

Abstract
Identification of novel targets for the development of more effective antimalarial drugs and vaccines is a primary goal of the Plasmodium genome project. However, deciding which gene products are ideal drug/vaccine targets remains a difficult task. Currently, a systematic disruption of every single gene in Plasmodium is technically challenging. Hence, we have developed a computational approach to prioritize potential targets. A pathway/genome database (PGDB) integrates pathway information with information about the complete genome of an organism. We have constructed PlasmoCyc, a PGDB for Plasmodium falciparum 3D7, using its annotated genomic sequence. In addition to the annotations provided in the genome database, we add 956 additional annotations to proteins annotated as “hypothetical” using the GeneQuiz annotation system. We apply a novel computational algorithm to PlasmoCyc to identify 216 “chokepoint enzymes.” All three clinically validated drug targets are chokepoint enzymes. A total of 87.5% of proposed drug targets with biological evidence in the literature are chokepoint reactions. Therefore, identifying chokepoint enzymes represents one systematic way to identify potential metabolic drug targets.
Four species of the Plasmodium genus cause human malaria. Among these, Plasmodium falciparum inflicts the most mortality. Over 1 million children under the age of five die due to malaria each year (World Health Organization 1993). Global and local climate changes, the emergence of insecticide resistant mosquitoes, and a steadily rising number of malaria parasites resistant to currently available antimalarial drugs result in a growing malaria threat. Estimates suggest that 40% of the world's population is at risk of malaria (Brown and Reeder 2002). Despite initially promising results with multicomponent recombinant protein vaccines targeted against the asexual blood stages (Genton et al. 2003) and vaccines directed against the sporozoite stage (Bojang et al. 2001), effective immunization against the disease is not yet available. The P. falciparum Genome Sequencing Project was established to facilitate the development of new drugs and vaccines (Hoffman et al. 1997).
With the malaria genome essentially complete (Gardner et al. 1998; Bowman et al. 1999; Gardner et al. 2002a,b; Hall et al. 2002; Hyman et al. 2002), we can study the organism from a whole-genome standpoint. PlasmoDB (http://www.plasmodb.org) is the official database of the malaria parasite genome project and contains the finished genome for P. falciparum strain 3D7 and its official annotation as provided by the members of the genome sequencing consortium. In addition, PlasmoDB provides additional GO annotations, provided by manual assignment or sequence analysis. The GO2EC mapping is used to assign EC numbers on the basis of GO annotations. EC numbers are also assigned manually.
Understanding the cellular mechanisms and interactions between cellular components is instrumental to the development of new effective drugs and vaccines. Functional annotations of gene products allow the assembly of metabolic pathways that illustrate how proteins work in concert to produce cellular compounds or to transmit information. The Malaria Metabolic Pathways (http://sites.huji.ac.il/malaria) illustrates current knowledge of malarial metabolism in diagrammatic form. PlasmoDB contains information about 18 different plasmodial pathways and allows for querying of proteins by pathway. Several pathway databases exist that describe the interconnection of metabolites and enzymes within an organism such as KEGG, WIT, and MetaCyc (Kanehisa and Goto 2000; Overbeek et al. 2000; Karp et al. 2002b).
The Pathway Tools software environment has been used to construct PGDBs for numerous prokaryotic and eukaryotic organisms (http://biocyc.org; Karp et al. 2002a). The underlying formal ontology defines an array of different concepts such as genes, proteins, compounds, reactions, and pathways in a frame-based representation (Karp 2000). Due to the number of interactions between these biological entities in an organism, it is difficult to manually track all cellular processes. The representation allows us to specify which protein a gene encodes, what modified forms of particular proteins exist, and how subunits assemble to form protein complexes.
The frames (DB objects) that encode proteins and protein complexes are identified as enzymes by defining database relationships that link them with frames that encode biochemical reactions. Each reaction frame identifies the substrates and products of a specific chemical reaction. The association between a protein and a specific reaction is captured in an enzymatic-reaction frame, which allows us to specify inhibitors and cofactors for a specific enzyme's catalysis of a specific reaction (Fig. 1). Encoding these relationships in a computational data structure allows us to perform systematic analyses over the entire system, including complex queries and checks for data inconsistency within the pathway database.
Figure 1.
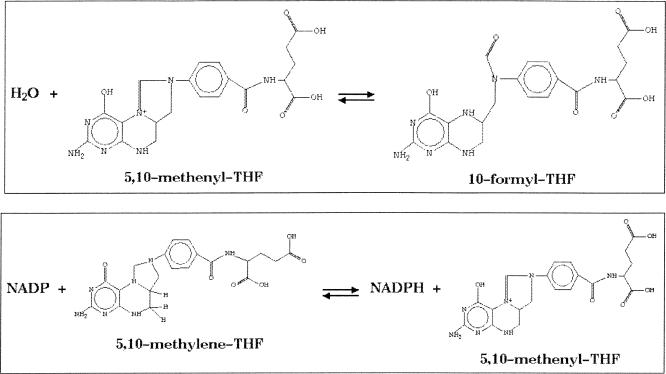
Both of these reactions are catalyzed by the same protein, MAL6P1.121. Therefore, MAL6P1.121 is associated with two distinct Enzymatic-Reaction frames, each linking the protein to one specific reaction. In this manner, inhibitors can be specified for a particular enzyme/reaction pair instead of for a reaction (which may be catalyzed by numerous enzymes with different inhibitor sensitivities) or for a protein (which may have multiple active sites).
