patients, bone loss stops as the bone is no longer exposed to bacterial stimuli. Such stimuli are normally absent from the skeleton bones, which are thus not likely to be affected. On the other hand, the demonstration that defective neutrophil recruitment can lead to IL-17–dependent immunopathology may shed light on the understanding of other mucosal diseases associated with LAD-I, such as colitis [47].
Fig. 2. Dysregulated overproduction of IL-17 in LAD-I causes periodontal bone loss and increased microbial burden.
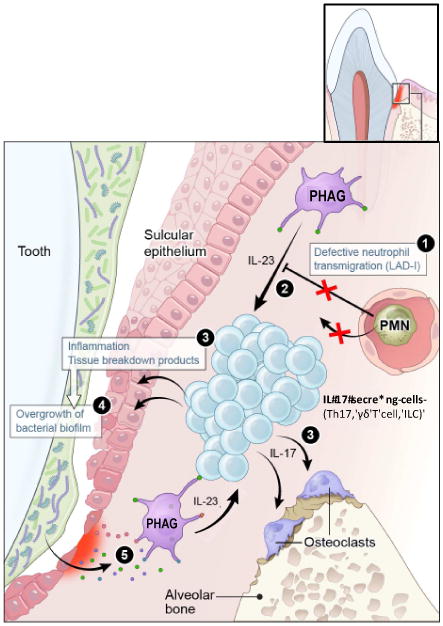
In normal individuals (i.e., with transmigration-competent neutrophils), recruited neutrophils regulate IL-23 production by tissue phagocytes (“PHAG”; e.g., macrophages) and hence the expression of IL-17 by adaptive and innate immune cells (e.g., Th17, γδ T cells, and innate lymphoid cells [ILC]) [11]. In contrast, LAD-I, which impairs neutrophil transmigration (1) leads to dysregulation of IL-23 (2) and hence overproduction of the inflammatory and bone-resorptive cytokine IL-17 (3). Inflammatory tissue breakdown products serve as nutrients for the local microbiome, thereby contributing to its overgrowth (4). Microbial products translocated into the lesions (for instance, LPS [44]) persistently stimulate the disinhibited IL-23–IL-17 axis (5) amplifying the destructive response. From ref. [18]. Used by permission.
Fig. 3. Subgingival microbiome in LAD-I.
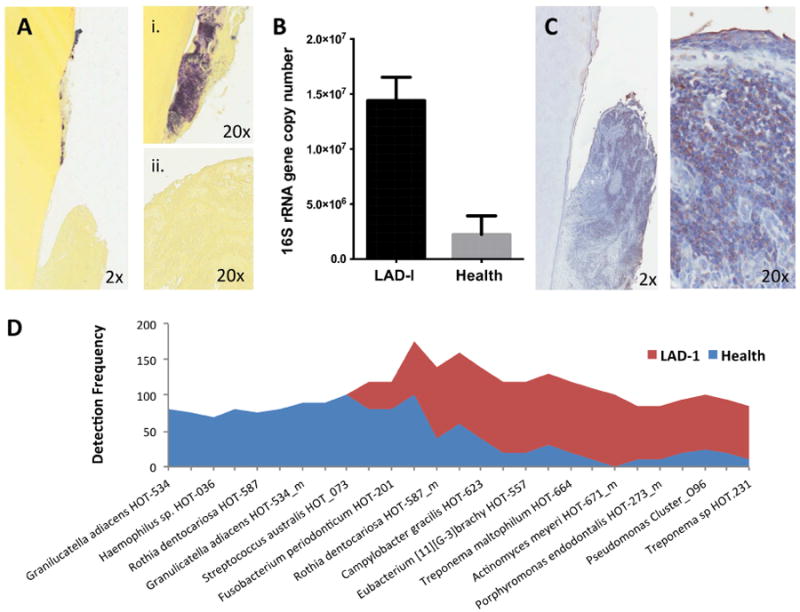
(A) Gram staining (Brown and Brenn) of extracted tooth (i) and adjacent soft tissues (ii) (2×-20× original magnification). (B) Total bacterial load (quantified with real-time PCR for 16S rRNA) for LAD-I and health. Bacterial load values are expressed as log (10) of 16S rRNA gene copy number (mean+SEM shown, p<0.05) [44]. (C) Immunohistochemistry for bacterial lipopolysaccharide (LPS) on extracted LAD tooth and surrounding tissues (2×-20× original magnification). (D) Highly prevalent taxa (species) in health and LAD-I. Detection frequency shown per group. From ref. [44]. Original, not previously published images were used in this review.
