

Abstract
Systems for managing genomic data must store a vast quantity of information. Ensembl stores these data in several MySQL databases. The core software libraries provide a practical and effective means for programmers to access these data. By encapsulating the underlying database structure, the libraries present end users with a simple, abstract interface to a complex data model. Programs that use the libraries rather than SQL to access the data are unaffected by most schema changes. The architecture of the core software libraries, the schema, and the factors influencing their design are described. All code and data are freely available.
The storage and manipulation of genome sequence and its associated information are at the heart of any genome informatics project. Such a project must provide persistent data storage and programmatic ways to access its information. Many bioinformatics applications use flat files as input and output, which has led to the development of several file-based methods of storage. For example, hierarchical directory structure has been used to organize the information (Wendl et al. 1998), and such a system was the basis of the project that developed into Ensembl. The Gene Ontology (Ashburner et al. 2000), and Pfam (protein families database of alignments), projects (Bateman et al. 1999) successfully use tools such as CVS (Concurrent Versions System) and RCS (Revision Control System) as layers of abstraction over file systems. Similarly, programmatic access to information has often grown organically, with individual programs or scripts interacting with the persistent data. One major drawback with an ad hoc scripting approach to persistent storage access is that it encourages the explosion of small, redundant scripts with no organized central path of access. Over any length of extended development, and as the number of personnel increases, such a system rapidly becomes unworkable.
At the onset of the Ensembl project there were two available bioinformatics frameworks with a well-structured approach to storing and manipulating genome data: ACeDB (Eeckman and Durbin 1995) and the NCBI toolkit (Wheeler et al. 2001). The ACeDB project was a source of many of our original ideas for modeling genome information, but we did not think that its binary-file-based method of persistent storage would scale to accommodate the human genome. The NCBI toolkit requires predominantly C-based programmatic access, and would have resulted in a longer development time and a steeper learning curve for biologists unfamiliar with the C language. In addition, the primary mechanism of persistence storage offered by the toolkit (ASN.1 binary files) still requires an indexing and large-scale storage system for efficiency. It was decided to use a relational database management system (RDBMS) because of its numerous benefits over a file-based approach. A relational database scales well, is accessible to users via a well-known query language (SQL), provides a means to index data for rapid queries, and allows many concurrent users to access the data at once.
Two reliable open-source RDBMSs were considered for persistent storage: MySQL and PostgreSQL. MySQL was chosen because of its faster performance and better long-string support, and a Perl application programming interface (API) was developed as the primary method of programmatic access.
Since the inception of the project, several other data storage and API solutions for genome information have become available. In particular, the GadFly project (Mungall et al. 2002) was started at approximately the same time as Ensembl, and we have had a productive exchange of ideas. The Grand Unified Schema (Bahl et al. 2003) also matured into a successful project. There are several effective in-house RDBMS projects for storing genome information, such as the Saccharomyces Genome Database (Weng et al. 2003) and Mouse Genome Informatics (Baldarelli et al. 2003). Although these projects provide excellent Web access to their resources, their schemas and code bases are either unavailable or their use is limited. The WormBase project has been migrating from a pure ACeDB system toward a mixed ACeDB and RDBMS (Stein et al. 2001). Several of these projects are now coordinating via the Generic Model Organism Database (Stein et al. 2002) consortium to provide common tools. The UCSC genome browser uses a relational database and a C-language implementation (Karolchik et al. 2003). Finally, there have been several commercial genome management products based on proprietary technology from Softberry, Celera, and Doubletwist.
Ensembl enjoyed interacting with many of these other developers, and freely shares all of its code and ideas. The rest of this article describes Ensembl's database and API, which have been the result of four years of development. Some decisions were well thought out and stood the test of time; others were due to the rapid pace of development, in particular at the start of the project. The Ensembl system has proven flexible enough to be adopted for many genome projects. In house, Ensembl is currently used to annotate or display nine species, and externally the Ensembl system has been extended for use with the genomes of many organisms including Arabidopsis, rice, and numerous pathogens.