RESULTS
The PlasmoCyc PGDB remains a work in progress and will change with additional curation and updates to the genome. Currently, it describes 5441 genes, of which 5366 code for polypeptides and 75 code for an RNA. Eight additional polypeptides are modified forms of gene products. An additional 21 protein complexes are formed for a total of 5395 distinct proteins.
A metabolic overview of our current understanding of P. falciparum is provided in Figure 2. There are currently 116 pathways, of which 23 were added manually using the Malaria Metabolic Pathways as a guide. These new pathways have Plasmodium in their names and are linked to their diagrams in the Malaria Metabolic Pathways resource.
Figure 2.
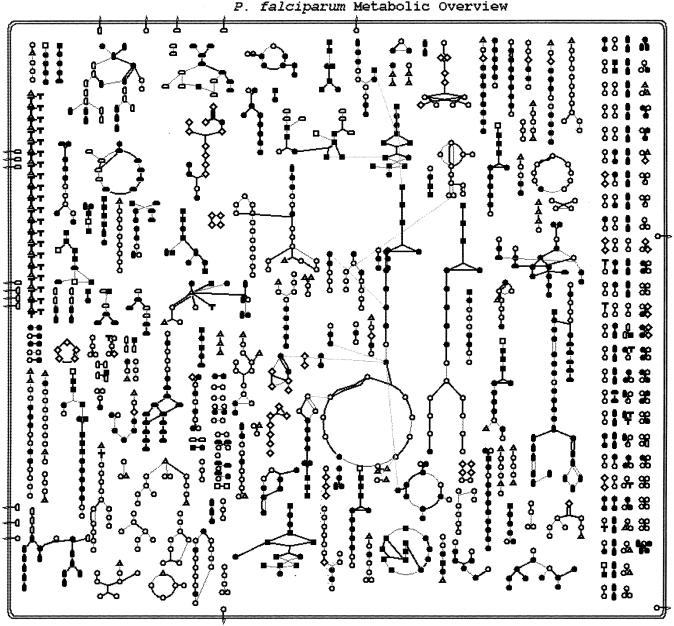
Overview of the metabolic map for P. falciparum. Each node represents a metabolite, with the type of metabolite indicated by the shape of the node as indicated by the legend. The lines connecting the metabolites represent reactions. Bold lines correspond to reactions with an identified enzyme; gray lines correspond to reactions without an identified enzyme.
A total of 697 distinct chemical reactions are present in PlasmoCyc, of which 416 are associated with an enzyme. During manual curation, we added 96 new reactions to PlasmoCyc. Sixty-eight of these reactions were associated with Enzyme Commission (EC) numbers. In many cases, new reactions associated with EC numbers were specific instances of abstract reactions or were alternative reactions for the EC number (e.g., a hexose kinase can catalyze several specific reactions). Many of the new reactions added to PlasmoCyc are GPI-anchoring reactions or ubiquitination reactions from newly added pathways. A large category of new reactions added to PlasmoCyc consists of reactions with important effects that cannot be captured by a simple chemical reaction, for example, dynein ATPase, which effects the movement of organelles along microtubules. Although PlasmoCyc can capture the chemical formulas of the reactions, there is not currently a method for describing the potentially critically important side effects in a computational framework.
Genomic evidence exists for 146 reactions that were not incorporated into any existing pathway from MetaCyc or the Malaria Metabolic Pathway resource. By computationally examining common substrates, we linked seven pairs of these orphan reactions together to form new pathways. A total of 559 reactions (80%) are assigned to metabolic pathways. Of these, 278 reactions have enzymes detected in the genome. The remaining 138 reactions in PlasmoCyc that are not in pathways all have enzymes detected in the genome.
By comparison, EcoCyc, a PGDB for Escherichia coli K-12 has 4421 genes that code for 4468 polypeptides (Karp et al. 2000). EcoCyc has 1072 reactions that are catalyzed by an enzyme in the genome, and of these reactions, 666 (62%) are assigned to pathways. Only 84 reactions, which are contained in pathways, do not have an identified enzyme. There are an additional 406 reactions that are catalyzed, but do not belong to any pathway. Key statistics for PlasmoCyc and EcoCyc are presented in Table 1.
Table 1.
A Comparison of the Statistics of the P. falciparum PGDB With Those of E. coli
| Organism | Genes | Polypeptides | Protein complexes | Enzymes | Compounds | Reactions | Enzymatic reactions | Pathways |
|---|---|---|---|---|---|---|---|---|
| P. falciparum | 5441 | 5366 | 18 | 737 | 525 | 696 | 861 | 122 |
| E. coli | 4421 | 4468 | 629 | 975 | 873 | 3090 | 1210 | 172 |
There are 281 reactions that are in pathways, but for which no enzyme has been detected in the genome. This implies that either there are (1) enzymes in the genome that have not been identified, (2) enzymatic functions that have not been assigned to identified proteins, (3) parasite importation of the enzymatic activity from the host, or (4) an alternate pathway in the organism that does not involve the reaction. If these missing enzymes are produced by P. falciparum, they must reside in regions of the genome still unfinished, be annotated as noncoding regions, or be missing the functional annotation for this enzymatic activity. If they are not produced by P. falciparum, they or their products may be imported from the host, a variant pathway that does not use the reaction may exist, or the pathway may not exist at all. The Pathway Tools software automatically generates a list of missing reactions, available on the PlasmoCyc webpage (http://plasmocyc.stanford.edu).