4. Characterization of the LAD-I periodontitis-associated subgingival microbiome
The subgingival microbiome of LAD-I patients has been recently characterized using a 16s rRNA gene-based microarray (HOMIM) which simultaneously detects more than 300 of the most prevalent oral bacterial species. Results of these analyses reveal that the tooth-associated microbial communities in LAD-I are distinct from those associated with health or aggressive periodontitis in the general population [44]. Unique characteristics of the LAD-I microbiome are its increased biomass (bacterial load) and the reduced number of species detected. An increase in microbial load has also been detected in the subgingival microbiome of chronic periodontitis patients, particularly at sites that are inflamed and bleeding [48]. However, chronic and aggressive forms or periodontitis in the absence of immune deficiency are characterized by increased diversity and richness in the subgingival biofilm and a significant increase in species detected within the periodontal microbial communities compared to those of health [48, 49]. Intriguingly, the LAD-I microbiome displays a decreased diversity of species detected even when compared to health and is dominated by a complete depletion of a large number of bacterial species associated with health. Classic health species from several genera such as Actinomyces, Rothia, Granulicatella and Streptococci [48] were undetectable in LAD-I (Fig. 3D).
LAD communities also do not resemble communities in chronic or aggressive periodontitis. The classical periodontitis-associated species Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia [49] were not detected at high levels in LAD-I compared to health [44]. Aggregatibacter actinomycetemcomitans, an organism associated with aggressive periodontitis [50], was also not detected in LAD-I periodontitis. Nevertheless, a number of species associated with chronic periodontitis such as Parvimonas micra HOT-111, Porphyromonas endodontalis HOT-273_m, Treponema maltophilum HOT-664, Treponema sp. HOT-257, Eubacterium [11][G-3] brachy HOT-557 and Bacteroidales [G-2] sp. HOT-274 [48], were detected at high levels in LAD-I (Fig. 3D).
Unique species detected in LAD-I included Pseudomonas aeruginosa, a bacterium not typically harbored in subgingival plaque but associated with severe infections in immunocompromised hosts including in LAD patients [51]. Leptotrichia buccalis as well as multiple other Leptotrichia spp. were also uniquely detected in LAD-I. While these bacteria are considered commensals, L. buccalis has been linked to bacteremia and severe illness in neutropenic patients [52]. Finally, Scardovia wiggsiae, a bacterium recently associated with severe childhood caries [53], was also detected in LAD-I but in the absence of caries in this population.
This unique composition of the LAD-I microbiome can conceivably be attributed to the severely dysregulated inflammation associated with LAD-I, although contributing effects by the antibiotics given to these patients cannot be ruled out. Nevertheless, it is important to point out that despite sporadic or consistent use of antibiotics in the LAD cohort, the biomass of the LAD microbiome remained very high (Fig. 3B) [44], suggesting a level of resistance to antibiotics in this complex biofilm. Moreover, it is important to note that the LAD-I subingival microbiome represents a continuous trigger for local immunopathology and periodontal destruction despite exposure to antibiotics.
5. Conclusion
Recent developments in the study of LAD-I periodontitis indicate that neutrophils are implicated in periodontal tissue destruction due to their absence, rather than through the usual bystander injury dogma that applies to many other neutrophil-associated inflammatory diseases including the chronic form of periodontitis [41, 54-58]. The implication of the dysregulated IL-23 and IL-17 response in the pathogenesis of LAD-I periodontitis can lead to innovative and effective host-modulation approaches that can revolutionize the periodontal treatment of affected individuals. The inhibition of the inflammatory IL-23/IL-17 response is likely to control the local microbiome as suggested by animal experiments showing that bacterial growth requires an active inflammatory response. These developments, however, do not rule out a role for the bacteria in the pathogenic process. Indeed, the subgingival communities of LAD-I appear to serve as initial triggers for local immunopathology through translocation of bacterial products into the underlying gingival tissue where they unleash the disinhibited IL-23–IL-17 axis.
Highlights (YMPAT-D-15-00150).