Design Process
The Ensembl database is used in two distinct phases and has two resultant patterns of usage. The first phase is the production of the data and involves a high volume of both reads and writes to the database. The second phase, the presentation of the data by a Web interface, requires rapid read-only access to the database. It was decided to serve both phases with the same schema and programming interface despite their divergent patterns of usage. A single code base has the advantages that there is less code to maintain, it removes the necessity of a postdata production denormalization, and it leads to more robust and flexible code. It does, however, prevent the use of certain database speed optimization methods and leads to a compromise between normalization of data, query optimization, and development time.
We avoided autogenerating the code or schema from a higher-level language (e.g., UML, XML) because we found that autogenerated systems were too slow and that they invariably required customization for particular use cases.
Database
All annotation and sequence data are stored in an MySQL RDBMS. The tables defined in the Ensembl schema can be divided into three functional categories: tables for the storage of DNA and assemblies, tables for the storage of computed features and genes, and tables containing miscellaneous information. Figure 1 provides a general outline of the database structure.
Figure 1.
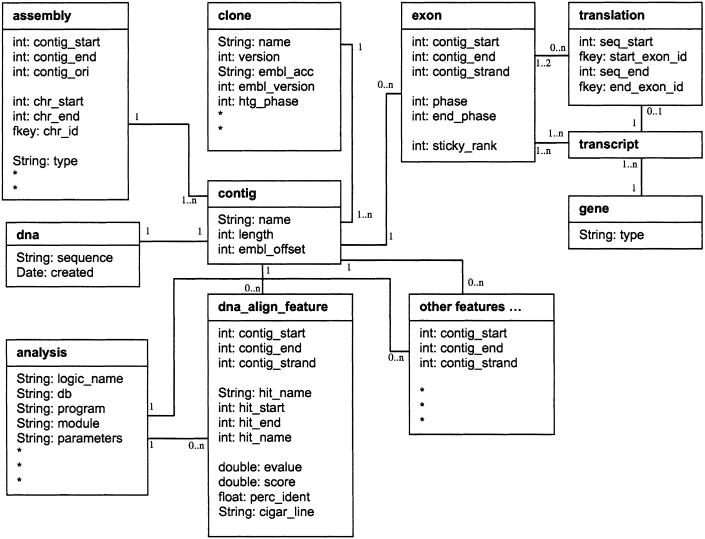
Entity relationship model of the Ensembl schema. Tables are represented as divided rectangles consisting of a boldface table name at the top and a list of table attributes and attribute types below. Internal identifiers and join tables are omitted. Relationships between tables are represented by lines labeled with the relationship type: 1___1...n one to one-or-many relationship; 1___1 one-to-one relationship; 1___0...n one to zero-or-many relationship; 1...n___1...n one-or-many to one-or-many relationship; 1...2___0...n one-or-two to zero-or-many relationship.
The basic unit of sequence is stored in the contig table. It contains information about contiguous sequence from BAC clone files in the EMBL database. The string representation of the DNA sequence for a contig is stored in the dna table. Each contig row references a row in the clone table that provides additional detail about the BAC clone. Unfinished clones are comprised of multiple contig rows; a finished clone consists of a single contig. The information needed to assemble chromosomal sequence from the set of contig sequences is stored in the assembly table.
Various features are positioned on the genome sequence and stored in database tables. All features define a genomic position through a reference to a contig and start and end coordinates on the contig. Example feature tables are dna_align_ feature and protein_align_feature for alignments from similarity searches, repeat_feature for repeats, marker_feature for marker positions, prediction_transcript for ab initio gene (transcript) predictions, and simple_feature for general annotations with genomic positions. Some features contain additional, nonpositional information in related tables. For example, marker features have details about the marker in the marker table, biological mapping information in marker_map_ location, and alternative names in marker_synonym.
An innovation in the storage of similarity search results is the compression of gapped alignment information in the form of dense character strings. This was originally developed as an output format from Exonerate (G. Slater, unpubl.) and is known by its original acronym “cigar” (concise idiosyncratic gapped alignment report). Alignment features store the full extent of the gapped alignment and a cigar line. Each cigar line consists of an alternating series of numbers and letters, for example, 40M2I12M4D, with the letters standing for Match, Insertion, or Deletion. The number preceding each letter dictates the length of the match, insertion, or deletion; used together with the feature's start and end coordinates, the complete alignment can be reconstructed. Prior to the adoption of cigar lines, alignments in Ensembl were stored as multiple ungapped features, with a single row for each matching region of the alignment. The storage of alignments as a single gapped row has reduced the size of Ensembl's similarity tables by 60%.