Pathway Details
There are 525 small molecule compounds represented in PlasmoCyc. A total of 470 compounds act as substrates in reactions, whereas the remaining compounds either act as inhibitors or structural building blocks for other compounds. A total of 334 specific compounds are substrates of reactions that have evidence for catalysis (either direct experimental evidence or the presence of a predicted enzyme in the genome) in the parasite. We manually entered 59 compounds from the literature not previously in the Pathway Tools framework. Thirty-five of these compounds are substrates in reactions, whereas 24 are inhibitors of enzymatic-reactions.
There are 816 enzymatic-reaction instances, more than the number of enzymes, because many enzymes catalyze more than one reaction. The Pathway Tools software assigned a majority of enzymatic-reactions, 501, automatically, 41 were assigned by automated enzyme name match, and 460 by EC number. An additional 315 enzymatic-reaction assignments were made manually. The method by which the enzymatic reaction was assigned is stored in the Basis-for-Assignment slot. In addition to the annotation provided by the malaria sequencing consortium, we used the GeneQuiz system to predict the functions of all proteins automatically (Andrade et al. 1999). GeneQuiz uses a variety of sequence-analysis techniques to compare the query sequence with a large database of protein and nucleotide sequences, protein structures, and sequence motifs. An automated reasoning module uses the output of these sequence analyses to assign functions to the query protein with a numeric confidence measure categorized into clear, tentative, and marginal. When the official annotation is hypothetical protein or ambiguous as to its metabolic function, we use the GeneQuiz annotation when it is clear or tentative. GeneQuiz annotations are used as the primary annotation for 956 proteins, and as the basis of 109 of the enzymatic reactions. The annotation by which its function was assigned is documented in each protein's comment. Each polypeptide is linked to both its PlasmoDB (Kissinger et al. 2002; Bahl et al. 2003) and GeneQuiz annotations. Where we manually annotate the enzymatic function of a protein using a literature reference, we include a link to the PubMed abstract of the reference (Fig. 3).
Figure 3.

The description of the polypeptide from gene PF11_0338, annotated as aquaglyceroporin. The unification links contain links to an article abstract in PubMed, and protein annotations in GeneQuiz and PlasmoDB.
Identification of Potential Drug Targets
To identify potential drug targets, we performed a chokepoint analysis of the metabolic network of P. falciparum 3D7. We define a “chokepoint reaction” as a reaction that either uniquely consumes a specific substrate or uniquely produces a specific product in the PlasmoCyc metabolic network (Fig. 4). We expect the inhibition of an enzyme that consumes a unique substrate to result in the accumulation of the unique substrate (potentially toxic to the cell) and the inhibition of an enzyme that produces a unique product to result in the starvation of the unique product (potentially crippling essential cell functions). Thus, we believe that chokepoint enzymes may be essential to the parasite and are therefore potential drug targets. Chokepoint analyses are particularly straightforward to perform with a computational representation of metabolism, due to the constraints placed on the representation, and would be difficult to perform on a flat list of enzymatic reactions (due to synonyms for reactions and compounds). There are certain reactions that we excluded from our chokepoint analysis, namely proteolytic reactions (as we do not capture the specificity of these reactions), reactions that do not have clearly defined substrates (e.g., protein disulfide isomerase), and reactions with important side effects (many ATPases). Although these enzymes could be good drug targets, we did not expect the rationale of our method to apply in these cases. We identified 216 of 303 distinct enzymatic activities (71.3%) as catalyzing chokepoint reactions, assuming each enzyme has only one active site, unless annotated as multifunctional. If an enzyme catalyzes at least one chokepoint reaction, we classify it as a potential drug target.
Figure 4.
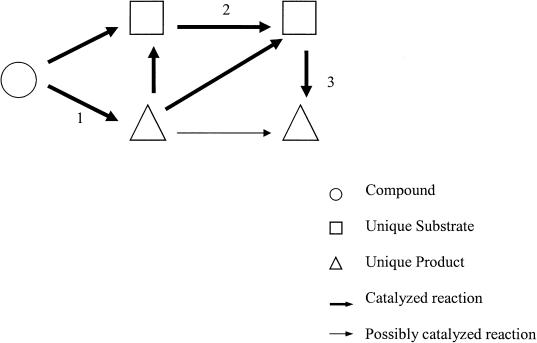
The thick arrows represent reactions that are catalyzed by enzymes, whereas the thin arrows represent reactions that are present, but with no evidence of the corresponding enzymes. When determining chokepoint reactions, we only consider the catalyzed reaction. (1) A chokepoint, because it produces a unique product; (2) a chokepoint, because it consumes a unique substrate; (3) a chokepoint, because it consumes a unique substrate and produces a unique product.
To assess the usefulness of identifying chokepoint enzymes for proposing drug targets, we compared chokepoints and nonchokepoints against proposed drug targets from the literature. We attempted a complete literature search for proposed malaria drug targets that were metabolic enzymes and met our criteria outlined above. We identified three targets of clinically proven drugs and 24 proposed drug targets with biological evidence (such as in vitro growth inhibition of the parasite with inhibition of the target). All targets of clinically proven malaria drugs, dihydrofolate reductase (Sixsmith et al. 1984), dihydropteroate synthase (Triglia and Cowman 1994), and 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Lell et al. 2003) are chokepoints in PlasmoCyc. Of the 24 proposed targets with biological evidence, 21 are chokepoints in PlasmoCyc (Table 2). A total of 87.5% of proposed drug targets with biological evidence are chokepoint reactions in PlasmoCyc. Of the chokepoint reactions, 24 (11.16%) were drug targets (validated or proposed with evidence) and of the nonchokepoint reactions, three (3.41%) were drug targets (proposed with evidence). The percentage of drug targets in chokepoints and nonchokepoints is statistically different at 95% confidence (P = 0.023, see Methods).