Leukocyte adhesion deficiency Type I (LAD-I) causes severe early-age periodontitis
LAD-I periodontitis is caused by dysregulation of the IL-23/IL-17 response
The host inflammatory response sustains and shapes a unique local microbiome
LAD-I periodontitis does not involve tissue-invasive infection
Translocated bacterial products may unleash the dysregulated IL-23–IL-17 axis
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, NIDCR (N.M.M.) and by U.S. Public Health Service grants DE015254, DE021685, DE024716, and AI068730 from NIDCR and NIAID (G.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012;91:914–20. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 2.Kebschull M, Demmer RT, Papapanou PN. “Gum bug leave my heart alone”: Epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010;89:879–902. doi: 10.1177/0022034510375281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–20. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 4.Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44. doi: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussbaum G, Shapira L. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol. 2011;38:49–59. doi: 10.1111/j.1600-051X.2010.01678.x. [DOI] [PubMed] [Google Scholar]
- 6.Waldrop TC, Anderson DC, Hallmon WW, Schmalstieg FC, Jacobs RL. Periodontal manifestations of the heritable Mac-1, LFA-1, deficiency syndrome. Clinical, histopathologic and molecular characteristics. J Periodontol. 1987;58:400–16. doi: 10.1902/jop.1987.58.6.400. [DOI] [PubMed] [Google Scholar]
- 7.Deas DE, Mackey SA, McDonnell HT. Systemic disease and periodontitis: manifestations of neutrophil dysfunction. Periodontol 2000. 2003;32:82–104. doi: 10.1046/j.0906-6713.2003.03207.x. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt S, Moser M, Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol. 2012;55:49–58. doi: 10.1016/j.molimm.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 9.van de Vijver E, van den Berg TK, Kuijpers TW. Leukocyte adhesion deficiencies. Hematol Oncol Clin North Am. 2013;27:101–16. doi: 10.1016/j.hoc.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 2014;93:231–7. doi: 10.1177/0022034513507956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17– driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra40. doi: 10.1126/scitranslmed.3007696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–94. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 13.Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. 2012;1250:50–5. doi: 10.1111/j.1749-6632.2011.06389.x. [DOI] [PubMed] [Google Scholar]
- 14.Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371:2039–41. doi: 10.1016/S0140-6736(08)60872-7. [DOI] [PubMed] [Google Scholar]
- 15.Dodge JA, Chigladze T, Donadieu J, Grossman Z, Ramos F, Serlicorni A, et al. The importance of rare diseases: from the gene to society. Arch Dis Child. 2011;96:791–2. doi: 10.1136/adc.2010.193664. [DOI] [PubMed] [Google Scholar]
- 16.Peltonen L, Perola M, Naukkarinen J, Palotie A. Lessons from studying monogenic disease for common disease. Hum Mol Genet. 2006;15 Spec No 1:R67–74. doi: 10.1093/hmg/ddl060. [DOI] [PubMed] [Google Scholar]
- 17.Fischer A. Human primary immunodeficiency diseases. Immunity. 2007;27:835–45. doi: 10.1016/j.immuni.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Moutsopoulos NM, Lionakis MS, Hajishengallis G. Inborn errors in immunity: unique natural models to dissect oral immunity. J Dent Res. 2015;94:753–8. doi: 10.1177/0022034515583533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etzioni A, Tonetti M. Leukocyte adhesion deficiency II-from A to almost Z. Immunol Rev. 2000;178:138–47. doi: 10.1034/j.1600-065x.2000.17805.x. [DOI] [PubMed] [Google Scholar]
- 20.Bunting M, Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving beta 2 integrins and selectin ligands. Curr Opin Hematol. 2002;9:30–5. doi: 10.1097/00062752-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 22.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–90. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013;34:1–6. doi: 10.1016/j.it.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meerschaert J, Furie MB. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J Immunol. 1995;154:4099–112. [PubMed] [Google Scholar]
- 25.Kuchroo VK, Martin CA, Greer JM, Ju ST, Sobel RA, Dorf ME. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–82. [PubMed] [Google Scholar]
- 26.Hyun YM, Chung HL, McGrath JL, Waugh RE, Kim M. Activated integrin VLA-4 localizes to the lamellipodia and mediates T cell migration on VCAM-1. J Immunol. 2009;183:359–69. doi: 10.4049/jimmunol.0803388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luscinskas FW, Ding H, Tan P, Cumming D, Tedder TF, Gerritsen ME. L- and P-selectins, but not CD49d (VLA-4) integrins, mediate monocyte initial attachment to TNF-alpha-activated vascular endothelium under flow in vitro. J Immunol. 1996;157:326–35. [PubMed] [Google Scholar]