The more complex structure of a gene is distributed over multiple tables. A gene from Ensembl's perspective is a set of transcripts that share at least one exon. This is a more limited definition than, for example, a genetic locus, but it describes a relationship that can be easily identified computationally. A row in the gene table relates to one or more transcript table rows, each of which references a list of exon_transcript table rows that describe the ordering of exons in each transcript. Exons and their associated genomic positions are stored in the exon table. Transcripts reference zero or one translation table rows that describe the composition of untranslated regions and coding sequences. Pseudogenes and ncRNAs are examples of transcripts without translations. For some genes, we provide a gene_description that is derived from a SWISS-PROT database entry with a sufficiently similar protein sequence. Evidence for predicted exons is provided by links to alignment features (in the dna/protein_align_feature tables) via entries in the supporting_feature table.
Exons are predicted with chromosomal positions but stored with contig positions. The chromosomal coordinate system changes with each new assembly of a genome, and is thus more volatile than the contig coordinate system. Storing exons in contig coordinates ensures that unchanged exons have unchanged coordinates. One drawback to this approach is that exons may span multiple contigs when converted from chromosomal coordinates. Exons that cross contig boundaries occupy multiple rows in the exon table and are distinguished from ordinary exons by a sticky_rank attribute. When these split exons are retrieved from the database they are reassembled into a single exon by the API software.
Across different releases of human genome assemblies and other sequence data, Ensembl provides changing gene predictions. To allow the user to track a particular gene prediction despite changing coordinates, all gene-related predictions are assigned stable identifiers. These are stored in the gene_stable_id, exon_stable_id, transcript_stable_id, and translation_ stable_id tables. Between two versions of a genome we determine the correspondence between the old and new predictions, taking into account changes in genomic position or sequence. New predictions with a sufficiently high similarity to a previously made prediction inherit the previous prediction's stable identifier.
The database follows some simple naming conventions to facilitate easier understanding and maintenance. Tables generally have integer primary keys that follow a column-naming convention of tablename_id, and foreign keys referencing other tables' primary keys use the same column names to be easily identifiable. Following this convention, every table with a locatable annotation has a contig_id column that is a foreign key referencing the contig_id primary key of the contig table. Additionally, each of the feature tables uses the same column names, contig_start, contig_end, and contig_strand, to describe the precise locations of features on a given contig.
Code
Ensembl's database access layer is written in Perl because of its numerous advantages as an implementation language. Perl is widely used in the bioinformatics and biology community, and it is a language well suited for writing Web applications. Another important factor was that Ensembl was originally created out of a Perl-based human annotation project, and parts of the existing software could be reused.
Adoption of Perl also enabled Ensembl to leverage the existence of the BioPerl project (Stajich et al. 2002). BioPerl provided a base for an initial object model and aided in the dumping and parsing of flat files. As Ensembl has become more complex over its lifetime, this dependence on BioPerl has slowly diminished. Currently there is very little BioPerl dependence inside Ensembl, and we are considering replacing the hard dependencies and producing a separate Ensembl-to-BioPerl bridge.
However, some aspects of Perl are not well suited for a software project of Ensembl's size. Whereas weak typing allows for rapid program development, absence of compile time checking of function prototypes and variable types is a steady source of runtime errors. Another disadvantage of Perl is its reference-count-based garbage collector, which effectively limits the use of circular references. Variables that are part of a circular reference structure are never garbage-collected and can introduce potentially serious memory leaks. Avoidance of circular reference memory leaks has necessitated some compromises to the overall system design. As described below, a Java API was developed to test ideas and allow gradual progression to a more strongly typed language.