Table 2.
Drug Targets Proposed in the Literature With Biological Evidence, Such as In Vitro Growth Inhibition When Enzyme Inhibited
| Target enzyme with evidence | EC number | Reference | Enzymes | Chokepoint | In human |
|---|---|---|---|---|---|
| 3-oxoacyl-[acyl-carrier protein] synthase | 2.3.1.41 | Waller et al. 2003 | PFB0505c | Yes | Yes |
| Carbamoyl-phosphate synthase | 6.3.5.5 | Flores et al. 1997 | PF13_0044 | Yes | Yes |
| Choline transport | Ancelin et al. 1986 | PFL0620c | Yes | No | |
| Chorismate synthase | 4.2.3.5 | McRobert and McConkey 2002 | MAL6P1.199 | Yes | No |
| DNA-directed DNA polymerase | 2.7.7.7 | Barker Jr. et al. 1996 | 11 proteins | Yes | Yes |
| Enoyl-[acyl-carrier protein] reductase (NADH) | 1.3.1.9 | Surolia and Surolia 2001 | MAL6P1.275 | Yes | No |
| Farnesyl-diphosphate famesyltransferase | 2.5.1.21 | Chakrabarti et al. 2002 | PF11_0483 | Yes | Yes |
| Fructose-bisphosphate aldolase | 4.1.2.13 | Wanidworanun et al. 1999 | PF14_0425 | Yes | Yes |
| Gamma-glutamylcysteine synthetase | 6.3.2.2 | Meierjohann et al. 2002 | PFI0925w | Yes | Yes |
| Histone deacetylase | Darkin-Rattray et al. 1996 | PF10_0078 | Yes | Yes | |
| PF14_0690 | |||||
| PF11_1260c | |||||
| Hypoxanthine phosphoribosyltransferase | 2.4.2.8 | Dawson et al. 1993 | PF10_0121 | Yes | Yes |
| IMP dehydrogenase | 1.1.1.205 | Webster et al. 1982 | PFI1020c | No | Yes |
| Lactoylglutathione lyase | 4.4.1.5 | Thornalley et al. 1994 | PF11_0145 | Yes | Yes |
| MAL6P1.50 | |||||
| Lysophospholipase | 3.1.1.5 | Zidovetzki et al. 1994 | PF07_0005 | Yes | No |
| PF07_0040 | |||||
| PF14_0738 | |||||
| PF14_0737 | |||||
| PF14_0017 | |||||
| NADH dehydrogenase (ubiquinone) | 1.6.5.3 | Krungkrai et al. 2002 | PFC0505c | Yes | No |
| Purine nucleoside phosphorylase | 2.4.2.1 | Kiscka et al. 2002 | PFE0660c | Yes | No |
| Ribonucleoside Reductase | 1.17.4.1 | Chakrabarti et al. 1993 | PF10_0154 | Yes | Yes |
| PF14_0352 | |||||
| PF14_0053 | |||||
| RNA Polymerase | 2.7.7.6 | Lin et al. 2002 | 24 proteins | Yes | Yes |
| S-adenosyl-L-homocysteine hydrolase | 3.3.1.1 | Shuto et al. 2002 | PFE1050w | Yes | Yes |
| S-adenosylmethionine decarboxylase | 4.1.1.50 | Wright et al. 1991 | PF10_0322 | Yes | Yes |
| Sphingomyelinase | 3.1.4.12 | Hanada et al. 2002 | PFL1870c | Yes | No |
| Succinate dehydrogenase | 1.3.99.1 | Suraveratum et al. 2000 | PFL0630w | No | Yes |
| PF10_0334 | |||||
| Thioredoxin reductase (NADPH) | 1.8.1.9 | Krnajski et al. 2002 | PFI1170c | No | Yes |
| Thymidylate synthase | 2.1.1.45 | Jiang et al. 2000 | PFD0830w | Yes | Yes |
The reaction and its EC number are given as well as the Protein Identifiers for the corresponding enzymes (for enzymatic reactions catalyzed by more than 10 proteins, see http://plasmocyc.stanford.edu). The In Human column denotes whether or not the enzymatic activity has a similar enzyme in human as determined by BLAST alignment with an expectation of less than 0.0001. Of these 24 reactions, 21 (87.5%) were identified as chokepoints.
Due to the high percentage of enzymes identified as chokepoints, we examined additional criteria in addition to being a chokepoint enzyme for identifying potential drug targets. An enzyme not having isozymes would make it more likely to be a good drug target, as one enzyme should be easier to inhibit than a family of enzymes. We classified all enzymes into those with isozymes and those without. A total of 230 of 303 enzymes (75.9%) do not have isozymes. Of the clinically validated drug targets, none of the three have isozymes, and among biologically validated drug targets, 19 (79.1%) have exactly one enzyme. If isozymes are not predictive for good drug targets, we expect 20.6 enzymes with no isozymes to be among validated and proposed drug targets, and we get 21 not statistically different than expected, meaning that whether or not an enzyme has isozymes is not predictive. Among the chokepoints, 167 (76.9%) of the active sites have exactly one enzyme, whereas among the nonchokepoints, 63 (73.6%) have exactly one enzyme.