- 28.Yusuf-Makagiansar H, Anderson ME, Yakovleva TV, Murray JS, Siahaan TJ. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev. 2002;22:146–67. doi: 10.1002/med.10001. [DOI] [PubMed] [Google Scholar]
- 29.Siegelman MH, Stanescu D, Estess P. The CD44-initiated pathway of T-cell extravasation uses VLA-4 but not LFA-1 for firm adhesion. J Clin Invest. 2000;105:683–91. doi: 10.1172/JCI8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinane DF, Hart TC. Genes and gene polymorphisms associated with periodontal disease. Crit Rev Oral Biol Med. 2003;14:430–49. doi: 10.1177/154411130301400605. [DOI] [PubMed] [Google Scholar]
- 31.Dababneh R, Al-Wahadneh AM, Hamadneh S, Khouri A, Bissada NF. Periodontal manifestation of leukocyte adhesion deficiency type I. J Periodontol. 2008;79:764–8. doi: 10.1902/jop.2008.070323. [DOI] [PubMed] [Google Scholar]
- 32.Sollecito TP, Sullivan KE, Pinto A, Stewart J, Korostoff J. Systemic conditions associated with periodontitis in childhood and adolescence. A review of diagnostic possibilities. Med Oral Patol Oral Cir Bucal. 2005;10:142–50. [PubMed] [Google Scholar]
- 33.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–53. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 35.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–73. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–94. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–9. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 38.Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. 2014;29:248–57. doi: 10.1111/omi.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abe T, Shin J, Hosur K, Udey MC, Chavakis T, Hajishengallis G. Regulation of osteoclast homeostasis and inflammatory bone loss by MFG-E8. J Immunol. 2014;193:1383–91. doi: 10.4049/jimmunol.1400970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–94. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- 41.Sima C, Glogauer M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: Current state of knowledge. Curr Oral Health Rep. 2014;1:95–103. [Google Scholar]
- 42.Zenobia C, Luo XL, Hashim A, Abe T, Jin L, Chang Y, et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. 2013;15:1419–26. doi: 10.1111/cmi.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122:327–36. doi: 10.1172/JCI57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moutsopoulos NM, Chalmers NI, Barb JJ, Abusleme L, Greenwell-Wild T, Dutzan N, et al. Subgingival microbial communities in leukocyte adhesion deficiency and their relationship with local immunopathology. PLoS Pathog. 2015;11:e1004698. doi: 10.1371/journal.ppat.1004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosshardt DD, Lang NP. The junctional epithelium: from health to disease. J Dent Res. 2005;84:9–20. doi: 10.1177/154405910508400102. [DOI] [PubMed] [Google Scholar]
- 46.Allam JP, Duan Y, Heinemann F, Winter J, Gotz W, Deschner J, et al. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol. 2011;38:879–86. doi: 10.1111/j.1600-051X.2011.01752.x. [DOI] [PubMed] [Google Scholar]
- 47.Uzel G, Kleiner DE, Kuhns DB, Holland SM. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology. 2001;121:958–64. doi: 10.1053/gast.2001.28022. [DOI] [PubMed] [Google Scholar]
- 48.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7:1016–25. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000. 2005;38:135–87. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- 50.Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol. 2007;45:3859–69. doi: 10.1128/JCM.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerald L, Mandell JEB, Raphael Dolin. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. In: Gerald L, Mandell JEB, Raphael Dolin, editors. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia: Elsevier; 2009. [Google Scholar]
- 52.Reig M, Baquero F, Garcia-Campello M, Loza E. Leptotrichia buccalis bacteremia in neutropenic children. J Clin Microbiol. 1985;22:320–1. doi: 10.1128/jcm.22.2.320-321.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanner AC, Kent RL, Jr, Holgerson PL, Hughes CV, Loo CY, Kanasi E, et al. Microbiota of severe early childhood caries before and after therapy. J Dent Res. 2011;90:1298–305. doi: 10.1177/0022034511421201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood. 2014;124:710–9. doi: 10.1182/blood-2014-03-453217. [DOI] [PubMed] [Google Scholar]
- 55.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–75. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 56.Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–66. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 58.Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol. 2014 doi: 10.1189/jlb.3VMR1014-468R. [DOI] [PMC free article] [PubMed] [Google Scholar]