Ensembl models real-world biological constructs as data objects. For example, Gene objects represent genes, Exon objects represent exons, and RepeatFeature objects represent repetitive regions. Data objects provide a natural, intuitive way to access the wide variety of information that is available in a genome. All information relating to a data object can be obtained by querying the object's methods. As an example, a Transcript object can provide the user with its identifier, its exons and its translation, and the like.
Data representation and database access are cleanly separated in the Ensembl API. Database access code resides exclusively in adaptor classes that create data objects. Each data object x (e.g., a Gene) has an adaptor class xAdaptor (e.g., GeneAdaptor) responsible for generating database queries on tables associated with x and instantiating objects of type x. Adaptors provide multiple ways to retrieve and store data objects from the database via methods that follow strict naming conventions. This separation of logic enables adaptor classes to share query generation code and insulates data objects from underlying schema changes. The modularity of this design also makes it easy to add new data objects to the system.
The decoupling of database logic additionally allows the transparent substitution of one data source for another. The abstraction of the data sources allows them to be interchanged to address particular flexibility and performance needs. This tactic is utilized by the Ensembl system to improve the retrieval speed of some features through the use of a query-optimized denormalized database (known as the “lite” database). Retrieval from the optimized database is facilitated by ProxyAdaptor classes that dynamically decide to forward requests to either the default data source or to the optimized data source.
The DBAdaptor is a specialized adaptor that maintains a connection to the database and acts as a factory for object adaptors. The centralized object adaptor creation code can ensure that only a single object adaptor of each type is created per database. This enables the object adaptors to cache instances of the features they retrieve and to improve overall performance.
A Container class alleviates memory leaks created by the circular references between the DBAdaptor and its object adaptors. Upon instantiation, DBAdaptor objects are transparently wrapped in a Container object that dynamically forwards method calls destined for the enclosed DBAdaptor. The Container object is external to the circular reference hierarchy, and it is responsible for breaking the circular references after it passes out of scope. Users of the API are, in most circumstances, unaware of the existence of the Container object, and it serves as an effective, hidden solution to the memory leak problem.
Specific regions of genomic sequence are represented as either Slice or RawContig objects. Feature objects with genomic locations can be retrieved using method calls on either of these sequence objects. A RawContig object represents a single contiguous piece of sequence and a single row in the contig table. A Slice object does not correspond directly to a particular table in the database, but represents a portion of chromosomal sequence assembled from smaller contigs that may contain gaps. All database access code resides in adaptor classes; Slice and RawContig methods merely delegate object and sequence retrieval to the responsible adaptors.
In the unfinished human genome, RawContig objects were the primary method of sequence and feature retrieval. As more assembled sequence data have become available, Slice objects have emerged as the predominant method of access. Even when Slices will be based on completely finished genomes with no underlying contigs, their usage will remain the same. This is one major advantage of an API-based approach to genomic data storage.
The primary coordinate system in which all database features are stored has remained contig-based despite the common usage of a chromosomal-based coordinate system. Rather than distributing coordinate transformation code throughout the code base, we introduced a general Mapper class that encapsulates coordinate transformation between two sequences. A more specific AssemblyMapper class utilizes the Mapper object and the contents of the assembly table to translate from contig-based coordinates to chromosomal coordinates and vice versa.
Object creation and coordinate transformation have an impact on the speed of the API. An application that uses the API will be slower than one that uses SQL to access the database directly. The Perl API requires ∼0.40 sec to retrieve 1000 features from a 1-MB region; the Java code version needs similar time. To retrieve the same data as arrays using SQL via Perl DBI requires ∼0.15 sec. For our dominant use cases we find this is an acceptable performance decrease.
The following code is a typical example for EnsEMBL database access.
# create a database connection my $db=Bio::EnsEMBL::DBSQL::DBAdaptor→new (-host ⇒ `ensembldb.ensembl.org', -user ⇒ `anonymous', -dbname ⇒ `homo_sapiens_core_19_34' ); # get a slice adaptor my $sliceadaptor=$db→get_SliceAdaptor(); # get a slice from 10MB to 20MB on Chromosome 1 my $slice=$slicedaptor→fetch_by_chr_start_end ( `1', 10_000_000, 20_000_000 ); # retrieve some features from the slice my $features=$slice→get_all_protein_align_features ( `SWALL' );
We aim to have the greatest possible ease of use for the Perl API. It should not just support our gene prediction process and Web display code, but it should also make it simple for researchers to perform their own genome analysis tasks. We have a tutorial document available online (http://www.ensembl.org/Docs/ensembl_tutorial.pdf) and open access to a MySQL server (ensembldb.ensembl.org); all source code is available through anonymous CVS.