Another feature we would expect a good drug target to have is a lack of similarity to any human enzyme. We used BLAST to align enzymes in PlasmoCyc against the peptide database of the Ensembl human genome (see Methods). We used a significance threshold of e-value <0.075. A total of 38 of 303 enzymes (12.5%) have no significant similarity to a human enzyme. Of the chokepoints, 30 enzymes (13.9%) did not have a human homolog. Of the nonchokepoints, eight enzymes (9.2%) did not have a human homolog. Of the clinically validated drug targets, only dihydrofolate reductase has significant sequence similarity to a human protein, thymidylate synthase (the plasmodial protein is bifunctional). Of the biologically validated proposed targets, eight (29.6%) do not have significant sequence similarity to any known human peptide versus 30 of the remaining enzymes having no significant sequence similarity. Not having significant sequence similarity is predictive for drug targets with P = 0.009. A total of 30 chokepoint enzymes have no significant sequence similarity to any known human enzyme.
PlasmoCyc is available for browsing and querying at http://plasmocyc.stanford.edu/. Queries available include searching for specific genes, proteins, compounds, reactions, or pathways. The pathway evidence report, list of missing enzymes, list of drug targets from the literature, and list of chokepoint reactions are also available on the Web site, and lists the number of enzymes that catalyze each reaction, as well as its sequence similarity to human proteins.
DISCUSSION
Although PlasmoCyc continues to evolve, it can already support useful analyses. The genomic sequence of P. falciparum 3D7 is promising from the standpoint of drug development, because it provides an electronic catalog of parasitic proteins, but it requires a pathway organization to facilitate easy analysis. Unlike other current malarial pathway resources, PlasmoCyc links information about substrates, reactions, and pathways in a computationally accessible format. Therefore, we can link reactions together by substrates and evaluate the metabolic network as a whole instead of on a pathway-by-pathway basis.
Our initial experiments indicate that whole-genome metabolic analysis can assist in drug-target identification. By identifying chokepoint reactions, we are trying to identify enzymes that are essential to the parasite's survival. All clinically proven drug targets are chokepoints, and 83% of drug targets with biological evidence are chokepoints. There is also an enrichment of drug targets in chokepoints as compared with nonchokepoints. This leads to the conclusion that the classification of an enzyme as a chokepoint has some bearing on whether or not it would make a good drug target. In addition, we find that lack of sequence similarity has some predictive value for whether or not a plasmodial enzyme is a biologically validated drug target. We identify 216 chokepoint reactions, and 29 of these have no sequence similarity to human enzymes with expectation values <0.1.
Our chokepoint analysis is limited because the annotation of the malaria genome is incomplete. Thus, there may be places in our current network that appear to be chokepoints, but are not due to an enzyme not yet annotated. In addition, our analysis is limited, because we have not considered the capabilities of the parasite to transport an accumulating metabolite out of the cell or a limiting metabolite into the cell. One reason that chokepoints may not be essential could be that they create unique intermediates to an essential product that are not essential themselves because of another pathway to the essential product. Finally, there could be chokepoint reactions that are not essential due to other pathways that achieve the same metabolic goal within the organism, such that blocking the reaction has no deleterious effects on the parasite.
The effectiveness of our initial chokepoint algorithm for target prediction can be improved with refinement of the underlying metabolic network. The structure of the metabolic network will improve with further annotation of the metabolic functions in the parasite, as well as the incorporation of additional types of information, expression data for the different cell cycle stages, and cellular localization of specific enzymes. Currently, we can identify proteins that catalyze more than one reaction, but cannot determine whether or not the same active site on the protein catalyzes all of the reactions. With more structural information, we could add a specification for active sites to PlasmoCyc to specify which active sites catalyze which reactions. By including more complex information, we will be able to more accurately define which enzymatic functions are present at which times in the lifecycle of the parasite.
There are further aspects on which we can narrow down our list of potential drug targets. The drug should adversely affect the parasite but not the human host; therefore, if the drug target has a homologous enzyme in human, it should not be essential or have differential inhibition in human (perhaps due to different protein structure or different regulation). Potential drug targets should be expressed in the human stages of the parasite. Our provisional targets need to be examined further, both computationally and experimentally for these additional features.
Using a computational framework that incorporates structured data to represent metabolic pathways allows inference over many more entities in a PGDB than a human mind can comprehend at one time. PlasmoCyc incorporates additional metabolic information with annotated sequence information to model the metabolism of P. falciparum 3D7. The pathways in PlasmoCyc are accessible to both user browsing and computational algorithms. The pathways we manually added to PlasmoCyc can be added to the reference pathway database to facilitate pathway prediction in related organisms and rapid rebuilds of the PGDB as individual gene annotations improve or as more complete versions of the genome become available. In addition, PlasmoCyc can store the certainty of functional annotations as well as curator comments, so that individual users can decide which reactions they believe to exist on the basis of current evidence.
By assembling individual protein functions into pathways, we derive a more complex understanding of an organism. PlasmoCyc links genomic data to protein annotation, to enzymatic-reactions, to pathways, and to additional sources of information. The addition of biological knowledge about when proteins are expressed (gene expression and proteomics experiments) and where the proteins are located (computational predictions and localization experiments) will help us refine PlasmoCyc by helping dissect out variant pathways from our current pathways and determining their biological context. With PlasmoCyc publicly available, the malaria community can participate in the curation process, initially by direct communication, but perhaps in the future through automated submissions. We plan to update PlasmoCyc by adding protein localization information on the basis of predicted and experimentally determined localization and by adding life-cycle stage-specific expres sion information on the basis of microarray experiments. The next version of PlasmoCyc should be released within the next year.
METHODS
An initial version of PlasmoCyc was built automatically using the PathoLogic program, which takes as input the annotated genome of an organism (i.e., the list of genes and proteins and their known or putative functions) and produces a PGDB containing pathways inferred to be in the organism on the basis of a reference database of previously described pathways. The genomic sequences for P. falciparum3D7 and their functional annotations were obtained from PlasmoDB (version 4.1).
We provided the genomic sequence, the mapping of each protein to a chromosome or contig, and the functional annotation of each protein. PathoLogic first maps proteins to specific reactions in the reference pathway database (MetaCyc) that contains reactions and pathways from many different organisms and most reactions specified in the EC hierarchy. Identification of a protein as an enzyme for a particular reaction is considered as evidence that the reaction occurs in the organism. Such identification can be made on the basis of EC number annotation. If no EC number is provided, then we assign proteins to reactions based on analysis of the text of the functional annotation. Whereas many enzymes can be assigned automatically in this fashion, some need to be assigned manually (using clues such as enzyme names ending in “-ase” or partial name matches). In some cases, the pathway database must be augmented with new reactions, on the basis of reports in the primary literature.
When there is evidence for a reaction in a reference pathway, PathoLogic infers that the pathway is present (except in cases where pathways are marked as variants of each other; in which case, the variant with the most evidence is inferred to be present). Due to this intentionally liberal computational inference of pathways, manual pruning of pathways is required to eliminate false-positive pathways. A pathway evidence report helps decide which pathways to prune. Pathway glyphs graphically summarize the evidence for a particular pathway in an organism. The pathway evidence for the pathway folate biosynthesis is shown in Figure 5. To prune out improbable pathways, we required that (1) No pathways with evidence for reactions that are unique to the pathway are removed; (2) if the set of reactions (for which there is evidence) in a pathway is a proper subset of the reactions (for which there is evidence) in another pathway, the pathway is deleted; (3) in the case that two pathways contain the same set of reactions, we prune out the pathway with more reactions for which there is no evidence.
Figure 5.

Pathway evidence report for folic acid biosynthesis pathway. The pathway glyph shows reactions with enzyme present in the genome and unique to the pathway (green), reactions with enzyme present but nonunique (present in other pathways—orange), reactions with enzyme absent and unique to the pathway (black), reactions with enzyme absent and present in other pathways (blue), and spontaneous reactions (pink). In this example, of the total of 15 reactions that make up the pathway, 10 have enzyme present, and only two of these participate in other pathways. The other pathways are listed at right.
PathoLogic can only infer the presence of pathways if they are in the reference pathway database. We added new pathways based on literature and the Malaria Metabolic Pathways resource manually. In addition, we looked at the proteins that weren't assigned to any reactions automatically and tried to assign them to reactions on the basis of the literature.
To access whether the distribution of validated drug targets across chokepoints and nonchokepoints is different with statistical significance, we determine the probability of having three or fewer proposed drug targets assigned to nonchokepoint enzymes under the null hypothesis of random assortment. The exact probability can be computed:
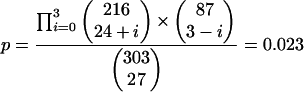 |
To determine sequence similarity between P. falciparum enzymes and human enzymes, we ran stand-alone BLAST downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/) against the Ensembl Human Genome Peptide database Homo_sapiens.NCBI34.pep.fa downloaded from (ftp://ftp.ensembl.org/pub/current_human/data/fasta/pep/), using default parameters. For enzymes with isozymes or subunit structure, any isozyme or subunit with homology classified the entire enzyme as having sequence similarity. To access whether the distribution of enzymes without sequence similarity to a human enzyme is different across validated drug targets and all other enzymes, we calculate a p-value as above, P = 0.032.
Acknowledgments
R.B.A. is supported by the Burroughs Wellcome Fund. I.Y. is supported by NIH 5T32GM07365. S.T. acknowledges the support of the Medical Research Council (UK). P.D.K. is funded by NIH grant R01-HG02729-01. Thanks to Hagai Ginsburg for insights about malaria metabolic pathways. Thanks to Christos Ouzounis for assistance in providing GeneQuiz annotations.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2050304. Article published online before print in April 2004.