Perl suffers from certain disadvantages as an implementation language for a large-scale project. Java overcomes many of these problems and has the benefits of compile time type checking, enforced interfaces, multi threading, better support for graphical user interfaces, and correct garbage collection of circularly referenced objects. We have written a Java version of the core Ensembl API that offers very similar data access logic to the Perl API. Additionally, the Java API separates interface from implementation for all standard data objects and adaptors and thus allows for transparent alternative implementations. It is used by the standalone genome browser Apollo, and it is used internally for the stable identifier mapping process and by various other projects such as Toucan (Aerts et al. 2003) and Sockeye (Montgomery et al. 2004).
The above Perl code example would look like the following written in Java:
public class FeatureRetrieval { public static void main( Strings[] args) { Driver d = new MySQLDriver( “ensembldb.ensembl.org”, “homo_sapiens_core_19_34”, “anonymous” ); Location loc = new AssemblyLocation ( “1”, 10000000, 20000000 ); List proteinAlignments = d.getDnaProteinAlignmentAdaptor(). fetch(loc, “SWALL” ); } }
Ensembl releases about 35 databases on a monthly basis. To ensure their relational integrity and to validate that they are populated with reasonable data, several SQL-based tests are performed using a Java quality assurance system named ensjhealthcheck. The system consists of groups of tests individually encapsulated into Java classes. It is relatively easy, even for Java newcomers, to write integrity checks that are automatically detected and used. Since the introduction of the ensj-healthcheck system, better consistency over schemas has been achieved, and it has become easier to detect data errors at an early stage. Newly detected errors are added to the existing suite of test cases and are prevented from reoccurring.
Biological Relevance
Genomic data pose serious challenges to biologists: both the mundane challenges of how to manipulate the data and the scientific challenges of how to find and use the information contained within it. In the case of mammalian genomes, the mundane aspects can come to dominate researchers' time. The system described by this article enables scientists to work effectively with the genome by providing an efficient means to access vast amounts of data. The other articles in this issue detail how this software system is already used to create and to display genomic information for biologists. The system has been designed for maximum utility by researchers. Ensembl's relational database allows the flexible retrieval of nonredundant information. For example, researchers can retrieve genes using several different constraints such as a genomic location or an HUGO identifier. An increasing number of laboratory biologists have Perl, Java, or Python programming skills. The software system developed as part of this project allows simple scripts to be developed by external researchers and eliminates the overhead required to construct a framework for access to this genomic information. High-throughput technologies used in smaller laboratory settings demand specialized data analysis software to be written to integrate experimental results with reference data sets such as the genome. For example, researchers investigating specific gene families can run primer design programs across the genome to provide a high-throughput system to survey mutations.
Future
The current Ensembl system is biased toward a clone-based genome project. Every genome imported into Ensembl requires entries in the clone, contig, chromosome, and assembly tables. Whole-genome shotgun assemblies do not naturally fit into this mould, either because they do not have clones or because the assembly may be fragmented into thousands of scaffolds instead of chromosomes.
The Ensembl sequence storage system will be improved by replacing the contig, clone, and chromosome tables with a single general sequence region table. A modified assembly table will describe the composition of arbitrary sequence regions rather than the makeup of chromosomes from contigs. Locatable features will be stored with coordinates relative to sequence regions and will not be limited to storage in the contig coordinate system. Effectively, bias toward particular coordinate systems will be removed; the system will become more flexible and will accommodate a wider variety of methods of sequencing and assembly.
The Ensembl system will also be extended to include support for alternative sequence regions. This will include the ability to represent structural haplotypes with highly divergent sequence (e.g., the MHC region) and pseudoautosomal regions on sex chromosomes.