References
- Ancelin, M.L. and Vial, H.J. 1986. Quaternary ammonium compounds efficiently inhibit Plasmodium falciparum growth in vitro by impairment of choline transport. Antimicrob. Agents Chemother. 29: 814-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade, M.A., Brown, N.P., Leroy, C., Hoersch, S., de Daruvar, A., Reich, C., Franchini, A., Tamames, J., Valencia, A., Ouzounis, C., et al. 1999. Automated genome sequence analysis and annotation. Bioinformatics 15: 391-412. [DOI] [PubMed] [Google Scholar]
- Bahl, A., Brunk, B., Crabtree, J., Fraunholz, M.J., Gajria, B., Grant, G.R., Ginsburg, H., Gupta, D., Kissinger, J.C., Labo, P., et al. 2003. PlasmoDB: The Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 31: 212-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker Jr., R.H., Metelev, V., Rapaport, E., and Zamecnik, P. 1996. Inhibition of Plasmodium falciparum malaria using antisense oligodeoxynucleotides. Proc. Natl. Acad. Sci. 93 514-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojang, K.A., Milligan, P.J., Pinder, M., Vigneron, L., Alloueche, A., Kester, K.E., Ballou, W.R., Conway, D.J., Reece, W.H., Gothard, P., et al. 2001. Efficacy of RTS,S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: A randomized trial. Lancet 358: 1927-1934. [DOI] [PubMed] [Google Scholar]
- Bowman, S., Lawson, D., Basham, D., Brown, D., Chillingworth, T., Churcher, C.M., Craig, A., Davies, R.M., Devlin, K., Feltwell, T., et al. 1999. The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature 400: 532-538. [DOI] [PubMed] [Google Scholar]
- Brown, G.V. and Reeder, J.C. 2002. Malaria Vaccines. Med. J. Australia 177: 230-231. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, D., Schuster, S.M. and Chakrabarti, R. 1993. Cloning and characterization of subunit genes of ribonucleotide reductase, a cell-cycle-regulated enzyme, from Plasmodium falciparum. Proc. Natl. Acad. Sci. 90: 12020-12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti, D., Da Silva, T., Barger, J., Paquette, S., Patel, H., Patterson, S., and Allen, C.M. 2002. Protein farnesyltransferase and protein prenylation in Plasmodium falciparum. J. Biol. Chem. 277: 42066-42073. [DOI] [PubMed] [Google Scholar]
- Darkin-Rattray, S.J., Gurnett, A.M., Myers, R.W., Dulski, P.M., Crumley, T.M., Allocco, J.J., Cannova, C., Meinke, P.T., Colletti, S.L., Bednarek, M.A., et al. 1996. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. 93: 13143-13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P.A., Cochran, D.A., Emmerson, B.T., and Gordon, R.B. 1993. Inhibition of Plasmodium falciparum hypoxanthine-guanine phosphoribosyltransferase mRNA by antisense oligodeoxynucleotide sequence. Mol. Biochem. Parasitol. 60: 153-156. [DOI] [PubMed] [Google Scholar]
- Flores, M.V., Atkins, D., Wade, D., O'Sullivan, W.J., and Stewart, T.S. 1997. Inhibition of Plasmodium falciparum proliferation in vitro by ribozymes. J. Biol. Chem. 272: 16940-16945. [DOI] [PubMed] [Google Scholar]
- Gardner, M.J., Tettelin, H., Carucci, D.J., Cummings, L.M., Aravind, L., Koonin, E.V., Shallom, S., Mason, T., Yu, K., Fujii, C., et al. 1998. Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science 282: 1126-1132. [DOI] [PubMed] [Google Scholar]
- Gardner, M.J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R.W., Carlton, J.M., Pain, A., Nelson, K.E., Bowman, S., et al. 2002a. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419: 498-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, M.J., Shallom, S.J., Carlton, J.M., Salzberg, S.L., Nene, V., Shoaibi, A., Ciecko, A., Lynn, J., Rizzo, M., Weaver, B., et al. 2002b. Sequence of Plasmodium falciparum chromosomes 2, 10, 11, 14. Nature 41: 531-534. [DOI] [PubMed] [Google Scholar]
- Genton, B., Anders, R.F., Alpers, M.P. and Reeder, J.C. 2003. The malaria vaccine development program in Papua New Guinea. Trends Parasitol. 19: 264-270. [DOI] [PubMed] [Google Scholar]
- Hall, N., Fung, E., White, O., and Berriman, M. 2002. Sequence of Plasmodium falciparum chromosomes 1, 3–9 and 13. Nature 419: 531-534. [DOI] [PubMed] [Google Scholar]
- Hanada, K., Palacpac, N.M., Magistrado, P.A., Kurokawa, K., Rai, G., Sakata, D., Hara, T., Horii, T., Nishijima, M., and Mitamura, T. 2002. Plasmodium falciparum phospholipase C hydrolyzing sphingomyelin and lysocholinephospholipids is a possible target for malaria chemotherapy. J. Exper. Med. 195: 23-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, S.L., Bancroft, W.H., Gottlieb, M., James, S.L., Burroughs, E.C., Stephenson, J.R., and Morgan, M.J. 1997. Funding from malaria genome sequencing. Nature 387: 647. [DOI] [PubMed] [Google Scholar]
- Hyman, R.W., Fung, E., Conway, D.J., Kurdi, O., Mao, J., Miranda, M., Nakao, B., Rowley, D., Tamaki, T., Wang, F., et al. 2002. Sequence of Plasmodium falciparum chromosome 12. Nature 419: 534-537. [DOI] [PubMed] [Google Scholar]
- Jiang, L., Lee, P.C., White, J., and Rathod, P.K. 2000. Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob. Agents Chemother. 44: 1047-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. and Goto, S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28: 27-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D. 2000. An ontology for biological function based on molecular interactions. Bioinformatics 16: 269-285. [DOI] [PubMed] [Google Scholar]
- Karp, P.D., Riley, M., Saier, M., Paulsen, I.T., Paley, S.M., and Pellegrini-Toole, A. 2000. The EcoCyc and MetaCyc databases. Nucleic Acids Res. 28: 56-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D., Paley, S., and Romero, P. 2002a. The Pathway Tools Software. Tenth International Conference on Intelligent Systems in Molecular Biology. Oxford University Press, Edmonton, Alberta, Canada.
- Karp, P.D., Riley, M., Paley, S.M., and Pellegrini-Toole, A. 2002b. The MetaCyc database. Nucleic Acids Res. 30: 59-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kicska, G.A., Tyler, P.C., Evans, G.B., Furneaux, R.H., Schramm, V.L., and Kim, K. 2002. Purine-less death in Plasmodium falciparum induced by immucillin-H, a transition state analogue of purine nucleoside phosphorylase. J. Biol. Chem. 277: 3226-3231. [DOI] [PubMed] [Google Scholar]
- Kissinger, J.C., Brunk, B.P., Crabtree, J., Fraunholz, M.J., Garjria, B., Milgram, A.J., Pearson, D.S., Schug, J., Bahl, A., Diskin, S.J., et al. 2002. The Plasmodium genome database. Nature 419: 490-492. [DOI] [PubMed] [Google Scholar]
- Krnajski, Z., Gilberger, T.W., Walter, R.D., Cowman, A.F., and Muller, S. 2002. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 277: 25970-25975. [DOI] [PubMed] [Google Scholar]
- Krungkrai, J., Kanchanarithisak, R., Krungkrai, S.R., and Rochanakij, S. 2002. Mitochondrial NADH dehydrogenase from Plasmodium falciparum and Plasmodium berghei. Exper. Parasitol. 100: 54-61. [DOI] [PubMed] [Google Scholar]
- Lell, B., Ruangweerayut, R., Wiesner, J., Missinou, M.A., Schindler, A., Baranek, T., Hintz, M., Hutchinson, D., Jomaa, H., and Kremsner, P.G. 2003. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 47: 735-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Q., Katakura, K., and Suzuki, M. 2002. Inhibition of mitochondrial and plastid activity of Plasmodium falciparum by minocycline. FEBS Lett. 515: 71-74. [DOI] [PubMed] [Google Scholar]
- McRobert, L. and McConkey, G.A. 2002. RNA interference (RNAi) inhibits growth of Plasmodium falciparum. Mol. Biochem. Parasitol. 119: 273-278. [DOI] [PubMed] [Google Scholar]
- Meierjohann, S., Walter, R.D., and Muller, S. 2002. Glutathione synthetase from Plasmodium falciparum. Biochem. J. 363: 833-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek, R., Larsen, N., Pusch, G.D., D'Souza, M., Selkov Jr., E., Kyrpides, N., Fonstein, M., Maltsev, N., and Selkov, E. 2000. WIT: Integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 28: 123-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto, S., Minakawa, N., Niizuma, S., Kim, H.S., Wataya, Y., and Matsuda, A. 2002. New neplanocin analogues. 12. Alternative synthesis and antimalarial effect of (6′R)-6′-C-methylneplanocin A, a potent AdoHcy hydrolase inhibitor. J. Med. Chem. 45: 748-751. [DOI] [PubMed] [Google Scholar]
- Sixsmith, D.G., Watkins, W.M., Chulay, J.D., and Spencer, H.C. 1984. In vitro antimalarial activity of tetrahydrofolate dehydrogenase inhibitors. Amer. J. Trop. Med. Hyg. 33: 772-776. [DOI] [PubMed] [Google Scholar]
- Suraveratum, N., Krungkrai, S.R., Leangaramgul, P., Prapunwattana, P., and Krungkrai, J. 2000. Purification and characterization of Plasmodium falciparum succinate dehydrogenase. Mol. Biochem. Parasitol. 105: 215-222. [DOI] [PubMed] [Google Scholar]
- Surolia, N. and Surolia, A. 2001. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 7: 167-173. [DOI] [PubMed] [Google Scholar]
- Thornalley, P.J., Strath, M., and Wilson, R.J. 1994. Antimalarial activity in vitro of the glyoxalase I inhibitor diester, S-p-bromobenzylglutathione diethyl ester. Biochem. Pharmacol. 47: 418-420. [DOI] [PubMed] [Google Scholar]
- Triglia, T. and Cowman, A.F. 1994. Primary structure and expression of the dihydropteroate synthetase gene of Plasmodium falciparum. Proc. Natl. Acad. Sci. 91: 7149-7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller, R.F., Ralph, S.A., Reed, M.B., Su, V., Douglas, J.D., Minnikin, D.E., Cowman, A.F., Besra, G.S., and McFadden, G.I. 2003. A type II pathway for fatty acid biosynthesis presents drug targets in Plasmodium falciparum. Antimicrob. Agents Chemother. 47: 297-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanidworanun, C., Nagel, R.L., and Shear, H.L. 1999. Antisense oligonucleotides targeting malarial aldolase inhibit the asexual erythrocytic stages of Plasmodium falciparum. Mol. Biochem. Parasitol. 102: 91-101. [DOI] [PubMed] [Google Scholar]
- Webster, H.K. and Whaun, J.M. 1982. Antimalarial properties of bredinin. Prediction based on identification of differences in human host-parasite purine metabolism. J. Clin. Invest. 70: 461-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization.1993. A global strategy for malaria control. World Health Organization, Geneva.
- Wright, P.S., Byers, T.L., Cross-Doersen, D.E., McCann, P.P., and Bitonti, A.J. 1991. Irreversible inhibition of S-adenosylmethionine decarboxylase in Plasmodium falciparum-infected erythrocytes: Growth inhibition in vitro. Biochem. Pharmacol. 41: 1713-1718. [DOI] [PubMed] [Google Scholar]
- Zidovetzki, R., Sherman, I.W., Prudhomme, J., and Crawford, J. 1994. Inhibition of Plasmodium falciparum lysophospholipase by anti-malarial drugs and sulphydryl reagents. Parasitology 108: 249-255. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://ftp.ncbi.nlm.nih.gov/blast/executables/; BLAST executables.
- http://plasmocyc.stanford.edu; Plasmo Cyc.
- http://biocyc.org; BioCyc.
- http://plasmodb.org; PlasmoDB.
- http://sites.huji.ac.il/malaria/; Malaria Parasite Metabolic Pathways, Hagai Ginsburg.