The generalization of the sequence and assembly storage and the addition of new features are expected to require only minor changes to the existing programming interface. Users will be shielded from the low-level database and code changes by the layers of abstraction in the existing system. The current API will continue to function in nearly all cases, but some functions and naming conventions will slowly replace the current ones. Retrieval speed should improve as features can be calculated and stored in the coordinate system in which they are predominantly requested.
There exists a myriad of ways to store and manipulate genome sequence; the Ensembl system is a robust and scaleable solution. It is reassuring that the core concepts from the project inception (e.g., genes, transcripts, and features) have remained intact, despite the progressive evolution toward the existing maintainable and scaleable architecture. A similar dynamic of evolutionary development is expected to occur over the next five years with the Ensembl database and API providing the central support for genome information.
Acknowledgments
The Ensembl group is funded primarily by the Wellcome Trust with additional funding from EMBL and NIH-NIAID. We thank the other members of the Ensembl group for their support and patience during the development of the system. We also thank Nicole Redaschi, Simon Mercer, and Chris Mungall for insightful design comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1857204.
References
- Aerts, S., Thijs, G., Coessens, B., Staes, M., Moreau, Y., and De Moor, B. 2003. Toucan: Deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 31: 1753-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl, A., Brunk, B. Crabtree, J., Fraunholz, M.J., Gajria, B., Grant, G.R., Ginsburg, H., Gupta, D., Kissinger, J.C., Labo, P., et al. 2003. PlasmoDB: The Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 31: 212-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldarelli, R.M., Hill, D.P., Blake, J.A., Adachi, J., Furuno, M., Bradt, D., Corbani, L.E., Cousins, S., Frazer, K.S., Qi, D., et al. 2003. Connecting sequence and biology in the laboratory mouse. Genome Res. 13: 1505-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Finn, R.D., and Sonnhammer, E.L. 1999. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27: 260-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckman, F.H. and Durbin, R. 1995. AceDB and macace. Methods Cell Biol. 48: 583-605. [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31: 51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, S.B., Astakhova, T., Bilenky, M., Birney, E., Fu, T., Hassel, M., Melsopp C., Rak, M., Robertson, A.G., Sleumer, M., et al. 2004. Sockeye: A 3D environment for comparative genomics. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Mungall, C.J., Misra, S., Berman, B.P., Carlson, J., Frise, E., Harris, N., Marshall, B., Shu, S., Kaminker, J.S., Prochnik, S.E., et al. 2002. An integrated computational pipeline and database to support whole-genome sequence annotation. Genome Biol. 3: RESEARCH0081. [DOI] [PMC free article] [PubMed]
- Stajich, J.E., Block, D., Boulez, K., Brenner, S.E., Chervitz, S.A., Dagdigian, C., Fuellen, G., Gilbert, J.G., Korf, I., Lapp, H., et al. 2002. The Bioperl Toolkit: Perl modules for the life sciences. Genome Res. 12: 1611-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L., Sternberg, P., Durbin, R., Thierry-Mieg, J., and Spieth, J. 2001. WormBase: Network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 29: 82-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L.D., Mungall, C., Shu, S., Caudy, M., Mangone, M., Day, A., Nickerson, E., Stajich, J.E., Harris, T.W., Arva, A., et al. 2002. The generic genome browser: A building block for a model organism system database. Genome Res. 12: 1599-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendl, M.C., Dear, S., Hodgson, D., and Hillier, L. 1998. Automated sequence preprocessing in large-scale sequencing projects. Genome Res. 8: 975-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, S., Dong, Q., Balakrishnan, R., Christie, K., Costanzo, M., Dolinski, K., Dwight, S.S., Engel, S., Fisk, D.G., Hong, E., et al. 2003. Saccharomyces Genome Database (SGD) provides biochemical and structural information for budding yeast proteins. Nucleic Acids Res. 31: 216-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, D.L., Church, D.M., Lash, A.E., Leipe, D.D., Madden, T.L., Pontius, J.U., Schuler, G.D., Schriml, L.M., Tatusova, T.A., Wagner, L., et al. 2001. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 29: 11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ensembl.org/Docs/ensembl_tutorial.pdf; Ensembl online tutorial document.